

Abstract
The microbiome has captured the attention of scientists from multiple research fields including ecology, immunology, microbiology and cancer biology. The microbial community living in the gastrointestinal tract is the most abundant and diverse niche of the human body and it is not surprising that microbiome research has predominantly focused upon this organ system. In this addendum, we summarize the latest developments in microbiome research on inflammatory bowel diseases and colorectal cancer. In addition, we highlight our recent findings that chronic intestinal inflammation modulates microbial community composition and the development of colorectal cancer. Our findings redefine the paradigm of inflammation-associated cancer by illuminating the key role of bacteria in development of colorectal cancer.
Keywords: microbiome, colon cancer, inflammation, bacterial toxin, Escherichia coli, colibactin
Introduction
In the 19th century, the famous “pathologist” Rudolf Virchow observed inflammatory cells (leukocytes) in neoplastic lesions.1 Although it was not possible at that time to determine whether the presence of these inflammatory cells was the consequence of tumor development or cause of tumor progression, this simple observation evolved into the current concept that the inflammatory microenvironment represents a risk factor for cancer development. A link is now firmly established between inflammation and cancer development, including gastric, liver and colorectal cancer (CRC),2 and important gains of knowledge have been made regarding the cellular and molecular mechanisms by which inflammation fosters the development of cancer. In the case of colitis-associated CRC, various inflammatory mediators (e.g., TNF, IL-17A, IL-23) and genotoxic substances [e.g., reactive oxygen and nitrogen species (ROS, RNS)] generated by different cellular systems (immune cells, mesenchymal cells) act in concert to introduce genetic and epigenetic modifications that together ultimately lead to carcinogenesis.3-5 Therefore, any events fueling and maintaining inflammation could be considered potential carcinogenic contributors.
In intestinal pathologies such as inflammatory bowel diseases (IBD), a key initiator and perpetuator of intestinal inflammation is the vast population of commensal microorganisms inhabiting the lower gastrointestinal tract, termed the gut microbiota. The current research paradigm for colitis-associated CRC is that the gut microbiota promotes the development of colitis, with intestinal inflammation leading to tumorigenesis. This paradigm does not take into account recent observations suggesting that host inflammation impacts upon community composition and functional capabilities of the gut microbiota.6,7 A recent study from our laboratory has demonstrated that the gut microbiota and microbial genes play an active role in the development of CRC.8
Inflammation, the Microbiota and Colorectal Cancer
Infectious microorganisms have been linked to various forms of cancer. For example, Helicobacter pylori infection is strongly associated with development of gastric cancer while hepatitis C virus is linked with hepatocellular carcinoma.2,9 In the case of CRC, infection with enteropathogenic strains of Streptococcus bovis has been clinically associated with this pathology,1,10,11 although the etiological nature of this association remains unclear. It is interesting to note that for years, the endogenous, non-pathogenic gut microbiota has vastly been ignored as an environmental contributor to CRC.
The colon, a primary site of carcinogenesis, contains an estimated microbial load of 1013–1014 and a collective genome evaluated at 3 × 106 genes. This microbial ecosystem resides in relative close proximity to the intestinal epithelium and contributes essential functions involved in the maintenance of host homeostasis such as synthesis of essential vitamins, generation of various nutrients from complex dietary carbohydrates, toning and education of the mucosal immune system as well as ecological competition to fence off invading pathogenic microbes.2,12,13 Researchers have begun to survey this vast ecosystem of microorganisms to identify changes associated with health and disease states, including IBD and CRC. Because culture conditions have not been established for most of the microorganisms colonizing our GI tract, investigators have turned to the power of genomics, in particular next-generation sequencing targeting ribosomal 16S bacterial genes, to profile the biome of CRC patients.3-5,14 Studies evaluating the microbiota of CRC case and control patients have provided new insights into the unbalanced nature of the CRC patient gut microbial community, a phenomenon refered to as dysbiosis. Applying microbiome analysis of intestinal tissues and fecal materials, researchers have identified microbial groups associated with CRC. Although these studies have not etiologically linked a clear cluster of microorganisms to the development of CRC, these investigations have reproducibly showed differences between the microbiome of CRC patients with that of healthy individuals. For example, the stool of CRC patients has been shown to harbor an increased abundance of the anaerobic bacterial belonging to the group Bacteroides-Prevotella compared with healthy controls.8,15 Other groups observed that the luminal compartment of CRC patients showed higher prevalence of the genera Enterococcus, Escherichia/Shigella, Klebsiella, Streptococcus and Peptostreptococcus compared with controls, while the family Lachnospiraceae containing butyrate-producing bacteria were less abundant.2,9,16 When investigators surveyed the intestinal mucosal surface of patients with adenoma, an increased abundance of Firmicutes, Bacteroidetes and Proteobacteria was observed compared with non-adenoma subjects.17 Using resected tissues from adenocarcinoma patients and adjacent non-malignant sites, the genera Coriobacteria, Roseburia, Fusobacterium and Faecalibacterium were increased in tumors compared with adjacent non-tumor control tissue.18 The fusobacterium genera is particularly intriguing since a similar expansion was reported by multiple groups in rectal swab samples and colonic tissues from CRC patients relative to healthy controls.19-21 Moreover, the presence of Fusobacterium nucleatum correlates with development of IBD.22 Although these investigative efforts have generated interesting observations regarding microbial dysbiosis in CRC patients, no clear picture has emerged from these studies regarding a common group of microorganisms associated with the pathology. This highlights the need to combine next-generation sequencing technology with conventional microbiology approaches (culture/isolation) and animal models to evaluate the functional impact of microorganisms on CRC initiation and progression.
E. coli, IBD and CRC
Using culture-based methods and immunohistological staining, researchers have made an observation that ileal and colonic mucosal surfaces of IBD and CRC patients harbor increased numbers of adherent-invasive Escherichia coli relative to healthy control individuals.23-25 Moreover, a number of studies employing metagenomic analyses have reported an expansion of Proteobacteria in patients with IBD, especially Enterobacteriaceae, to which the E. coli species belong.26 However, a higher prevalence of Proteobacteria/Enterobacteriaceae/E. coli in inflamed tissues could be a consequence, rather than a cause of intestinal inflammation.
Inflammation Alters the Composition of the Colonic Microbiota
In a recent study, we sought to better understand the interplay between inflammation, CRC and the gut microbiota.8 We had previously shown that microbial status influences the development of inflammation and tumorigenesis in the azoxymethane (AOM)/Il10−/− model of colitis-associated CRC.27 In this model, interleukin-10-deficient (Il10−/−) mice develop chronic intestinal inflammation due to lack of the immunosuppressive cytokine IL-10 on effector T cells. Chronic inflammation promotes tumorigenesis that is initiated via injection of the colon specific carcinogen AOM.
We hypothesized that inflammation would alter microbial community composition in AOM/Il10−/− colitis-susceptible mice relative to wildtype (WT) healthy controls. Il10−/− and WT 129/SvEv mice born in germ-free (GF) conditions were transferred to our specific pathogen free (SPF) facility to time the onset of inflammation. Although germ-free mice have been shown to have an immature immune system, this approach is ideal for microbiome studies since it nullifies microbiota “legacy” effects, such as mother-to-pup transmission, that can occur among mice born in SPF conditions.28 Stool and distal colon samples, representative of the luminal and mucosally-adherent microbiota respectively, were collected from Il10−/− (colitis), AOM-treated Il10−/− (colitis/cancer), WT (healthy) and AOM-treated WT (healthy) mice at 20 weeks post-transfer to SPF conditions. Microbial populations were evaluated using Illumina HiSeq2000 sequencing of the hypervariable V6 region of the bacterial 16S rRNA gene. Next-generation sequencing revealed that both the luminal and mucosally-adherent microbiota significantly differed between Il10−/− and WT mice. Colitis-susceptible Il10−/− mice exhibited a reduction in luminal microbial richness, relative to healthy WT mice (Fig. 1), similar to what has been observed in human IBD patients.29,30 We were surprised to find no difference in microbial community composition or richness between Il10−/− mice with colitis vs. colitis/cancer. These data suggest that inflammation, not cancer, is the driving force behind dysbiosis observed in Il10−/− mice.

Figure 1. Dysbiosis in Il10−/− mice is driven by inflammation rather than cancer. WT and Il10−/− mice were transferred from germ-free to specific pathogen free (SPF) conditions. In SPF conditions, WT mice remain healthy, but 100% of Il10−/− mice develop colitis. Half of each cohort was treated with AOM, which induces no cancer in WT mice but cancer in ~60–80% of Il10−/− mice. When the microbiota of each cohort was assessed by Illumina sequencing of 16S ribosomal genes, we found it did not differ by cancer status (Il10−/− vs. AOM/Il10−/−). In contrast, inflammatory status (Il10−/− vs. WT) was associated with alterations in microbial community composition, including an expansion of Enterobacteriaceae bacteria.
Increased abundance of Proteobacteria has been linked to human IBD and gastrointestinal cancer,9,23-25,31-33 which led us to hypothesize that the inflammatory microenvironment of the Il10−/− colon supported the expansion of this particular bacterial group. Indeed, we observed an increased abundance of Proteobacteria in the Il10−/− microbiota, relative to healthy WT controls. Within Proteobacteria, the Gammaproteobacteria class, Enterobacteriales order and Enterobacteriaceae family were all significantly more abundant in Il10−/− mice. Adherent-invasive Escherichia coli have been associated with human IBD and CRC,23-25 and PCR designed to specifically amplify E. coli revealed a 100-fold increase in the microbiota of Il10−/− mice, relatively to WT controls.
Inflammation is Not Sufficient to Promote CRC
To determine if commensal E. coli play a causative role in CRC development, we mono-associated GF Il10−/− mice with either the mouse E. coli strain NC101 or the human commensal Enterococcus faecalis strain OG1RF. As expected,34 both commensal strains caused aggressive colitis in Il10−/− mice with similar levels of colon tissue inflammatory cytokines and infiltrating immune/inflammatory cells. However, only E. coli was able to reproducibly induce cancer in AOM-treated Il10−/− mice, with an ~80% penetrance of invasive mucinous adenocarcinoma. WT mice mono-associated with E. coli NC101 developed neither inflammation nor tumors, suggesting that some level of baseline genetic susceptibility to disease (for example, inflammation) is necessary for E. coli NC101-induced tumorigenesis in this model. From these observations, we concluded that inflammation alone is insufficient to induce CRC and that microbial entities play a key role in the pathology.
E. coli NC101 Harbors the Genotoxic pks Island, Which is Associated with Human IBD and CRC
We hypothesized that E. coli NC101 has carcinogenic capabilities that E. faecalis lacks. Several members of the family Enterobacteriaceae, in particular E. coli strains of phylotype B2, harbor a ~54kb polyketide synthases (pks) pathogenicity island that encodes the multi-enzymatic machinery for a putative peptide-polyketide genotoxin called Colibactin.35-38 Basic Local Alignment Seach Tool (BLAST), PCR amplification and sequencing revealed the presence of pks in E. coli NC101 and the absence of other E. coli genotoxins including Cif, CNF and CDT. Pks was not detected in E. faecalis or the non-colitogenic E. coli strain K12. To determine if pks could be associated with human IBD or CRC, we screened mucosa-associated E. coli strains from colorectal tissue biopsies of 35 patients with IBD, 21 with CRC and 24 non-IBD/non-CRC controls.23 Samples were unavailable from IBD-associated CRC patients, as IBD patients typically undergo preventative colectomy upon initial diagnosis of colorectal dysplasia. Only 20.8% of non-IBD/non-CRC controls harbored pks+ E. coli, in contrast to a significantly greater proportion of IBD and CRC patients – 40% of IBD patients and 66.7% of CRC patients. These data suggest that pks+ bacteria are associated with chronic intestinal inflammation and CRC and indicate that pks may play an active role in promoting tumorigenesis.
Deletion of pks Reduces the Tumorigenicity of E. coli NC101
To functionally link pks with cancer-promoting activites, we created an isogenic E. coli NC101 strain deficient in pks (NC101Δpks). Pks from extra-intestinal pathogenic strains of E. coli, and more recently from the probiotic E. coli strain Nissle 1917, elicit DNA damage in mammalian cells.35,36,39 We observed that pks+ E. coli NC101 but not NC101Δpks, induced the surrogate markers of DNA damage γH2AX and cell cycle arrest in IEC-6 intestinal epithelial cells. This demonstrated that pks+ bacteria alone can induce DNA damage in the absence of a carcinogen such as AOM.
To determine if pks can promote tumorigenesis in vivo, we mono-associated GF Il10−/− mice with E. coli NC101 or E. coli NC101Δpks both with and without AOM treatment. Interestingly, the presence of Pks did not affect the ability of E. coli NC101 to induce histologic inflammation, cytokine expression, or infiltration of immune cells in the colon of Il10−/− mice with colitis (12 weeks, no AOM) or colitis/cancer (14 and 18 weeks, +AOM). However, deletion of pks reduced macroscopic tumor burden and histologic carcinoma invasion. At 14 weeks, five of eight AOM-treated Il10−/− mice mono-associated with E. coli NC101 exhibited high grade dysplasia (HGD) or invasive carcinoma, in contrast to only one of eight NC101Δpks mono-associated mice who developed HGD. At 18 weeks, all nine AOM-treated Il10−/− mice mono-associated with E. coli NC101 developed invasive carcinoma, with four of nine exhibiting full carcinoma invasion through the muscularis propria and serosa. Zero of five NC101Δpks mono-associated mice exhibited full carcinoma invasion at 18 weeks. In the absence of AOM, Il10−/− mice mono-associated for 21 weeks with either E. coli NC101 or NC101Δpks developed only mild dysplasia that by histologic assessment, was less advanced in NC101Δpks mono-associated animals. These findings suggest that pks accelerates the carcinogenic process without affecting the extent of inflammation.
The complex interplay between inflammation and bacteria is highlighted in an experiment where Il10−/−;Rag2−/− mice were monoassociated with E. coli NC101. Whereas AOM-treated Il10−/− mice mono-associated with E. coli NC101 developed inflammation and CRC, mono-associated Il10−/−;Rag2−/− mice remained healthy (Marcus Mühlbauer and C. Jobin personal observation). Together with the observation that E. faecalis failed to promote cancer development despite high colitogenic ability, our findings strongly point to a novel mechanism where inflammation acts in concert with specific microbial entities/genes to induce CRC.
Deletion of pks Reduces the Genotoxicity of E. coli NC101
We had observed that IEC-6 intestinal epithelial cells exposed to E. coli NC101 exhibited increased markers of DNA damage that were significantly reduced in IEC-6 cells exposed to E. coli NC101Δpks. To evaluate the effect of pks on DNA damage in vivo, we measured colonocyte γH2AX foci in non-tumor tissue of AOM-treated Il10−/− mice mono-associated with E. coli NC101 or NC101Δpks for 14 weeks. We detected a significant reduction in γH2AX+ colonocytes from mice mono-associated with E. coli NC101 vs. NC101Δpks. These data imply that pks-elicited DNA damage contributes to colorectal carcinogenesis. Inflammation is required for this phenomenon, as WT mice exhibit minimal DNA damage (~80% reduction in in γH2AX+ colonocytes relative to Il10−/− mice) when mono-associated with E. coli NC101.
Although we demonstrated that E. coli NC101 pks induces a DNA damage response in vivo and in vitro, the mechanism by which this bacterium promotes CRC is unclear. We performed laser capture microdissection of tumor and non-tumor tissues from mice colonized with E. coli NC101 vs. NC101Δpks and assessed mutation hotspots of common CRC oncogenes and tumor suppressors such as Ctnnb1, Kras, Braf and Tp53 using a PCR/sequencing approach. While Ctnnb1 mutations could be detected in the AOM/DSS positive control samples, we were unable to detect mutations in the tested regions/genes of AOM/Il10−/− mice (Arthur and Jobin, personal observation). In absence of a whole-genome approach, the location and frequency of mutations induced by E. coli NC101 in the AOM/Il10−/− model remains unclear. Pks can induce aneuploidy and mutations at the hprt reporter locus in vitro,36 suggesting that E. coli NC101 pks could very likely contribute to tumorigenesis by inducing cancer-promoting mutations in vivo. Furthermore, it appears that both inflammation and microbial factors such as pks act together to create a host microenvironment that promotes carcinogenesis.
Conclusions and Future Directions
Together, our findings indicate that inflammation targets the microbiota and fosters the expansion of bacteria with pro-carcinogenic activities that influence the development of CRC (Fig. 2). However, numerous outstanding questions persist regarding the relationship between inflammation and E.coli-mediated carcinogenic response (Box 1). Bioactive products of intestinal bacteria perform a wide variety of functions that may influence cancer development, such as activating environmental carcinogens, degrading protective mucins, oxidizing or reducing dietary compounds and inducing DNA damage and genotoxicity.40 In the case of pks+E. coli, carcinogenesis is presumably accelerated through a DNA-damaging agent, termed Colibactin, that is generated through the enzymatic activities encoded in the pks island. Colibactin, however, has not yet been purified and therefore its ability to damage DNA cannot be directly tested at this time. Purifying this bioactive compound will be essential for understanding the mechanism by which pks exerts its genotoxic function.
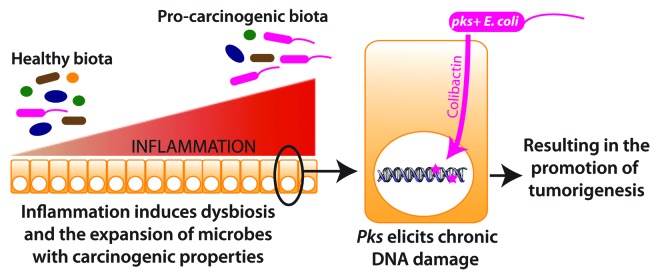
Figure 2. Model for enhanced tumorigenicity by pks+ E. coli.
Box 1.
Outstanding questions
1). How mechanistically does inflammation impact upon microbial composition?
2). What is the biochemical identity of Colibactin? Does pks produce additional bioactive molecules?
3). How is the expression of pks and the production of its bioactive molecules regulated? Does inflammation play a role?
4). What is the mechanism by which pks induces DNA damage?
5). What is the microbiological and ecological role of pks?
In addition to altering the relative amount of specific microbes residing in the intestine, inflammation may influence gene expression and functional capabilities of members within this population. New technologies are rapidly evolving (metagenomics, RNaseq, metabolomics, etc.) and in the near future, will allow us to assess the functional capacity of single commensal microbes as well as complex microbial populations in vivo. This will allow us to evaluate not just which microbes are present, but what activites they are performing and to what extent they contribute to health and disease. For example, inflammation in E. coli NC101 mono-associated Il10−/− mice affects bacterial fitness by upregulating genes involved in adherence and stress responses.41 Gene expression studies have revealed no change in the expression of pks genes under various in vitro conditions37; however pks gene expression has yet to be evaluated in vivo. It will be especially important to determine if inflammation plays a role in pks gene expression and Colibactin production/activity.
It is important to note that intestinal inflammation is not a phenomenon restricted to chronic inflammatory disorders, but can occur in response to acute injury, infection and cancer. Inflammatory features including increased expression of pro-inflammatory cytokines and lipid mediators, infiltration of immune/inflammatory cells, mucus depletion and barrier dysfunction have been observed in and surrounding colon tumors.42,43 Mucus depletion and increased barrier permeability could allow bacteria and bacterial products to more readily access the intestinal epithelium and underlying tissue. The mechanism by which pks induces host DNA damage appears to require cell-cell contact,35,36 thus greater accessibility to the host epithelium could provide the close proximity needed to deliver genotoxic substances that may accelerate tumorigenesis. Recent evidence has suggested that the pks-encoded ClbP functions as a peptidase that cleaves an inactive pre-Colibactin to its active form in the periplasm.44 Nonetheless, how Colibactin is delivered to mammalian cells to exert its genotoxicity, and why cell-cell contact is required remains unknown.
In summary, our work defines a new paradigm where chronic inflammation alone is not sufficient to promote CRC and identifies the microbiota as a target of inflammation. As inflammation changes microbial composition and induces the expansion of microbes with cancer-promoting activities, the host is faced with a double hit system in which both endogenous inflammatory mediators and bacterial-derived mediators influence CRC development. The continous mining of the microbiota in health and disease will likely reveal novel therapeutic targets and treatment strategies for intestinal pathologies such as IBD and CRC.
Acknowledgments
This study received National Institutes of Health grants DK047700 and DK073338; the University of North Carolina at Chapel Hill; American Cancer Society PF-12–263–01-MPC.
Disclosure of Potential Conflicts of Interest
No potential conflict of interest was disclosed.
Footnotes
Previously published online: www.landesbioscience.com/journals/gutmicrobes/article/24220
References
- 1.Heidland A, Klassen A, Rutkowski P, Bahner U. The contribution of Rudolf Virchow to the concept of inflammation: what is still of importance? J Nephrol. 2006;19(Suppl 10):S102–9. [PubMed] [Google Scholar]
- 2.Chiba T, Marusawa H, Ushijima T. Inflammation-associated cancer development in digestive organs: mechanisms and roles for genetic and epigenetic modulation. Gastroenterology. 2012;143:550–63. doi: 10.1053/j.gastro.2012.07.009. [DOI] [PubMed] [Google Scholar]
- 3.Balkwill F, Mantovani A. Inflammation and cancer: back to Virchow? Lancet. 2001;357:539–45. doi: 10.1016/S0140-6736(00)04046-0. [DOI] [PubMed] [Google Scholar]
- 4.Ullman TA, Itzkowitz SH. Intestinal inflammation and cancer. Gastroenterology. 2011;140:1807–16. doi: 10.1053/j.gastro.2011.01.057. [DOI] [PubMed] [Google Scholar]
- 5.Lin W-W, Karin M. A cytokine-mediated link between innate immunity, inflammation, and cancer. J Clin Invest. 2007;117:1175–83. doi: 10.1172/JCI31537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Morgan XC, Tickle TL, Sokol H, Gevers D, Devaney KL, Ward DV, et al. Dysfunction of the intestinal microbiome in inflammatory bowel disease and treatment. Genome Biol. 2012;13:R79. doi: 10.1186/gb-2012-13-9-r79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sokol H, Lay C, Seksik P, Tannock GW. Analysis of bacterial bowel communities of IBD patients: what has it revealed? Inflamm Bowel Dis. 2008;14:858–67. doi: 10.1002/ibd.20392. [DOI] [PubMed] [Google Scholar]
- 8.Arthur JC, Perez-Chanona E, Mühlbauer M, Tomkovich S, Uronis JM, Fan TJ, et al. Intestinal inflammation targets cancer-inducing activity of the microbiota. Science. 2012;338:120–3. doi: 10.1126/science.1224820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Marshall BJ. The 1995 Albert Lasker Medical Research Award. Helicobacter pylori. The etiologic agent for peptic ulcer. JAMA. 1995;274:1064–6. doi: 10.1001/jama.274.13.1064. [DOI] [PubMed] [Google Scholar]
- 10.Klein RS, Recco RA, Catalano MT, Edberg SC, Casey JI, Steigbigel NH. Association of Streptococcus bovis with carcinoma of the colon. N Engl J Med. 1977;297:800–2. doi: 10.1056/NEJM197710132971503. [DOI] [PubMed] [Google Scholar]
- 11.Boleij A, van Gelder MMHJ, Swinkels DW, Tjalsma H. Clinical Importance of Streptococcus gallolyticus infection among colorectal cancer patients: systematic review and meta-analysis. Clin Infect Dis. 2011;53:870–8. doi: 10.1093/cid/cir609. [DOI] [PubMed] [Google Scholar]
- 12.Hooper LV, Gordon JI. Commensal host-bacterial relationships in the gut. Science. 2001;292:1115–8. doi: 10.1126/science.1058709. [DOI] [PubMed] [Google Scholar]
- 13.Lozupone CA, Stombaugh JI, Gordon JI, Jansson JK, Knight R. Diversity, stability and resilience of the human gut microbiota. Nature. 2012;489:220–30. doi: 10.1038/nature11550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kuczynski J, Lauber CL, Walters WA, Parfrey LW, Clemente JC, Gevers D, et al. Experimental and analytical tools for studying the human microbiome. Nat Rev Genet. 2012;13:47–58. doi: 10.1038/nrg3129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sobhani I, Tap J, Roudot-Thoraval F, Roperch JP, Letulle S, Langella P, et al. Microbial dysbiosis in colorectal cancer (CRC) patients. PLoS One. 2011;6:e16393. doi: 10.1371/journal.pone.0016393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang T, Cai G, Qiu Y, Fei N, Zhang M, Pang X, et al. Structural segregation of gut microbiota between colorectal cancer patients and healthy volunteers. ISME J. 2012;6:320–9. doi: 10.1038/ismej.2011.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sanapareddy N, Legge RM, Jovov B, McCoy A, Burcal L, Araujo-Perez F, et al. Increased rectal microbial richness is associated with the presence of colorectal adenomas in humans. ISME J. 2012;6:1858–68. doi: 10.1038/ismej.2012.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Marchesi JR, Dutilh BE, Hall N, Peters WH, Roelofs R, Boleij A, et al. Towards the human colorectal cancer microbiome. PLoS One. 2011;6:e20447. doi: 10.1371/journal.pone.0020447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kostic AD, Gevers D, Pedamallu CS, Michaud M, Duke F, Earl AM, et al. Genomic analysis identifies association of Fusobacterium with colorectal carcinoma. Genome Res. 2012;22:292–8. doi: 10.1101/gr.126573.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Castellarin M, Warren RL, Freeman JD, Dreolini L, Krzywinski M, Strauss J, et al. Fusobacterium nucleatum infection is prevalent in human colorectal carcinoma. Genome Res. 2012;22:299–306. doi: 10.1101/gr.126516.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chen W, Liu F, Ling Z, Tong X, Xiang C. Human intestinal lumen and mucosa-associated microbiota in patients with colorectal cancer. PLoS One. 2012;7:e39743. doi: 10.1371/journal.pone.0039743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Strauss J, Kaplan GG, Beck PL, Rioux K, Panaccione R, Devinney R, et al. Invasive potential of gut mucosa-derived Fusobacterium nucleatum positively correlates with IBD status of the host. Inflamm Bowel Dis. 2011;17:1971–8. doi: 10.1002/ibd.21606. [DOI] [PubMed] [Google Scholar]
- 23.Martin HM, Campbell BJ, Hart CA, Mpofu C, Nayar M, Singh R, et al. Enhanced Escherichia coli adherence and invasion in Crohn’s disease and colon cancer. Gastroenterology. 2004;127:80–93. doi: 10.1053/j.gastro.2004.03.054. [DOI] [PubMed] [Google Scholar]
- 24.Darfeuille-Michaud A, Neut C, Barnich N, Lederman E, Di Martino P, Desreumaux P, et al. Presence of adherent Escherichia coli strains in ileal mucosa of patients with Crohn’s disease. Gastroenterology. 1998;115:1405–13. doi: 10.1016/S0016-5085(98)70019-8. [DOI] [PubMed] [Google Scholar]
- 25.Swidsinski A, Khilkin M, Kerjaschki D, Schreiber S, Ortner M, Weber J, et al. Association between intraepithelial Escherichia coli and colorectal cancer. Gastroenterology. 1998;115:281–6. doi: 10.1016/S0016-5085(98)70194-5. [DOI] [PubMed] [Google Scholar]
- 26.Mukhopadhya I, Hansen R, El-Omar EM, Hold GL. IBD-what role do Proteobacteria play? Nat Rev Gastroenterol Hepatol. 2012;9:219–30. doi: 10.1038/nrgastro.2012.14. [DOI] [PubMed] [Google Scholar]
- 27.Uronis JM, Mühlbauer M, Herfarth HH, Rubinas TC, Jones GS, Jobin C. Modulation of the intestinal microbiota alters colitis-associated colorectal cancer susceptibility. PLoS One. 2009;4:e6026. doi: 10.1371/journal.pone.0006026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ubeda C, Lipuma L, Gobourne A, Viale A, Leiner I, Equinda M, et al. Familial transmission rather than defective innate immunity shapes the distinct intestinal microbiota of TLR-deficient mice. J Exp Med. 2012;209:1445–56. doi: 10.1084/jem.20120504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ott SJ, Musfeldt M, Wenderoth DF, Hampe J, Brant O, Fölsch UR, et al. Reduction in diversity of the colonic mucosa associated bacterial microflora in patients with active inflammatory bowel disease. Gut. 2004;53:685–93. doi: 10.1136/gut.2003.025403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Manichanh C, Rigottier-Gois L, Bonnaud E, Gloux K, Pelletier E, Frangeul L, et al. Reduced diversity of faecal microbiota in Crohn’s disease revealed by a metagenomic approach. Gut. 2006;55:205–11. doi: 10.1136/gut.2005.073817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Garrett WS, Gallini CA, Yatsunenko T, Michaud M, DuBois A, Delaney ML, et al. Enterobacteriaceae act in concert with the gut microbiota to induce spontaneous and maternally transmitted colitis. Cell Host Microbe. 2010;8:292–300. doi: 10.1016/j.chom.2010.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Luperchio SA, Schauer DB. Molecular pathogenesis of Citrobacter rodentium and transmissible murine colonic hyperplasia. Microbes Infect. 2001;3:333–40. doi: 10.1016/S1286-4579(01)01387-9. [DOI] [PubMed] [Google Scholar]
- 33.Shen XJ, Rawls JF, Randall T, Burcal L, Mpande CN, Jenkins N, et al. Molecular characterization of mucosal adherent bacteria and associations with colorectal adenomas. Gut Microbes. 2010;1:138–47. doi: 10.4161/gmic.1.3.12360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kim SC, Tonkonogy SL, Albright CA, Tsang J, Balish EJ, Braun J, et al. Variable phenotypes of enterocolitis in interleukin 10-deficient mice monoassociated with two different commensal bacteria. Gastroenterology. 2005;128:891–906. doi: 10.1053/j.gastro.2005.02.009. [DOI] [PubMed] [Google Scholar]
- 35.Nougayrède JP, Homburg S, Taieb F, Boury M, Brzuszkiewicz E, Gottschalk G, et al. Escherichia coli induces DNA double-strand breaks in eukaryotic cells. Science. 2006;313:848–51. doi: 10.1126/science.1127059. [DOI] [PubMed] [Google Scholar]
- 36.Cuevas-Ramos G, Petit CR, Marcq I, Boury M, Oswald E, Nougayrède JP. Escherichia coli induces DNA damage in vivo and triggers genomic instability in mammalian cells. Proc Natl Acad Sci U S A. 2010;107:11537–42. doi: 10.1073/pnas.1001261107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Homburg S, Oswald E, Hacker J, Dobrindt U. Expression analysis of the colibactin gene cluster coding for a novel polyketide in Escherichia coli. FEMS Microbiol Lett. 2007;275:255–62. doi: 10.1111/j.1574-6968.2007.00889.x. [DOI] [PubMed] [Google Scholar]
- 38.Putze J, Hennequin C, Nougayrède JP, Zhang W, Homburg S, Karch H, et al. Genetic structure and distribution of the colibactin genomic island among members of the family Enterobacteriaceae. Infect Immun. 2009;77:4696–703. doi: 10.1128/IAI.00522-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Olier M, Marcq I, Salvador-Cartier C, Secher T, Dobrindt U, Boury M, et al. Genotoxicity of Escherichia coli Nissle 1917 strain cannot be dissociated from its probiotic activity. Gut Microbes. 2012;3:501–9. doi: 10.4161/gmic.21737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Arthur JC, Jobin C. The struggle within: microbial influences on colorectal cancer. Inflamm Bowel Dis. 2011;17:396–409. doi: 10.1002/ibd.21354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Patwa LG, Fan T-J, Tchaptchet S, Liu Y, Lussier YA, Sartor RB, et al. Chronic intestinal inflammation induces stress-response genes in commensal Escherichia coli. Gastroenterology. 2011;141:1842–51, e1-10. doi: 10.1053/j.gastro.2011.06.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rhodes JM, Campbell BJ. Inflammation and colorectal cancer: IBD-associated and sporadic cancer compared. Trends Mol Med. 2002;8:10–6. doi: 10.1016/S1471-4914(01)02194-3. [DOI] [PubMed] [Google Scholar]
- 43.Grivennikov SI, Wang K, Mucida D, Stewart CA, Schnabl B, Jauch D, et al. Adenoma-linked barrier defects and microbial products drive IL-23/IL-17-mediated tumour growth. Nature. 2012;491:254–8. doi: 10.1038/nature11465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cougnoux A, Gibold L, Robin F, Dubois D, Pradel N, Darfeuille-Michaud A, et al. Analysis of structure-function relationships in the colibactin-maturating enzyme ClbP. J Mol Biol. 2012;424:203–14. doi: 10.1016/j.jmb.2012.09.017. [DOI] [PubMed] [Google Scholar]