

Abstract
Easy access to next generation sequencing has enabled the rapid analysis of complex microbial populations. To take full advantage of these technologies, animal models enabling the manipulation of human microbiomes and the study of the impact of such perturbations on the host are needed. To this aim we are developing experimentally tractable and clinically relevant pig models of the human adult and infant gastro-intestinal tract. The intestine of germ-free piglets was populated with human adult or infant fecal microbial populations, and the piglets were maintained on solid or milk diet, respectively. Amplicons of 16S rRNA V6 region were deep-sequenced to monitor to what extent the transplanted human microbiomes changed in the pig. Within 24 h of transfer of human fecal microbiome to pigs, bacterial microbiomes rich in Proteobacteria emerged. These populations evolved toward a more diverse composition rich in Bacteroidetes and Firmicutes. In the experiment where infant microbiome was used, the phylogenetic composition of the transplanted bacterial population converged toward that of the human inoculum. A majority of sequences belonged to a relatively small number of operational taxonomic units, whereas at the other end of the abundance spectrum, a large number of rare and transient OTUs were detected. Analysis of fecal and colonic microbiomes originating from the same animal indicate that feces closely replicate the colonic microbiome. We conclude that the pig intestine can be colonized with human fecal microbiomes to generate a realistic model of the human GI tract.
Keywords: pig model, intestinal microbiome, 16S rRNA amplicon sequencing, principal coordinate analysis, microbial diversity
Introduction
Intestinal microbiomes play an important role in human health. Dysbiotic intestinal bacterial populations have been linked to a variety of health conditions,1,2 even with conditions not directly associated with the gastro-intestinal (GI) tract such as asthma,3 diabetes,4 disorders of the immune system,5,6 arthritis7 and pregnancy.8 Research on intestinal microbiota and their interaction with the host often focuses on human subjects or uses germ-free mice inoculated with human fecal microbiota. The search for associations between human phenotypes and microbiome is hampered by the many confounding variables inherent to human cohorts and by the small number of subjects typically included in such studies. Humans are genetically diverse, are exposed to many environmental factors and consume different diets, all factors which directly or indirectly impact the intestinal ecology. To circumvent these limitations and to study the effect of specific alterations of the intestinal microbiome, germ-free rodents have been populated with microorganisms originating from human feces.9-14 Rodent models, and especially murine models, offer many advantages over other species. The fact that many strains are inbred and the availability of genetically modified lines facilitate research aiming at elucidating the interaction between microbiome, genetic background and disease.15 On the other hand, rodent models are limited by the many physiological differences between rodents and primates. Most importantly, mice and rats typically do not display clinical manifestations seen in human enteric diseases.
The need for a clinically relevant model of the human GI tract is driving the search for non-rodent models. The advantage of pigs as an alternative model of human diseases lies in similar clinical manifestations and their susceptibility to many enteric pathogens afflicting humans. Efforts to develop porcine models benefit from extensive research on the use of pigs as source of tissues and organs for xenotransplantation.16 This research expands our knowledge on this species and has led to the production of transgenic pigs.17 It has also promoted the production of immune reagents to study the porcine immune response.
Piglets derived by C-section are colostrum-deprived and hence, unlike humans and rodents, are born agammaglobulinemic, with no interfering maternal antibodies. As with human infants, the pig's immune system at birth is fully developed but not fully functional, maturing within 7–10 d. Relevant features of the immune system include a distribution of Peyer’s patches which is similar to that seen in humans and play a central role in antigen sampling by M cells. Pigs are also immunologically similar to humans in their pattern of lymphocyte distribution in the body and at the mucosal surfaces. Intra-epithelial lymphocytes are also broadly similar to those of humans, mice and rats, with a majority of cytotoxic suppressor T cells and fewer T helper cells. The presence of gut microflora in these animals accelerates gut maturity and promotes a robust immune response.18-20 Experiments have been conducted in the pig to study the effect of gut microflora on the immune system, both in conventional and germ-free animals.21 The protective effect of probiotics on subsequent colonization with pathogenic bacteria has been demonstrated in this species.20,22
From a practical point of view, piglets learn to drink sterile milk formula directly from a trough. At three weeks of age they can be weaned onto solid diet. Litters of 8–12 piglets provide a good sample size. Newborn piglets weigh about 1 kg, which facilitates handling, bleeding, surgery, repeated sampling, measuring of temperature, heartbeat, respiratory rates, collection of intestinal content for microbiome analyses and of intestinal epithelial cells for analyzing the transcriptional response. Piglets derived prematurely can be used to study complications of pre-term births.
Here we describe results obtained from three experiments comprising eight germ-free piglets inoculated with human adult or infant fecal microbiome. The transplanted microbial populations were monitored for up to 35 d. Evidence supporting the successful transplantation of human microbiota is presented. The implications of these observations for the development of better models of the human GI tract are discussed.
Results
Overview
A summary of the pig experiments is shown in Table 1. A total of three experiments were performed; two experiments using fecal extracts from human adults and one with fecal extract from a 3 mo old breast-fed baby. Small portions of these extracts were stored at -80°C to enable multiple experiments with the same stock. In experiment 1, adult microbiota was transplanted into two pigs 8 d of age maintained on Similac milk replacer for the 20-d duration of the experiment. Experiment 2 and 3 were designed to emulate the infant and adult GI tract, respectively. In experiment 2, infant microbiota was inoculated on day 5 or 30 into four germ-free piglets maintained on Similac, whereas in experiment 3 two pigs were inoculated with adult microbiota on day 23 of age, three days after weaning them onto solid porcine diet.
Table 1. Summary of pig experiments.
| experiment 1 | experiment 2 | experiment 3 | ||||||
|---|---|---|---|---|---|---|---|---|
| inoculum |
adult |
infant |
adult |
|||||
| pig ID |
4 |
5 |
7 |
8 |
9 |
10 |
1 |
2 |
| age inoculateda |
8 |
8 |
5 |
5 |
30 |
30 |
23 |
23 |
| diet |
Similac |
Similac |
Similac |
Similac |
Similac |
Similac |
solid |
solid |
| day euthanized | 28 | 28 | 41 | 15 | 41 | 41 | 36 | 36 |
a age in days
Phylogeny
The phylum-level classifications of time series originating from five pigs from experiment 1−3 are shown in Figure 1. In general, a reduction in the proportion of Proteobacteria and expansion of Firmicutes was observed. This trend was particularly apparent in experiment 2, which likely reflects the relative high proportion of Proteobacteria in the infant inoculum, and the fact that fecal samples collected 24 h post-inoculation were analyzed in this experiment. The phylogenetic profile of the earliest sample from three other pigs included in this experiment were also rich in Proteobacteria (98%, 78%, 84%). The histograms for those animals are not shown because of the short time series (see Table 1). Samples collected on the day following inoculation were not available for experiment 1 and 3. We notice in experiment 2 and 3 a close resemblance in the taxonomic profile of the fecal and colonic microbiome, labeled “gut” in Figure 1. The minor differences between fecal and colonic profile are of the same magnitude as between replicate human profiles shown in experiment 2 and are thus likely to represent technical variability. Unclassified sequence reads were particularly abundant in experiment 3. As further discussed below, this observation is probably related to sequencing strategy based on 60-nt reads. Providing a more detailed view of the evolution of the transplanted human fecal microbiomes, genus-level taxonomic classifications for the same five pigs as shown in Figure 1 are shown in Table S1. The table shows raw counts of sequences assigned to each genus.

Figure 1. Phylum-level taxonomy of fecal and colonic bacterial populations from pigs inoculated with human fecal microbiomes. Experiment 1−3 refer to the three experiments described in the text and in Table 1. Phyla are color-coded as shown in the key. A total of 104 V6 16S sequence reads per sample were classified. The taxonomic time series of two pigs from experiment 1 and 3 are shown. A single time series is shown for experiment 2. The x axis shows pig age in days. Bars labeled “H” represent human inocula; samples obtained directly from the colon are labeled “gut.” The two left-most bars in experiment 2 are from independent replicate analyses of the infant fecal sample used as inoculum.
Analysis of sequence evenness and diversity
To compare the evenness of intestinal microbiota before and after transplant into pigs, rank-abundance plots for each experiment were drawn. These plots showed some loss of evenness, particularly in experiments 1 and 3, where fecal microbiome from adults was transferred to milk-fed pigs (Fig. 2). The pig microbiomes which showed little or no loss of evenness were in experiment 2, in which the microbiome of a breast-fed child was transplanted to pigs fed milk replacer. In experiment 3, adult microbiome transplanted into weaned pigs experienced some loss of evenness.
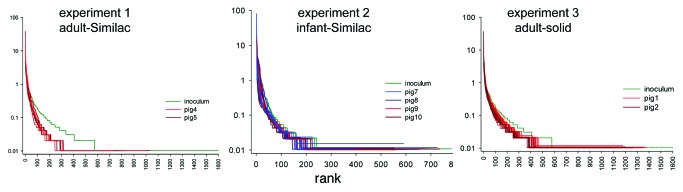
Figure 2. Rank-abundance plots of 16S sequence reads. Experiment 1−3 refer to the three experiments described in the text. Green indicates human sample. Samples collected from a same animal on subsequent days are coded with the same color as indicated in the keys. Some loss of diversity is apparent, particularly in experiment 1.
Alpha diversity was estimated using the Shannon index (Fig. 3). Consistent with the rank-abundance plots, experiment 2 shows that the pig microbiomes recovered to their original diversity after about 5 weeks in the pig. In contrast, in experiment 1 and 3 the pig microbiomes lost diversity, a trend that did not reverse itself over the duration of the experiment. As diversity in experiment 2 increased toward the end of the 41-d time series, it is conceivable that the same would have occurred in experiment 1 and 3 had the time series been extended to 41 d. The change in diet experienced by the microbiome in these two experiments could also have contributed to a loss in bacterial diversity.
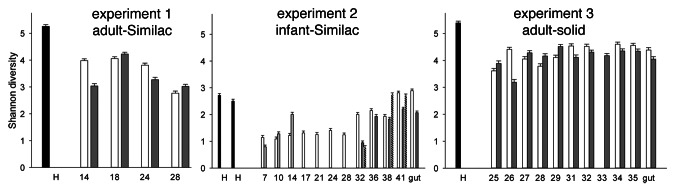
Figure 3. Diversity of pig intestinal microbiome OTUs. Diversity was estimated using the Shannon diversity index calculated with the natural log. Experiment 1−3 refer to the three experiments described in the text. The x axis shows pig age in days. Black bars indicate human inocula. Bars are shaded according to pig; experiment 1 and 3 included two pigs each, shown with white and gray bars, respectively. Four pigs were used in experiment 2, as indicated with white (pig 7), light gray (pig 8), dark gray (pig 9) and hatched bars (pig 10).
Analysis of Unifrac distances
PCoA was used to visualize weighted pairwise Unifrac distances. A separate analysis was performed for each of the 3 experiments to assess the divergence of the microbiome following transplant into pigs. To avoid data point compression, data from samples collected within 24 h of inoculation were excluded, as these populations were characterized by a high abundance of Proteobacteria. To visualize the trajectory of the transplanted fecal bacterial populations, data points from subsequent days were linked (Fig. 4). The PCoA plots illustrate the extent to which the transplanted fecal microbiomes evolved in the pig GI tract over a period of up to 35 d. Consistent with the evenness and diversity analyses shown in Figures 2 and 3, the data points from experiment 1 reveal a relatively large distance between inoculum and transplanted populations. Pig microbiomes collected at four time points over a period of 15 d did not converge toward the composition of the human inoculum. These experiment 1 trajectories contrast with those observed in experiment 2. In this experiment bacterial populations transplanted into four pigs converged toward the human inoculum, consistent with a similar diversity of the human inoculum and the pig microbiome 41 d post-inoculation. The trajectories for pigs 8 and 10 are only three and two days long, respectively, which makes it difficult to assess whether the microbiome would ultimately have converged toward the human inoculum. Pig 8 was euthanized on day 15 due to deteriorating health and for pig 10 only two fecal samples and the gut content were sequenced. In experiment 3, adult microbiome transplanted into 23-d old pigs fed a solid diet did not follow an obvious trajectory in the course of the time series lasting 12 d. Weighted Unifrac distances from the inoculum 2 d post-inoculation were 0.476 and 0.408 for pig 1 and pig 2, respectively. On day 35 of age weighted distances from the inoculum were almost unchanged at 0.421 and 0.451, for pig 1 and 2, respectively.

Figure 4. Principal Coordinate Analysis of 16S sequences from human fecal microbiota before and after transplant into germ-free pigs. The analysis is based on pairwise weighted Unifrac distances. Human microbiota are shown in green. Data points from each pig, color-coded as shown in the keys, are connected in chronological order. Numbers indicate age in days of first and last sample. Full symbols indicate microbiota from the feces, empty symbols samples recovered from the colon. The proximity of the gut microbiota and the fecal microbiota obtained on the final day of the experiment (day 41, 38 and 35, respectively) indicate a close similarity between colon and fecal microbiota.
A significant advantage of animal models over human subjects is the possibility to easily collect samples directly from the colon. We took advantage of this possibility to compare microbiomes originating from the feces excreted prior to euthanasia and collected from the colon a short time after. Samples from the colon were obtained from four of the eight animals examined here, as indicated with an open circle in Figure 4 and as labeled with “gut” in Figures 1 and 3. In all cases the colonic samples closely resembled the fecal microbiomes from the same animal in terms phylogeny (Fig. 1), diversity (Fig. 3) and Unifrac distance (Fig. 4).
Discussion
An ideal model of the human GI tract faithfully replicates the ecology of the gut. Such a model would be susceptible to diseases afflicting humans, develop similar symptoms, mimic the adults or infant GI tract and would be experimentally tractable and affordable. The pig model we present above fulfills several criteria; specifically it is experimentally tractable and it is a clinically relevant model of the human GI tract. We aim to use this model to explore the interaction between microbiome and host in healthy and diseased animals. Such a model will enable research on how to cure or alleviate disease of the GI tract.
For obvious reasons, much of the research community is using rodents as a model to study the human GI tract.11,13,23-25 Although the pig model we describe here is not perfect, we believe that it offers significant advantages over rodents for basic and translational research. Foremost is the physiological similarity between the human and porcine GI tract. Pigs have not only been shown to be susceptible to many enteric human pathogens,26-32 but present symptoms characteristic of human infections.33 The results described here showing that a human intestinal ecosystem can be transplanted into pigs expands the potential role of pigs in research beyond the germ-free or gnotobiotic model. The data presented here indicate that pigs are a particularly good model for studying the development of the infant microbiome. For instance, the infant model would enable studies on how the exposure to various components of the human microbiome affects development
of the innate immune system. The research we have presented paves the way for experiments aiming at elucidating a more complex and realistic model of the GI tract involving the host, the microbiome and enteric pathogens. We intend to perform future research with the same human donor samples which have been preserved at -80°C. Although we have not tested to what extent storage conditions have affected the composition of the microbiome, we believe that the advantage of using a standardized stocks of adult and infant microbiomes overrides any concerns about the impact of storage on the microbial population.
Our sequencing strategy is based on the Illumina platform and differs from more commonly used pyrosequencing methods.34 The main difference to pyrosequencing is the shorter length of sequence reads. The Illumina platform was chosen for practical reasons, specifically because its higher throughput enables the analysis of an essentially unlimited number of samples collected from multiple animals over an entire experimental period. The fact that the V6 amplicon sequences are compatible with phylogenetic classification and diversity analysis is due to several factors. In the first place, the V6 loop targeted in our protocol has a higher information entropy than any other region in the 16S gene.35 Second, we used a 16S database of reference sequences trimmed to 100 nt centered on the V6 loop. This approach improves classification. A systematic evaluation of the effect of the sequence database showed that customized reference databases, as used here, can reduce the number of unclassified reads.36 We verified the observation by Werner et al. using the conventional full-length RDP template database37 or the V6 database. In agreement with Werner et al.36 we found a reduction in the proportion of unclassified reads when using a trimmed template file. Experiments in which classification based on the V6-V9 region was compared with a truncated template corresponding to the V6 region support the view that a short hypervariable region can be used instead of longer regions. This analysis revealed that classification was more likely to be impacted by primer bias than by read length.38
A system-wide analysis of the gut environment and its response to perturbation will require expanding the analyses beyond 16S amplicon sequencing to the microbial metagenome and the transcriptome of the gut epithelium. Including the epithelial transcriptome will reveal if and how the host responds to perturbations of the intestinal ecosystem, for instance as a result of a change in diet, as a result of infection or treatment with anti-microbials. Surprisingly, we found only one report of such a study which used microarrays to compare the epithelial transcriptome of germ-free piglets and piglets populated with porcine microbiome. This study observed transcriptional changes of genes involved in epithelial cell turnover, mucus synthesis and in the regulation of the immune system.39 Because of the larger size of the pig intestine, as compared with the mouse, we anticipate that the pig model will enable a more comprehensive analysis of the gut environment, including microorganisms which live in close association with the epithelium40,41 and may not be adequately represented in fecal samples.
Although the number of experiments we have performed to date is small and does not support robust inferences on the putative impact of diet on microbiome diversity, our observations (see Fig. 2 and 3) are consistent with surveys in humans42,43 and experiments in rodents.11,44,45 If confirmed with additional experiments in pigs, the putative impact of diet on the composition of the microbiome would support the view that perturbations of the human GI tract environment can be replicated in microbiomes transplanted into pigs.
In contrast to a study of the evolution of native microbiome in mice over a period of one year,25 our study did not identify a measurable stabilization on the microbiome over time. In our experiments, weighted Unifrac distances typically decreased during the first few days following inoculation, but no obvious trend emerged thereafter. We hypothesize that the relatively short duration of the pig experiments was not sufficient for the gut microbiome to stabilize. Because our animals are housed in microbiological isolators46 for the duration of the experiment, extending the experiment beyond 5 or 6 weeks is not feasible. Experiments are needed to assess whether transferring pigs colonized with human microbiome to a normal environment will affect the composition of the microbiome. If, once established in the pigs' GI tract, the humanized microbiome becomes resistant to invasion by environmental bacteria, experiments of longer duration would become possible. Such experiments could enable research on slow-evolving or chronic conditions and reveal to what extent intestinal dysbiosis is associated with chronic conditions.
Materials and Methods
Animals and human fecal samples
Piglets were derived by C-section and housed in sterile isolators.46,47 Piglets were fed three times daily with human infant milk formula (Similac, Abbott). They were checked for bacterial contamination with daily aerobic culture on Brucella Blood Agar. In one experiment (experiment 3) pigs were weaned on day 20 by feeding ad libidum sterile Laboratory Porcine Grower Diet with 16% protein (LabDiet).
Feces were collected from 10 adult human donors (5 males and 5 females) aged 50 to 70 y and from a 3-mo old breast-fed healthy baby. To minimize the loss of anaerobic microorganisms, feces were transferred into completely filled airtight containers. Samples were homogenized in 9 volumes of reduced PBS containing 10% glycerol. The homogenates were filtered through a 425-µm pore mesh, dispensed into 15-ml centrifuge tubes and stored at -80°C. The samples from the adult donors were combined into a single sample.
On the day of inoculation with human fecal microbiota (Table 1), piglets were first given 10 ml of 0.2 M carbonate buffer pH 9.5 orally, followed by 3 ml of stool homogenate with a feeding needle inserted into the esophagus. The animals were monitored for the duration of the experiment for signs of disease, including diarrhea, dehydration, dyspnea, weakness, lethargy, anorexia, etc. Weights were recorded every other day and fecal samples collected for bacterial culture, for measuring cytokines and for extracting DNA for high-throughput sequencing. Piglets were euthanized with an intramuscular injection of 1 ml 100 mg/ml ketamine and 0.1 ml 100 mg/ml xylazine per kg body weight, followed by an intra-cardial injection of 1 ml of 390 mg/ml SomnaSol/kg (Butler and Schein). Following euthanasia, gut contents were recovered from the spiral colon by gently squeezing the colon.
DNA extraction and 16S library preparation
DNA was extracted from 200 µl of fecal slurry. Fecal samples were first subjected to two cycles of freeze-thawing (-80°C/37°C). DNA was extracted from these samples with the HighPure PCR Template Preparation kit (Roche Diagnostics). The final DNA extract was dissolved in 20 µl buffer. A volume of 1 µl of DNA was amplified in a primary PCR with primers flanking the approximately 60-nucleotide variable region of the V6 loop of the bacterial 16S rRNA gene.48 The following primers were used: 5′ CAACGCGAAGAACCTTACC 3′ and 5′ CGACAGCCATCGANCACCT 3′. The primary amplification consisted of 15 cycles of 94°C for 30 sec, 55°C of 30 sec and 68°C for 90 sec. A secondary PCR was used to amplify 1 µl of primary amplicon and generate a library of PCR products fused to Illumina adaptors and tagged with a 6-nucleotide barcode unique to each sample. The downstream (reverse) primer used in the secondary reaction incorporates in 5′ to 3′ orientation the Illumina flow-cell binding sequence, the barcode, the complement of the standard Illumina multiplex index read primer and the downstream conserved sequence flanking the V6 region. The secondary PCR consisted of three cycles of 94°C for 30 sec, 55°C of 30 sec, 68°C for 90 sec followed by 11 cycles of 94°C for 30 sec, 68°C for 30 sec. ExoSap was used to remove excess primers from the final PCR and the quality of the amplicons verified by electrophoresis on a 2% agarose gel. The expected amplicon size is approximately 200 nt. The pooled barcoded libraries were sequenced in a HiSeq2000 Illumina sequencer at the Tufts Genomics core facility (tucf.org). An average of 3.96 mil0lion 100-nucleotide (nt) reads were obtained for each sample (range 1.4 × 106−8.1 × 106 reads) (Table 2). Quality control for Illumina sequencing included a PhiX phage library sequenced with each reaction. This control was used to estimate phasing and matrix correction parameters for base calling. It also provided a reliable statistic of run error rate.
Table 2. Summary of sequencing results.
| samples | |||||
|---|---|---|---|---|---|
| |
adult (1, 3)a |
infantb(2) |
pig (1) |
pig (2) |
pig (3) |
| number of samples sequenced |
1 |
1 |
8 |
23 |
22 |
| number of pigs |
|
|
2 |
4 |
2 |
| sequences meeting quality criteria |
9713 |
9200 |
9814 (81.4)c |
9306 |
9581 (359) |
| unique sequences | 16.7% | 8.4% | 9.1% (1.2) | 5.8%(1.4) | 13.2% (0.9) |
a experiment number is indicated in parenthesis (see text); pooled adult fecal sample was used for experiment 1 and 3.
b mean of two replicates (two samples amplified and sequenced independently)
c standard deviation is shown in parenthesis
Data analysis
For most analyses the open-source software mothur version 1.25 was used.49 Sequences were initially parsed according to barcode and trimmed to eliminate the downstream primer sequence, leaving sequences of ~60 nt length in average (2.5 percentile = 56 nt; 97.5% percentile = 63 nt). A random subsample of 10,000 sequences was used for downstream analyses. Sequences were classified using the RDP classifier program implemented in mothur using the bayesian method.50 To optimize the taxonomic classification,36 a training set template file trimmed to a 100-nt region centered on the V6 region was generated from the 8422-sequence RDP training set version 6.37 The minimum bootstrap value for taxonomic assignment was set at 70%.
The phylogenetic distance between samples was quantified with the weighted UniFrac distance metric51 as implemented in mothur. Random subsamples of 10,000 sequences used for this analysis were screened to identify sequences which did not meet the following quality criteria: minimum length 50 nt, maximum length 70 nt, start and end of sequence within start and end position of 90% of the sequences. Between 92.0 and 98.1% of the sequences met these criteria and were included in the computation of Unifrac distance. Lower-triangle matrices of pairwise UniFrac distances were imported into GenAlex52 and distances between samples visualized using the Principal Coordinate Analysis (PCoA) calculator. The evenness of human and pig samples were compared using mothur's rarefaction.single command and visualized on rank-abundance plots.53 The Shannon diversity index was used to estimate α diversity.
Supplementary Material
Acknowledgments
We thank David Lazinski for sharing his unpublished PCR protocol for building 16S libraries, Kip Bodi from the Tufts University Genomics Core and his team for sequencing and advising on bioinformatics analyses and Alex Walker for preparing DNA libraries. Our thanks to Patty Boucher and Rachel Sora for caring for the animals and to Huyen Bum Kim for critical reading of the manuscript. Financial support to S.T. from the NIAID (grants AI088748 and AI094459), Progenics and Pfizer (ASPIRE award WS1953405) is gratefully acknowledged.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Footnotes
Previously published online: www.landesbioscience.com/journals/gutmicrobes/article/23867
References
- 1.Turnbaugh PJ, Hamady M, Yatsunenko T, Cantarel BL, Duncan A, Ley RE, et al. A core gut microbiome in obese and lean twins. Nature. 2009;457:480–4. doi: 10.1038/nature07540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hakansson A, Molin G. Gut microbiota and inflammation. Nutrients. 2011;3:637–82. doi: 10.3390/nu3060637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Azad MB, Kozyrskyj AL. Perinatal programming of asthma: the role of gut microbiota. Clin Dev Immunol. 2012;2012:932072. doi: 10.1155/2012/932072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Qin J, Li Y, Cai Z, Li S, Zhu J, Zhang F, et al. A metagenome-wide association study of gut microbiota in type 2 diabetes. Nature. 2012;490:55–60. doi: 10.1038/nature11450. [DOI] [PubMed] [Google Scholar]
- 5.Wen L, Ley RE, Volchkov PY, Stranges PB, Avanesyan L, Stonebraker AC, et al. Innate immunity and intestinal microbiota in the development of Type 1 diabetes. Nature. 2008;455:1109–13. doi: 10.1038/nature07336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brown K, DeCoffe D, Molcan E, Gibson DL. Diet-induced dysbiosis of the intestinal microbiota and the effects on immunity and disease. Nutrients. 2012;4:1095–119. doi: 10.3390/nu4081095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gomez A, Luckey D, Yeoman CJ, Marietta EV, Berg Miller ME, Murray JA, et al. Loss of sex and age driven differences in the gut microbiome characterize arthritis-susceptible 0401 mice but not arthritis-resistant 0402 mice. PLoS One. 2012;7:e36095. doi: 10.1371/journal.pone.0036095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Koren O, Goodrich JK, Cullender TC, Spor A, Laitinen K, Bäckhed HK, et al. Host remodeling of the gut microbiome and metabolic changes during pregnancy. Cell. 2012;150:470–80. doi: 10.1016/j.cell.2012.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Samuel BS, Gordon JI. A humanized gnotobiotic mouse model of host-archaeal-bacterial mutualism. Proc Natl Acad Sci U S A. 2006;103:10011–6. doi: 10.1073/pnas.0602187103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Martin FP, Wang Y, Sprenger N, Yap IK, Lundstedt T, Lek P, et al. Probiotic modulation of symbiotic gut microbial-host metabolic interactions in a humanized microbiome mouse model. Mol Syst Biol. 2008;4:157. doi: 10.1038/msb4100190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Turnbaugh PJ, Ridaura VK, Faith JJ, Rey FE, Knight R, Gordon JI. The effect of diet on the human gut microbiome: a metagenomic analysis in humanized gnotobiotic mice. Sci Transl Med. 2009;1:ra14. doi: 10.1126/scitranslmed.3000322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Faith JJ, Rey FE, O’Donnell D, Karlsson M, McNulty NP, Kallstrom G, et al. Creating and characterizing communities of human gut microbes in gnotobiotic mice. ISME J. 2010;4:1094–8. doi: 10.1038/ismej.2010.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Goodman AL, Kallstrom G, Faith JJ, Reyes A, Moore A, Dantas G, et al. Extensive personal human gut microbiota culture collections characterized and manipulated in gnotobiotic mice. Proc Natl Acad Sci U S A. 2011;108:6252–7. doi: 10.1073/pnas.1102938108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.McNulty NP, Yatsunenko T, Hsiao A, Faith JJ, Muegge BD, Goodman AL, et al. The impact of a consortium of fermented milk strains on the gut microbiome of gnotobiotic mice and monozygotic twins. Sci Transl Med. 2011;3:10ra106. doi: 10.1126/scitranslmed.3002701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Carvalho FA, Koren O, Goodrich JK, Johansson ME, Nalbantoglu I, Aitken JD, et al. Transient inability to manage proteobacteria promotes chronic gut inflammation in TLR5-deficient mice. Cell Host Microbe. 2012;12:139–52. doi: 10.1016/j.chom.2012.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ekser B, Ezzelarab M, Hara H, van der Windt DJ, Wijkstrom M, Bottino R, et al. Clinical xenotransplantation: the next medical revolution? Lancet. 2012;379:672–83. doi: 10.1016/S0140-6736(11)61091-X. [DOI] [PubMed] [Google Scholar]
- 17.Phelps CJ, Koike C, Vaught TD, Boone J, Wells KD, Chen SH, et al. Production of alpha 1,3-galactosyltransferase-deficient pigs. Science. 2003;299:411–4. doi: 10.1126/science.1078942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Butler JE, Weber P, Sinkora M, Baker D, Schoenherr A, Mayer B, et al. Antibody repertoire development in fetal and neonatal piglets. VIII. Colonization is required for newborn piglets to make serum antibodies to T-dependent and type 2 T-independent antigens. J Immunol. 2002;169:6822–30. doi: 10.4049/jimmunol.169.12.6822. [DOI] [PubMed] [Google Scholar]
- 19.Haverson K, Rehakova Z, Sinkora J, Sver L, Bailey M. Immune development in jejunal mucosa after colonization with selected commensal gut bacteria: a study in germ-free pigs. Vet Immunol Immunopathol. 2007;119:243–53. doi: 10.1016/j.vetimm.2007.05.022. [DOI] [PubMed] [Google Scholar]
- 20.Scharek L, Guth J, Reiter K, Weyrauch KD, Taras D, Schwerk P, et al. Influence of a probiotic Enterococcus faecium strain on development of the immune system of sows and piglets. Vet Immunol Immunopathol. 2005;105:151–61. doi: 10.1016/j.vetimm.2004.12.022. [DOI] [PubMed] [Google Scholar]
- 21.Wilson S, Norton P, Haverson K, Leigh J, Bailey M. Development of the palatine tonsil in conventional and germ-free piglets. Dev Comp Immunol. 2005;29:977–87. doi: 10.1016/j.dci.2005.03.008. [DOI] [PubMed] [Google Scholar]
- 22.Foster N, Lovell MA, Marston KL, Hulme SD, Frost AJ, Bland P, et al. Rapid protection of gnotobiotic pigs against experimental salmonellosis following induction of polymorphonuclear leukocytes by avirulent Salmonella enterica. Infect Immun. 2003;71:2182–91. doi: 10.1128/IAI.71.4.2182-2191.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Reeves AE, Theriot CM, Bergin IL, Huffnagle GB, Schloss PD, Young VB. The interplay between microbiome dynamics and pathogen dynamics in a murine model of Clostridium difficile Infection. Gut Microbes. 2011;2:145–58. doi: 10.4161/gmic.2.3.16333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gillilland MG, 3rd, Erb-Downward JR, Bassis CM, Shen MC, Toews GB, Young VB, et al. Ecological succession of bacterial communities during conventionalization of germ-free mice. Appl Environ Microbiol. 2012;78:2359–66. doi: 10.1128/AEM.05239-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schloss PD, Schubert AM, Zackular JP, Iverson KD, Young VB, Petrosino JF. Stabilization of the murine gut microbiome following weaning. Gut Microbes. 2012;3:383–93. doi: 10.4161/gmic.21008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yuan L, Ward LA, Rosen BI, To TL, Saif LJ. Systematic and intestinal antibody-secreting cell responses and correlates of protective immunity to human rotavirus in a gnotobiotic pig model of disease. J Virol. 1996;70:3075–83. doi: 10.1128/jvi.70.5.3075-3083.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Widmer G, Akiyoshi D, Buckholt MA, Feng X, Rich SM, Deary KM, et al. Animal propagation and genomic survey of a genotype 1 isolate of Cryptosporidium parvum. Mol Biochem Parasitol. 2000;108:187–97. doi: 10.1016/S0166-6851(00)00211-5. [DOI] [PubMed] [Google Scholar]
- 28.Andrutis KA, Riggle PJ, Kumamoto CA, Tzipori S. Intestinal lesions associated with disseminated candidiasis in an experimental animal model. J Clin Microbiol. 2000;38:2317–23. doi: 10.1128/jcm.38.6.2317-2323.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Steele J, Feng H, Parry N, Tzipori S. Piglet models of acute or chronic Clostridium difficile illness. J Infect Dis. 2010;201:428–34. doi: 10.1086/649799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sheoran A, Wiffin A, Widmer G, Singh P, Tzipori S. Infection with Cryptosporidium hominis provides incomplete protection of the host against Cryptosporidium parvum. J Infect Dis. 2012;205:1019–23. doi: 10.1093/infdis/jir874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Brady MJ, Radhakrishnan P, Liu H, Magoun L, Murphy KC, Mukherjee J, et al. Enhanced Actin Pedestal Formation by Enterohemorrhagic Escherichia coli O157:H7 Adapted to the Mammalian Host. Front Microbiol. 2011;2:226. doi: 10.3389/fmicb.2011.00226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jeong KI, Zhang Q, Nunnari J, Tzipori S. A piglet model of acute gastroenteritis induced by Shigella dysenteriae Type 1. J Infect Dis. 2010;201:903–11. doi: 10.1086/650995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Meurens F, Summerfield A, Nauwynck H, Saif L, Gerdts V. The pig: a model for human infectious diseases. Trends Microbiol. 2012;20:50–7. doi: 10.1016/j.tim.2011.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Margulies M, Egholm M, Altman WE, Attiya S, Bader JS, Bemben LA, et al. Genome sequencing in microfabricated high-density picolitre reactors. Nature. 2005;437:376–80. doi: 10.1038/nature03959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Andersson AF, Lindberg M, Jakobsson H, Bäckhed F, Nyrén P, Engstrand L. Comparative analysis of human gut microbiota by barcoded pyrosequencing. PLoS One. 2008;3:e2836. doi: 10.1371/journal.pone.0002836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Werner JJ, Koren O, Hugenholtz P, DeSantis TZ, Walters WA, Caporaso JG, et al. Impact of training sets on classification of high-throughput bacterial 16s rRNA gene surveys. ISME J. 2012;6:94–103. doi: 10.1038/ismej.2011.82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cole JR, Wang Q, Cardenas E, Fish J, Chai B, Farris RJ, et al. The Ribosomal Database Project: improved alignments and new tools for rRNA analysis. Nucleic Acids Res. 2009;37(Database issue):D141–5. doi: 10.1093/nar/gkn879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Smith BC, McAndrew T, Chen Z, Harari A, Barris DM, Viswanathan S, et al. The cervical microbiome over 7 years and a comparison of methodologies for its characterization. PLoS One. 2012;7:e40425. doi: 10.1371/journal.pone.0040425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chowdhury SR, King DE, Willing BP, Band MR, Beever JE, Lane AB, et al. Transcriptome profiling of the small intestinal epithelium in germfree versus conventional piglets. BMC Genomics. 2007;8:215. doi: 10.1186/1471-2164-8-215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nava GM, Carbonero F, Croix JA, Greenberg E, Gaskins HR. Abundance and diversity of mucosa-associated hydrogenotrophic microbes in the healthy human colon. ISME J. 2012;6:57–70. doi: 10.1038/ismej.2011.90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Maynard CL, Elson CO, Hatton RD, Weaver CT. Reciprocal interactions of the intestinal microbiota and immune system. Nature. 2012;489:231–41. doi: 10.1038/nature11551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fallani M, Young D, Scott J, Norin E, Amarri S, Adam R, et al. and Other Members of the INFABIO Team Intestinal microbiota of 6-week-old infants across Europe: geographic influence beyond delivery mode, breast-feeding, and antibiotics. J Pediatr Gastroenterol Nutr. 2010;51:77–84. doi: 10.1097/MPG.0b013e3181d1b11e. [DOI] [PubMed] [Google Scholar]
- 43.Wu GD, Chen J, Hoffmann C, Bittinger K, Chen YY, Keilbaugh SA, et al. Linking long-term dietary patterns with gut microbial enterotypes. Science. 2011;334:105–8. doi: 10.1126/science.1208344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Turnbaugh PJ, Bäckhed F, Fulton L, Gordon JI. Diet-induced obesity is linked to marked but reversible alterations in the mouse distal gut microbiome. Cell Host Microbe. 2008;3:213–23. doi: 10.1016/j.chom.2008.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hildebrandt MA, Hoffmann C, Sherrill-Mix SA, Keilbaugh SA, Hamady M, Chen YY, et al. High-fat diet determines the composition of the murine gut microbiome independently of obesity. Gastroenterology. 2009;137:1716–24, e1-2. doi: 10.1053/j.gastro.2009.08.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tzipori S, Rand W, Griffiths J, Widmer G, Crabb J. Evaluation of an animal model system for cryptosporidiosis: therapeutic efficacy of paromomycin and hyperimmune bovine colostrum-immunoglobulin. Clin Diagn Lab Immunol. 1994;1:450–63. doi: 10.1128/cdli.1.4.450-463.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tzipori S. Cryptosporidiosis: laboratory investigations and chemotherapy. Adv Parasitol. 1998;40:187–221. doi: 10.1016/S0065-308X(08)60121-9. [DOI] [PubMed] [Google Scholar]
- 48.Baker GC, Smith JJ, Cowan DA. Review and re-analysis of domain-specific 16S primers. J Microbiol Methods. 2003;55:541–55. doi: 10.1016/j.mimet.2003.08.009. [DOI] [PubMed] [Google Scholar]
- 49.Schloss PD, Westcott SL, Ryabin T, Hall JR, Hartmann M, Hollister EB, et al. Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl Environ Microbiol. 2009;75:7537–41. doi: 10.1128/AEM.01541-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wang Q, Garrity GM, Tiedje JM, Cole JR. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl Environ Microbiol. 2007;73:5261–7. doi: 10.1128/AEM.00062-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lozupone C, Hamady M, Knight R. UniFrac--an online tool for comparing microbial community diversity in a phylogenetic context. BMC Bioinformatics. 2006;7:371. doi: 10.1186/1471-2105-7-371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Peakall R, Smouse PE. GENALEX 6: genetic analysis in Excel. Population genetic software for teaching and research. Mol Ecol Notes. 2006;6:288–95. doi: 10.1111/j.1471-8286.2005.01155.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Whittaker RH. Dominance and Diversity in Land Plant Communities: Numerical relations of species express the importance of competition in community function and evolution. Science. 1965;147:250–60. doi: 10.1126/science.147.3655.250. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.