

Abstract
PDCD4 (programmed cell death protein 4), a suppressor of gene transcription and translation, plays a crucial inhibitory role in several types of human tumors. However, its underlying mechanisms remain unclear. Autophagy, an evolutionarily conserved catabolic process, maintains cellular homeostasis under stress conditions such as starvation and plays a crucial role in tumor initiation and progression. We report here that PDCD4 inhibits autophagy in multiple cell types both in vitro and in vivo, which in turn contributes to its tumor suppressor activity. Importantly, PDCD4 inhibits the expression of an essential autophagy related gene, ATG5 and the formation of an ATG12–ATG5 complex, and its ma3 domains are required for PDCD4-mediated inhibition of autophagy. Unlike most tumor suppressors that act as positive or dual regulators of autophagy, our findings indicate that PDCD4 negatively regulates autophagy by targeting ATG5, which provides a novel mechanism of tumor suppression.
Keywords: PDCD4, tumor suppressor, autophagy, ATG5, proliferation
Introduction
PDCD4 (programmed cell death 4) is an important tumor suppressor, inhibiting carcinogenesis, tumor progression and invasion,1 which has been characterized to inhibit translation to exert its tumor suppressive function. It inhibits translation by binding to eukaryotic translation initiation factor (EIF) 4A via two ma3 domains that are highly homologous to EIF4G2-4 or combining directly with target gene (MYB/c-MYB) coding region.5 It also suppresses transcription-activator protein JUN/AP-1-dependent transcription6 via downregulating MAP4K.7 On the other hand, Pdcd4 transgenic mice showed significant resistance to tumor induction,8 whereas pdcd4−/− mice developed spontaneous lymphomas.9 In addition, loss or reduction of PDCD4 expression was observed in multiple types of human tumor cell lines and primary tumors such as lung tumor,10 colorectal tumor,11 glioma,12 ovarian13 and liver tumor,14 and was also implicated in the development and progression of these human tumors. Furthermore, PDCD4 suppresses the malignant phenotype of tumors15 and enhances the chemosensitivity of tumors.16 Overall, these are indications that PDCD4 is a tumor suppressor.
However, the mechanisms by which PDCD4 inhibits tumors remain unclear and conflicted in different tumors. PDCD4 was first discovered in a screen for genes activated during apoptosis,17 suggesting its proapoptotic role. Indeed, research since then has demonstrated that PDCD4 promotes tumor cell apoptosis in hepatocellular,14 ovarian,15 glioma,18 breast19 and gastric tumor.20 However, it has been observed that PDCD4 plays an antiapoptotic role in HeLa cells via suppressing proCASP3 mRNA translation21 and has no impact on apoptosis in colon carcinoma.7 Furthermore, PDCD4 plays inhibitory functions22 or no effect7 on the cell cycle in some tumor cell lines. This suggests that the apoptotic and cell cycle roles of PDCD4 may be limited to certain cell types,23 which cannot explain the suppression of PDCD4 on multiple tumors.
(Macro)autophagy, a conserved catabolic process whereby cellular proteins and organelles are engulfed by autophagosomes, digested in lysosomes, and recycled to sustain cellular homeostasis, has dual roles in tumors.24 Some data support the idea that autophagy is classified as an antioncogenic mechanism;25 however, accumulating evidence strongly suggests that autophagy enhances tumorigenesis and protects tumor cells from death.26 Autophagy, as compensatory response under stresses, supplies recovered energy for tumor cells for survival and the reprogramming of their metabolism to accommodate rapid cell growth and proliferation.27 Many tumor suppressors, such as TP53,28 PTEN,29 DAPK130 and TSC1/TSC231 are involved in regulation of autophagy. Our aim is to determine whether tumor suppressor PDCD4 also affects autophagy and, if so, the mechanism(s) it impacts.
The process of autophagosome formation is regulated by several autophagy-related (ATG) genes, such as ATG5, ATG6 (also known as BECN1), ATG8 (microtubule-associated protein 1 light chain 3, LC3) and ATG12. ATG5 is a protein involved at the early stage of autophagosome formation, which is conjugated with ATG12 to form ATG12–ATG5 complex that contributes to elongation and closure of the autophagosomes in the generation of lipidated forms of LC3 (ATG8) family proteins32 and their localization to the autophagosome membranes.33 BECN1, which acts upstream of autophagosome formation, governs the autophagy process by regulating the activity of the PIK3C3/VPS34-dependent generation of phosphatidylinositol-3-phosphate (PtdIns3P)34 and the subsequent recruitment of additional ATG proteins for initiating autophagosome formation.35 Since PDCD4 is an inhibitor of transcription and translation, we hypothesize that PDCD4 may interfere in the formation of the autophagosome by targeting autophagy-associated genes.
In our current study, the results indicate that PDCD4 inhibits autophagy via selectively attenuating expression of ATG5. Interestingly, we detected that PDCD4 inhibits autophagy in both tumor and nontumor cells, suggesting the inhibitory effect of PDCD4 on autophagy is universal. Consequently, the PDCD4–attenuated autophagy is a novel mechanism by which PDCD4 function as a tumor suppressor.
Results
PDCD4 suppresses starvation-induced autophagy in multiple cell types in vitro
First, we detected the effect of PDCD4 on autophagy by overexpression and knockdown in vitro. Our previous findings showed that ovarian tumor-derived cell line Skov3 and glioma-derived cell line U87 expressed low levels of PDCD4. PDCD4 was overexpressed in these two cell lines by transfecting cells with PDCD4 plasmids (pPDCD4) and empty vectors (Mock) as controls, and then subjecting the cells to 2 h of starvation to induce autophagy. As shown in Figure 1A, autophagy was suppressed by overexpressed PDCD4 both in Skov3 and U87 cell lines, as evidenced by the decreases in the amount of LC3B-II and increases in the amount of SQSTM1/p62, two important markers of autophagy.36 Furthermore, the decrease of LC3B-II levels led by forced PDCD4 expression were still observed both in Skov3 and U87 cell lines in the presence of pepstatin A and E64d (Fig. 1A). Unlike in U87 cells, in Skov3 cells PDCD4 overexpression led to a much larger increase in SQSTM1 levels after inhibition of lysosomal degradation, suggesting the effect of PDCD4 on SQSTM1 appeared dependent on cell types. Accordingly, inhibition of PDCD4 on autophagy was also verified by immunofluorescence to show the formation of autophagosomes labeled by anti-LC3 antibody, which demonstrated a decrease in the number of autophagosomes (Fig. 1B and C) (***p < 0.001) in cells overexpressing PDCD4. Furthermore, silencing of PDCD4 expression was done in hepatic tumor cell line HepG2 and cervical tumor cell line HeLa with high levels of PDCD4 expression. In contrast, knockdown of PDCD4 protein expression by two different PDCD4-specific siRNA sequences significantly led to the increases in the amount of LC3B-II and decreases in the amount of SQSTM1, both of which were accumulated after inhibition of lysosomal degradation by pepstatin A and E64d. (Fig. 1D). Correspondingly, immunofluorescence staining showed that silencing of PDCD4 led to activated autophagosomes formation in both HepG2 and HeLa cell lines (Fig. 1E and F) (***p < 0.001). In addition, inhibition of PDCD4 on autophagy was also observed in other cell lines, such as ovarian tumor-derived Caov3 (Fig. S1), mouse lymphocytes from spleen, primary peritoneal macrophages, monocyte macrophage leukemia-derived cell line RAW264.7 (Fig. S2A–S2E) and human embryonic kidney 293 cells (HEK293 cell) under various stimuli including starvation, LPS, or rapamycin (Fig. S2A–S2D). Taken together, the observation that PDCD4 inhibits autophagy in multiple cell types suggests that the inhibitory effect of PDCD4 is universal.

Figure 1. PDCD4 suppresses starvation-induced autophagy in multiple cell types in vitro. (A–C) Skov3 and U87 cells were transfected using pDsRed2-N1 (Mock) and pDsRed2-N1-PDCD4 (pPDCD4) plasmids, 24 h later and starvation for 2 h in the absence or presence of 10 E64d and pepstatin A. (A) Western blot analysis demonstrated PDCD4, SQSTM1, LC3B-I/LC3B-II expression in Skov3, U87 cells. All the western blot data were quantified by using Quantity One software. (B) The cells were inspected under confocal laser microscopy to detect LC3 puncta by immunofluorescence. (C) LC3 puncta number per cell was quantified using the Image J program, and 20 to 50 cells chosen in random were counted. The results shown are means ± SD; **p < 0.01, ***p < 0.001. (D–F) HepG2 and HeLa cells were transfected with two siRNA targeting PDCD4 (siP1, siP2) or control (siC). Twenty-four hours after siRNA interference, cells were starved for 2 h in the absence or presence of pepstatin A and E64d. (D) PDCD4, SQSTM1 and LC3B-I/LC3B-II protein levels were analyzed by western blot analysis. All the western blot data were quantified. (E and F) The cells were inspected under confocal laser microscopy to detect LC3 puncta by immunofluorescence. LC3 puncta per cell were quantified as described above. The results shown are means ± SD; ***p < 0.001
PDCD4 suppresses autophagy in murine xenograft tumors
To confirm the inhibitory effect of PDCD4 on autophagy in vivo, murine xenograft tumors were established as described in “Materials and Methods” and treated with mock plasmid and PDCD4 plasmid by intratumor injection. As our previous research shown, tumors in the PDCD4 group grew lower than that in the mock group and weight and volume of tumors in the PDCD4 group were also smaller than that in the mock group at end of experiment.16 Here, western blot analysis revealed that the level of LC3B-II protein expression was significantly decreased and the amount of SQSTM1 was increased in the PDCD4 group (pPDCD4) compared with the mock group (Mock) (Fig. 2A, *p < 0.05). The result was confirmed by immunofluorescence. As shown in Figure 2B, a decrease in the number of autophagosomes was observed in the PDCD4 injection group (pPDCD4) compared with the mock (Mock) group. In addition, experiments of colocalization between PDCD4 and LC3 were performed. As shown in Figure S3, the site where PDCD4 expression was higher showed lower LC3B-II expression, whereas the place where PDCD4 expression was lower revealed higher LC3B-II expression, which is consistent with inhibition of PDCD4 on autophagy in vitro. The present study provides further insights into the inhibitory role of PDCD4 in autophagy in vivo.

Figure 2. PDCD4 suppresses autophagy in murine xenograft tumors. Xenograft tumor was established and treated with N.S (Con), mock vector (Mock), PDCD4 plasmid (pPDCD4), respectively. (A) PDCD4, SQSTM1, LC3B-I/LC3B-II expression were analyzed by western blot. All the western data were quantified. The results shown are means ± SD; n = 4, n = 5; *p < 0.05. (B) Expression of PDCD4 was detected by immunohistochemistry under an optical microscope. LC3 puncta was detected by immunofluorescence under confocal laser microscopy. LC3 puncta was quantified as described above. The results shown are means ± SD; **p < 0.01.
PDCD4-attenuated autophagy is required for inhibition of tumor cell growth
We investigated the contribution of PDCD4-mediated autophagy to tumor cell growth. First, two Skov3 stable screened cells Skov3-Mock (Mock) and Skov3-PDCD4 (PDCD4) were employed. As shown in Figure 3A, Skov3-Mock cells with high levels of autophagy grew significantly faster than Skov3-PDCD4 cells with attenuated autophagy in serum-free medium over the course of 24 h. However, the blockage of autophagy by 3-MA (3-methyladenine), an important agent to block autophagy (Fig. 3B), retarded Skov3-Mock cell viability (Fig. 3C), suggesting attenuated autophagy is required for inhibition of tumor cell survival. Furthermore, we confirmed the role of PDCD4-attenuated autophagy in HepG2 cells. As shown in Figure 3D, knockdown of PDCD4 expression by siRNA caused an increase of cell viability and autophagy (Fig. 3D and E). Consistently, the elevated cell viability of HepG2 was inhibited by 3-MA-mediated blockage of autophagy (Fig. 3F). To confirm the data, si ATG5 treatment was performed in order to deplete autophagy, as shown in Figure 3G, silencing of ATG5 caused depletion of autophagy both in Skov3 and HepG2 cells, and the depletion of autophagy could be enhanced by overexpression of PDCD4 in Skov3 cells and retarded by silencing of PDCD4 in HepG2 cells. Correspondingly, the cell viability after treatment with siATG5 was similar to that of cells treated with 3-MA (Fig. 3H and I). Collectively, these data indicated that PDCD4-attenuated autophagy is required for inhibition of tumor cell proliferation.

Figure 3. PDCD4-attenuated autophagy is required for inhibition of tumor cell growth. (A–C) Skov3 stable cell lines (Mock, PDCD4) were cultured in serum-free medium. (A) Cell viability was measured by CCK8 kit for 24, 48, 72 and 96 h. (B) LC3 puncta was detected treated with 3-MA or not cultured in serum-free medium for 24 h. (C) Cell viability was measured with or without 3-MA to block autophagy for 24 h and 48 h. (D–F) HepG2 cells were transfected with siRNA targeting PDCD4 (siP) or control (siC) cultured in serum-free medium. The cell viability was determined using CCK8 assay and LC3 puncta level was detected by immunofluorescence with or without 3-MA treatment as described above. (G–I) Skov3 cells were treated with pEGFP-C1 (Mock) or pEGFP-C1-PDCD4 (pPDCD4) plasmids; HepG2 cells were transfected with siRNA targeting PDCD4 (siP) or control (siC) respectively. All the cells were cotransfected with siATG5 (siATG5) or control (siC) and cultured in serum-free medium for 24 h and 48 h. (G) 24 h later western blot analysis showed PDCD4, ATG5, and LC3B-I/LC3B-II expression (Skov3: left; HepG2: right), and the data were quantified by using Quantity One program. Twenty-four hours and 48 h later, CCK8 analysis showed cell viability in Skov3 (H) and HepG2 (I) cells. The results shown are means ± SD; *p < 0.05, **p < 0.01, ***p < 0.001.
PDCD4 inhibits autophagy-related ATG5 protein expression in vitro and in vivo.
To explore the mechanism by which PDCD4 inhibits autophagy, we further investigated the effect of PDCD4 on autophagy-related genes. BECN1, an important molecule for initiation of autophagosome formation, was first detected in PDCD4-forced and -silenced cells. As shown in Figure 4A and B and Figure S4, BECN1 was not affected by either overexpressed or silenced PDCD4 under autophagy-inducing conditions (starvation, LPS and rapamycin) (Fig. S4), indicating that PDCD4 inhibits autophagy in a BECN1-independent manner. As shown in Figure 3G, the depletion of autophagy caused by siATG5 could be enhanced by overexpression of PDCD4 and retarded by silencing of PDCD4, so ATG5 was taken into consideration as the potential target gene of PDCD4. As shown in Figure 4A, we found that forced PDCD4 expression in Skov3 remarkably inhibited formation of the ATG12–ATG5 complex (conj-ATG5 detected by anti-ATG5 and conj-ATG12 by anti-ATG12 antibodies) and free ATG5. Conversely, silencing of PDCD4 expression in HepG2 upregulated the ATG12–ATG5 complex (conj-ATG5 and, conj-ATG12) and free ATG5. (Fig. 4B). Interestingly, free ATG12 in Skov3 with exogenous PDCD4 or in HepG2 with high levels of endogenous PDCD4 was higher than their controls, suggesting that ATG12 is not responsible for downregulation of the ATG12–ATG complex. These results suggested that PDCD4 inhibits ATG5, but not either BECN1 or ATG12.

Figure 4. PDCD4 selectively inhibits autophagy-related ATG5 protein expression in vitro and in vivo. (A) Skov3 cells were transfected with PDCD4 plasmid (pPDCD4) and control (Mock) treated by starvation for 0, 4 or 8 h, then PDCD4, BECN1, conjugated ATG5 (conj ATG5), free ATG5, conjugated ATG12 (conj ATG12), free ATG12 and LC3B-I/LC3B-II were analyzed by western blot analysis. Data were quantified. (B) HepG2 cells were transfected with siRNA targeting PDCD4 (siP) or control (siC), and starved for 0, 4 or 8 h, and then PDCD4, BECN1, conjugated ATG5 (conj ATG5), free ATG5, conjugated ATG12 (conj ATG12), free ATG12 and LC3B-I/LC3B-II were analyzed by western blot analysis. Data were quantified by Quantity One. (C) Samples from human liver, ovarian cancers, and glioma were divided in two groups (PDCD4low and PDCD4high) according to PDCD4 expression and from each group, ten sections were chosen randomly and stained with an anti-ATG5 antibody for IHC analysis and pathologically graded, guided by a pathologist. The relationship between ATG5 and PDCD4 expression was analyzed in liver cancer (left), ovarian cancer and glioma (right). (D) Xenograft tumors paraffin sections of nude mice treated by injection with N.S (Con), mock vector (Mock), PDCD4 plasmid (pPDCD4) respectively were inspected under optical microscope to detect ATG5 expression by IHC. The results shown are means ± SD; *p < 0.05, **p < 0.01, ***p < 0.001.
Previous studies have shown that there is an observable lack of PDCD4 in several types of malignant tumors, including gliomas12 and ovarian tumors,13 hepatocellular carcinomas.14 To obtain more clinical evidence that PDCD4 inhibits ATG5, we compared the relationship of PDCD4 and ATG5 expression in clinical samples from human liver (n = 20), ovarian cancers (n = 20) and gliomas (n = 20). We found that the ATG5 expression in three kinds of tumors with negative or low PDCD4 expression (PDCD4low, n = 10) was significantly higher than that with higher PDCD4 expression (PDCD4high, n = 10). Moreover, the relationship between PDCD4 and ATG5 expression was analyzed in murine xenograft tumors. As shown in Figure 4D, ATG5 expression was significantly reduced in the PDCD4 group (pPDCD4) compared with the NS (Con) or mock (Mock) group. In addition, colocalization between PDCD4 and ATG5 showed (Fig. S5) that PDCD4 expression had an opposite relationship with ATG5 expression in the same part of the section. Taken together, our results indicated that PDCD4 inhibits ATG5 protein expression in vitro and in vivo.
PDCD4 inhibits ATG5 translation and ATG5-mediated autophagy via its ma3 domains
It has been known that PDCD4 is an inhibitor of protein transcription or translation.37 To determine whether inhibition of PDCD4 on ATG5 occurs on a transcriptional level or translational level, we detected the effect of PDCD4 on ATG5 mRNA and protein by western blot and real-time PCR in the same system. As shown in Figure 5A and B, ATG5 protein was suppressed significantly by forced PDCD4 expression, while ATG5 mRNA was unaffected by PDCD4 expression, suggesting the inhibition of PDCD4 on ATG5 occurs translationally, but not transcriptionally. Previous research has indicated that PDCD4 inhibits protein translation by binding to the RNA-helicase EIF4A via two ma3 domains.2-4 To determine whether PDCD4 inhibits ATG5 expression by this mechanism, a mutant PDCD4 expression vector (mutPDCD4) based on the pEGFP-C1-PDCD4 plasmid, which carries several amino acid substitutions of residues in its ma3 domains resulting in failure to bind with EIF4A, was constructed according to methods described previously.38 HEK293 cells were transfected with pEGFP-C1-PDCD4 and pEGFP-C1-mutPDCD4 plasmids respectively, and an RNA-immunoprecipitation assay was used. The lysates of HEK293 cells transfected with pEGFP-C1-PDCD4 or pEGFP-C1-mutPDCD4 were coprecipitated with EGFP-specific antibody and with isotype IgG as control, and mRNA in the coprecipitated complex was detected using real-time PCR. As shown in Figure 5C, ATG5 mRNA, but not that for ATG12, was enriched in the EGFP-specific immunoprecipitate compared with the IgG precipitate from wild-type PDCD4-transfected cells (***p < 0.001). Nevertheless, ATG5 mRNA was not detected in the EGFP4-specific immunoprecipitate from mutant PDCD4-treated cells. This demonstrates that PDCD4 inhibits ATG5 by binding of PDCD4-ma3 with EIF4A-ATG5 mRNA. Next, western blot analysis demonstrated only wild-type PDCD4, but not mutant PDCD4, remarkably inhibited expression of conjugated ATG5, conjugated ATG12, free ATG5 and LC3B-II, but elevated the expression of SQSTM1 and free ATG12 (Fig. 5D), which was verified by immunofluorescence: a decrease in the number of autophagosomes (Fig. 5E) was caused by only wild-type PDCD4, but not mutant PDCD4. Moreover, HEK293 cells were cotransfected with both pEGFP-C1-ATG539 and pEGFP-C1-PDCD4, and, as shown in Figure 5F, autophagy caused by forced ATG5 expression was suppressed remarkably by PDCD4 overexpression. Finally, Skov3 cells were transfected with wild-type and mutant PDCD4 vectors, and, as shown in Figure 5G, only wild-type, but not mutant PDCD4, suppressed Skov3 cell viability. All these indicated the inhibitory effect of PDCD4 on ATG5 protein expression, and that autophagy depended on the ma3 domains, on which PDCD4 functions are based, to allow PDCD4 to function as a tumor suppressor.

Figure 5. PDCD4 inhibits ATG5 translation and ATG5-mediated autophagy via its ma3 domains. (A and B) HEK293 cells were transfected using pEGFP-C1 (Mock), pEGFP-C1-PDCD4 (pPDCD4), 24 h after transfection, (A) PDCD4, ATG5, BECN1, LC3B-I/LC3B-II expression were analyzed by western blot analysis 2 h after starvation treatment. (B) Relative mRNA levels of PDCD4, ATG5 were examined using specific primers for PDCD4, ATG5. (C–E) HEK293 cells were transfected using pEGFP-C1 vector (Mock), pEGFP-C1-PDCD4 (pPDCD4), or pEGFP-C1-mutPDCD4 (mutPDCD4) plasmid, 24 h after transfection. (C) Cell lysates were subjected to RNA-IP with EGFP-specific antibodies or IgG (right). RNA-IP with anti-SNRNP70 or IgG (negative control) acts as positive control (left). Purified RNA was then analyzed by real-time PCR using specific primer for ATG5, ATG12 and positive control U1 snRNA. (D) EGFP, SQSTM1, conjugated ATG5 (conj ATG5), free ATG5, conjugated ATG12 (conj ATG12), free ATG12 and LC3B-I/LC3B-II expression were analyzed by western blot analysis 2 h after starvation treatment. Data were quantified by Quantity One. (E) LC3 puncta was detected by immunofluorescence under confocal laser microscopy and quantified as described above. (F) HEK293 cells were treated with pEGFP-C1 (Mock) or pEGFP-C1-ATG5 (pATG5) plasmids, and were cotransfected with pEGFP-C1-PDCD4 (pPDCD4) or control (Mock) and cultured in EBSS for 2 h. PDCD4, ATG5, and LC3B-I/LC3B-II were analyzed by western blot analysis. Data were quantified. (G) Skov3 cells were transfected with pEGFP-C1 vector (Mock), pEGFP-C1-PDCD4 (pPDCD4), or pEGFP-C1-mutPDCD4 (mutPDCD4) vectors respectively, and subjected to CCK8 analysis as described above. The results shown are means ± SD; *p < 0.05; **p < 0.01, ***p < 0.001;
Discussion
Accumulating evidence has demonstrated that tumor suppressors are involved in the regulation of autophagy. Although a few tumor suppressors, such as TP5340 and DAPK1,41 have been described as both positive and negative regulators, most, such as PTEN,29 CDKN2A42 and SH3GLB1,43 are positive regulators. Here, we demonstrate, for the first time, that tumor suppressor PDCD4 is only a negative regulator of autophagy by inhibiting a key autophagy effector protein, ATG5 and then downregulating ATG12–ATG5 conjugation via a ma3 domain-dependent manner. Importantly, the inhibitory role of PDCD4 in autophagy is a universal phenomenon, independent of the cell types and stresses it is exposed to. In addition, our data show that PDCD4-attenuated autophagy is required for inhibition of tumor cell viability, supporting the idea that autophagy promotes tumor growth. So our finding provides novel insights into the mechanism of tumor suppression on autophagy, and autophagy negative regulation.
PDCD4 as a tumor suppressor has been well characterized in Pdcd4-transgenic or knockdown mice and in multiple human primary tumors as described above. The effect of PDCD4 on apoptosis and cell cycle has been investigated using in vitro and in vivo models. However, its effect on autophagy remains unknown. Here, we find that forced PDCD4 expression in cultured cell lines markedly inhibits autophagy specific marker LC3B-II expression and formation of autophagosomes in vitro and in vivo, while silencing of PDCD4 upregulates them (Figs. 1 and 2). Importantly, we showed that inhibition of PDCD4 on autophagy is a universal phenomenon, independent of cell types and stimulations. PDCD4 not only inhibits the autophagy of multiple tumor-derived cells (such as HepG2, HeLa, Skov3, Caov3, U87 and RAW264.7) but also HEK293 cells, and even normal primary immune cells (mouse primary peritoneal macrophages, lymphocytes in spleen) (Fig. 1; Figs. S1 and S2), which is different from the effect of PDCD4 on apoptosis, cell cycle and chemosensitivity that has cell specificity. In spite of this, the phenomenon that PDCD4 regulated SQSTM1 appeared dependent on cell types. Unlike in U87 and others, in Skov3 cells PDCD4 overexpression led to a much larger increase in SQSTM1 levels after inhibition of lysosomal degradation (Fig. 1A), suggesting PDCD4 might also increase autophagic flux in Skov3. Since SQSTM1 is not only a target of autophagy but also an important negative regulator of autophagy,44 the effect of PDCD4 on SQSTM1 in various cells needs to be further investigated. In addition, we speculate this is associated with the inhibitory effect of PDCD4 on ATG5, which is an essential autophagy effector molecule and widely exists in multiple cells. However, the detailed mechanism of this feature remains to be investigated.
We have now explored the mechanisms by which PDCD4 attenuates autophagy and indicated that PDCD4 suppresses a key autophagy effector, ATG5. Further, we find that PDCD4-mediated inhibition of ATG5, and that attenuation of autophagy depends on its ma3 domains. The process of autophagosome formation is regulated by several autophagy genes. It has previously been observed that Atg5- or Becn1-deficient mouse embryonic stem cells have decreased numbers of autophagic vesicles, which suggests that BECN1 and ATG5 are essential for autophagy.45 The antiapoptotic BCL2 negatively regulates autophagy by sequestering BECN1 from the complex, thereby restricting the production of PtdIns3P and formation of the autophagosome.46 However, our previous research indicates that PDCD4 has no significant effect on BCL2 expression16 and here results further showed that PDCD4 had neither direct impact on BECN1 expression, suggesting PDCD4 suppresses autophagy in a BECN1-independent manner. Interestingly, several lines of evidence indicate that PDCD4 inhibits formation of ATG12–ATG5 complex by targeting ATG5 (Fig. 4). First, PDCD4 overexpression markedly suppresses the ATG5 (conj ATG5) and free ATG5 protein expression, accompanied by free ATG12 increase, while its silencing reversed this effect in multiple cultured cells (Fig. 4A and B); Second, inhibition of PDCD4 on ATG5 protein expression is detected in murine xenograft tumors and a reverse of PDCD4 with ATG5 was found in clinical tumor samples, including hepatocellular, ovarian tumor and glioma (Fig. 4C and D). Third, suppression of PDCD4 on ATG5 occurs on proteins translationally but not on the transcription level since PDCD4 has no effect on ATG5 mRNA (Fig. 5A). Fourth, PDCD4 is thought to suppress translation of mRNAs containing structured 5′-UTRs by interacting with translation initiation factor EIF4A and inhibiting its helicase activity via its ma3 domains.38 Here, we have identified ATG5 mRNA but not ATG12 mRNA as a translational target of PDCD4 by RNA-immunoprecipitation assay and this action depends on ma3 domains of PDCD4 via ma3 mutation assay. This demonstrates that PDCD4 inhibits ATG5 to attenuate autophagy by binding of PDCD4-ma3 with EIF4A-ATG5 mRNA. Since many genes are involved in the regulation of autophagy, we cannot deny that other genes also contribute to PDCD4-attenuated autophagy in addition to ATG5. We can conclude that ATG5 is an essential gene required to for PDCD4-attenuated autophagy.
Although it is still a matter of debate whether autophagy is ultimately a protective response or a detrimental process, it seems clear that autophagy can lead to adverse effects in cells under certain pathological conditions.26 Therefore, autophagy signaling and functions are necessities, but ones that must be under tight negative regulation. PDCD4 is ubiquitously expressed in all normal tissue cells, suggesting it plays an important role in regulating normal level of autophagy. However, under pathological conditions, such as tumor, loss or decrease of PDCD4 expression fails to control normal autophagy and is involved in the progression of tumors. It has previously been demonstrated that calpain negatively regulates ATG5 on post-translational level by mediating the cleavage of ATG5.47 We now report that PDCD4 negatively regulates ATG5 on mRNA translation via binding of ma3 domain with EIF4-ATG5 mRNA, suggesting PDCD4 and calpain may cooperate to regulate ATG5 and then control the level of autophagy. Calpain-mediated cleavage of ATG5 switches autophagy to apoptosis. Further work is required to understand how PDCD4 and calpain collaborate to control autophagy and apoptosis.
In addition, TP53 is an important regulator for autophagy.40 Therefore, the relationship of PDCD4 to TP53 in regulating autophagy will be an interesting issue. Our data show that PDCD4 is able to inhibit autophagy either in cell lines with wild-type TP53 (HeLa,48 U87,49 HepG250) or a cell line without TP53 (Skov351), suggesting that inhibition of PDCD4 on autophagy is TP53-independent. However, detailing the relationship of PDCD4 and TP53 in autophagy should be addressed in the future by more experiments.
Collectively, we add a novel insight into mechanism of tumor suppressor on autophagy negative regulation. We demonstrate that, as shown in Figure 6, tumor suppressor PDCD4 is a negative regulator of autophagy by inhibiting ATG5 and then downregulating ATG12–ATG5 conjugation via its ma3 domains. Ubiquitous expression of PDCD4 and ATG5 in normal cells suggests that PDCD4-mediated ATG5 inhibition is important for negative regulation of normal autophagy and control of cell growth. Loss or decrease of PDCD4 in tumor leads to abnormal autophagy and rapid cell proliferation.
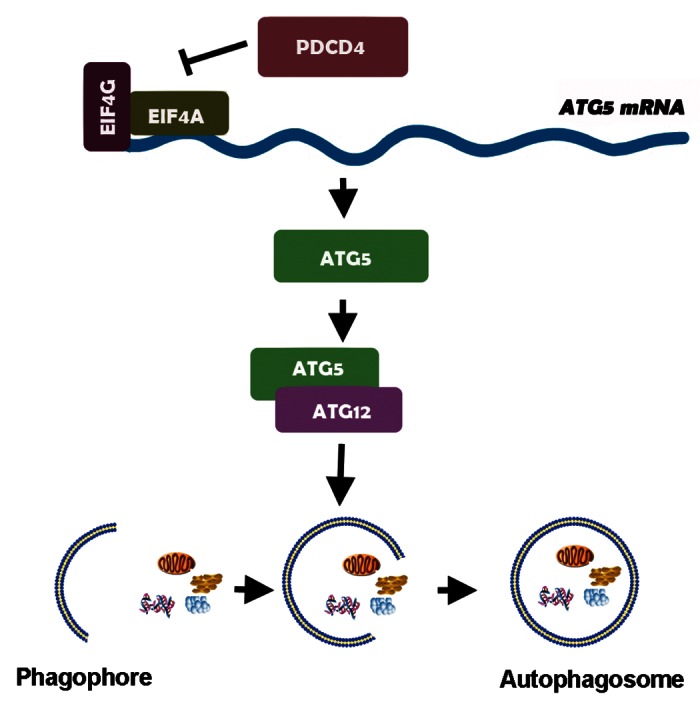
Figure 6. PDCD4 is a negative regulator of autophagy by selectively inhibiting expression of autophagy-related gene ATG5. Tumor suppressor PDCD4 is a negative regulator of autophagy by inhibiting a key autophagy effector protein, ATG5 and then downregulating ATG12–ATG5 conjugation via its ma3 domains, which contributes to elongation and closure of the autophagosomes in the generation of lipidated forms of LC3 (ATG8) family of proteins.
Materials and Methods
Cell culture
Human ovarian cancer-derived cell lines Skov3, Caov3, human glioma-derived cell line U87, human hepatocellular carcinoma-derived cell line HepG2, human cervical cancer-derived cell line HeLa, normal human embryonic cell line HEK293 cell, mice monocyte macrophage leukima-derived cell line RAW264.7 were purchased from the Shanghai Cell Bank of the Chinese Academy of Sciences (Shanghai, China) and the China Center for Type Culture Collection (Wuhan, China). Skov3 cells were grown in McCoy’s 5A (Sigma-Aldrich, M4892) supplemented with 10% FBS (Gibco, 10082147); RAW264.7, HEK293 and RAW264.7 were grown in DMEM (Gibco, 11965-092) supplemented with 10% FBS and all others were grown in RPMI 1640 medium (Gibco, 11875-093) supplemented with 10% FBS. The stably expressing PDCD4 cells (Skov3-PDCD4) and control cells (Skov3-Mock) were established as described previously15 and were cultured in medium with 300 μg/mL G418 (Merck, 345812). Cells were subjected to EBSS (Sigma-Aldrich, E7510-100ML) to induce autophagy with or without the presence of pepstatin A (Sigma-Aldrich, P5318) plus E64d (Sigma-Aldrich, E8640). In some experiments, 3-MA (3-methyladenine) (Sigma-Aldrich, M9281) was used to block autophagy.
Peritoneal macrophages from pdcd4−/− and sex-matched C57BL/6 mice at 8-weeks-old were cultivated with DMEM (Gibco) supplemented with 10% FBS and incubated for 2 h to allow macrophage adherence. The nonadherent cells were removed by three washes with DMEM. The adherent cells were harvested and the purity of macrophages was detected by flow cytometry following staining with PECy5-conjugated anti-F4/80 Ab (eBioscience, 11-4801-82) (purity > 95%). One × 106 purified macrophage cells were incubated for an additional 24 h, and the next day, were stimulated with starvation (EBSS), LPS (Sigma-Aldrich, L2880) or rapamycin (Sigma-Aldrich,R8781).
Cancer samples
Glioma and ovarian cancer samples came from patients who underwent surgical operations at Qilu Hospital and the Second Hospital, Shandong University from 2001 to 2007. The detailed information had been described previously.12,13 Primary hepatocellular carcinoma (n = 30) samples were obtained from patients aged between 30 and 82 y who underwent operations at the Qilu Hospital of Shandong University from January 2005 to October 2006. PDCD4 expression was detected by IHC and was graded as previously described.12,13 According to the expression intensity, samples were divided into two groups: PDCD4low and PDCD4high, from each group, ten sections were chosen randomly and stained with an anti-ATG5 antibody (Abcam, ab78073) by IHC to show ATG5 expression.
Recombinant vector, transfection and siRNA interference
pDsRed2-N1 and pDsRed2-N1-PDCD4 plasmids were kindly provided by Dr. Ozaki (Department of Internal Medicine, Saga Medical School, Saga University, Saga, Japan). pEGFP-C1 or pEGFP-C1-PDCD4 plasmids were constructed and kindly provided by Dr. Olubunmi Afonja (New York University, New York, NY, USA). pEGFP-C1-mutPDCD4 vector encoding a mutant of human PDCD4 by changing Glu249, Asp253, Asp414 and Asp418 into Ala was constructed as previously described.38pEGFP-C1-ATG5 was a gift of Ohsumi (Addgene plasmid #22952).39 Transfection of expressive vectors was performed using Lipofectamine 2000 according to the manufacturer’s protocols (Invitrogen,18292-011).
siRNA interference was performed as described previously.15 Two different Silencer Select Predesigned siRNA targeting PDCD4 and nonspecific negative control were purchased from Ambion. For siRNA-PDCD4–1: 5′GAGAUGGAAUUUUAUGUAATT3′, and 5′UUACAUAAAAUUCCAUCUCCA3′; for siRNA-PDCD4–2: 5′GG-CUGGAAUAAUUUCCAAATT3′, and 5′UUUGGAAAUUAUUCCAGCCTT3′. The siRNAs were transfected using siPORT NeoFX Transfection Agent (Ambion, AM4510) according to the manufacturer’s protocol. In some experiments, only siRNA-PDCD4-2 was applied. The Silencer Select Predesigned siRNA targeting ATG5 and nonspecific negative control were purchased from GenePharma (China, Shanghai). These were, for siRNA-ATG5: 5′ CCAUCAAUCGGAAACUCAUTT 3′; and 5′ AUGAGUUUCCGAUUGAUGGTT 3′. siRNA interference was performed using Lipofectamine 2000 according to the manufacturer’s protocols.
Western blots
The extraction and detection of protein from cultured cells and xenocraft tumor tissues was as described.15 50 μg quantity of protein was separated on SDS-PAGE and transferred onto PVDF membranes. Membranes were then blocked with 2% bovine serum albumin (BSA) in TBST containing 0.1% Tween-20 for 1 h. Membranes were probed overnight at 4°C with the following primary antibodies: rabbit monoclonal antibodies against human PDCD4 (9535S), LC3 (2775S), ATG5 (conj form, free form) (2630), ATG12 (conj form, free form) (4180S), and BECN1 (3738) (all 1:1000; Cell Signaling Technology, CST), mouse monoclonal antibody against ACTIN (1:2000; Santa Cruz, sc-7210), followed by secondary antibody conjugated (Beyotime, A0208) with peroxidase for 1 h at room temperature. After washing, signals were visualized by Super Signal West Pico Chemiluminescent Substrate (Pierce Biotechnology). Western blots were performed at least three times for each sample.
Immunohistochemistry (IHC)
Immunohistochemistry was performed as previously described.12 The slides were blocked for endogenous peroxidase activity, preincubated with goat serum, and then stained with poly-antibody goat-rabbit anti-ATG5 (1:300) (Abcam, ab78073) or anti-PDCD4 (1:700) by incubating for 1 h at room temperature. Secondary staining with HRP-conjugated anti-rabbit IgG was performed using a MaxVision Kit and a DAB Peroxidase Substrate Kit (Maixin, Kit-0017). Negative controls for the specificity of immunohistochemical reactions were performed by replacing the primary antibody with IgG of non-immunized rabbit (Beyotime, A7016). Immunohistochemistry was performed twice for each sample.
Immunofluorescence (IF)
Tumor cells were cultured on Glass Bottom Cell Culture Dishes (NEST, 801002) for detection by laser scanning confocal microscope (3.0 × 105 cells per 35-mm dish) and then subjected to treatments as indicated. Cells were fixed with 4% paraformaldehyde and permeabilized with 0.1% Triton X-100 for 15 min. After incubation for 1 h with anti-LC3 antibodies (1:300) (CST, 2775S) and washing with PBS, cells were incubated for 1 h with Alexa 488-conjugated (1:1000) (Beyotime, P0176) or Alexa 555-conjugated (1:500) (Beyotime, P0176) secondary antibodies, washed with PBS. Nuclei were stained by 4′, 6-diamidino-2-phenylindole (DAPI) (Beyotime, C1005) for 5min. Xenograft tumors paraffin sections were pretreated as described in IHC. After preincubation with goat serum, sections were stained with anti-LC3 (1:100) by incubating overnight at 4°C, followed by incubation with Alexa 488-conjugated (1:1000) secondary antibodies for 1 h, washed with PBS. Nuclei were stained by DAPI for 5min. Microscopy was done on a confocal laser microscopy (Carl Zeiss, LSM780).
Cell proliferation assay
Stable Skov3 cell lines were seeded in 96-well plates at a density of 3000 cells per well. HepG2 cells were seeded at a density of 5000 cells per well to overnight at 37°C, subjected to siRNA interference. All the cells were cultured with serum-free media instead of the serum-containing medium for indicated time points and OD value was measured at 450 nm using Cell Counting Kit-8 (Dojindo, CK04) at various time points according to the guidance of the manufacturer. Cell viability was calculated according to the formula: experimental OD value/control OD value × 100%.
RNA isolation and real-time PCR
Total RNA was extracted from cells using Trizol Reagent (Invitrogen). Real-time PCR was performed using UltraSYBR Mixture (CWBIO, CW0956) and specific primer pairs. The sequences of the sense and antisense primers were as follows: PDCD4: 5′ TGTAAACCCTGCAGATCCTGATAA 3′ and 5′TGGAGGATGCTGAAATCCAA 3′; ATG5: 5′-TGTGCTTCGAGATGTGTGGTT-3′ and 5′-ACCAACGTCAAATAGCTGACTC-3′; ATG12: 5′ AGTAGAGCGAACACGAACCA 3′ and 5′ GGAAGGAGCAAAGGACTGAT 3′; ACTB: 5′-AGTTGCGTTACACCCTTTC -3′, and 5′- CCTTCACCGTTCCAGTTT -3′. Data of relative molecule expression was presented by real-time quantitative PCR using the ∆∆Ct model. Using the 2^∆∆Ct method, our data were reported as the fold change in experimental group normalized to an endogenous reference gene (ACTB) and relative to control group.
In vivo studies using a xenograft animal model
Xenograft animal model was performed as previously described.16 Briefly, Nude mice, 6 to 8 weeks old, were inoculated s.c. into the left flank with 5 × 106 Skov3 cells in 100 μL PBS. When reached approximately 100 mm3 in size 1 week later, tumors were treated with 20 μg Mock plasmid and PDCD4 plasmid in a total volume of 50 μL by intra-tumor injection every 4 d for a total of four times. All mice were killed 1 week after completion of treatment and part of the tumors were then fixed in 4% formalin and processed for paraffin embedding for immunohistochemistry and immunofluorescence; others were milled, and lysed in RIPA Lysis Buffer (Beyotime, P0013B) for western blot analysis. The protocol was approved by the Animal Care and Utilization Committee of Shandong University. This study is also in full compliance with the guidelines for the welfare of animals in experimental neoplasia.
RNA-immunoprecipitation
RNA immunoprecipitation experiments were performed using a RIP kit (Millipore, 17-701,) according to the manufacturer’s protocol. Briefly, 2 × 107 HEK293 cells were plated on 10-cm dishes, 24h after transfection using pEGFP-C1-PDCD4 or pEGFP-C1-mutPDCD4, subjected to starvation for 2 h to induce ATG5 expression. After washing, cells were lysed with RIP lysis buffer, 10% of the supernatant (cytoplasmic fraction) was saved as input samples and the remaining material was used for immunoprecipitation to overnight at 4°C, using special antibody against EGFP (CWBIO, CW0258) or IgG (negative control) or SNRNP70 (positive control) which had been preincubated with magnetic beads at room temperature for 30 min. Protein binding with PDCD4 or IgG or SNRNP70 was digested by Proteinase K at 55°C for 30 min, and mRNA was extracted and purified with phenol: chloroform: isoamyl alcohol. cDNA synthesis and real-time PCR were performed as described above. RNA binding with positive control SNRNP70 by the primer for U1snRNA: 5′-GGGAGATACCATGATCACGAAGGT-3′ and 5′-CCACAAATTATGCAGTCGAGTTTCCC-3′.
Statistical analysis
Statistical significance was evaluated with data from at least three independent experiments. Statistical analysis was performed using the unpaired t-test or one-way ANOVA (spss 11.0; SPSS). Data are presented as the mean ± SEM. For all statistical tests, significance was established at p < 0.05.
Supplementary Material
Acknowledgments
This research was supported by the National “973” program of China (2011CB503906), the National Natural Science Foundation of China (81172863, 81128013, 81202069 and 81072407), Natural Science Foundation of Shandong (Z2008C02).
Glossary
Abbreviations:
- PDCD4
programmed cell death 4
- EIF4A/4G
eukaryotic translation initiation factor 4A/4G
- JUN/AP-1
jun proto-oncogene/activator protein-1
- MAP4K
mitogen-activated protein kinase kinase kinase kinase
- MYB/c-MYB
v-myb myeloblastosis viral oncogene homolog
- CASP3
caspase 3, apoptosis-related cysteine peptidase
- PTEN
phosphatase and tensin homolog
- DAPK1
death-associated protein kinase 1
- TSC1/TSC2
tuberous sclerosis 1/tuberous sclerosis 2
- ATG
autophagy-related gene
- BECN1
Beclin 1
- LC3
microtubule-associated protein 1 light chain 3
- PIK3C3
phosphatidylinositol 3-kinase, catalytic subunit type 3
- PtdIns3P
phosphatidylinositol-3-phosphate
- SQSTM1/p62
sequestosome 1
- LPS
lipopolysaccharide
- 3-MA
3-methyladenine
- CDKN2A
cyclin-dependent kinase inhibitor 2A
- SH3GLB1
SH3-domain GRB2-like endophilin B1
- N.S.
normal saline
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Supplemental Materials
Supplemental materials may be found here: www.landesbioscience.com/journals/autophagy/article/24069
Footnotes
Previously published online: www.landesbioscience.com/journals/autophagy/article/24069
References
- 1.Lankat-Buttgereit B, Göke R. The tumour suppressor Pdcd4: recent advances in the elucidation of function and regulation. Biol Cell. 2009;101:309–17. doi: 10.1042/BC20080191. [DOI] [PubMed] [Google Scholar]
- 2.Göke A, Göke R, Knolle A, Trusheim H, Schmidt H, Wilmen A, et al. DUG is a novel homologue of translation initiation factor 4G that binds eIF4A. Biochem Biophys Res Commun. 2002;297:78–82. doi: 10.1016/S0006-291X(02)02129-0. [DOI] [PubMed] [Google Scholar]
- 3.Yang HS, Jansen AP, Komar AA, Zheng X, Merrick WC, Costes S, et al. The transformation suppressor Pdcd4 is a novel eukaryotic translation initiation factor 4A binding protein that inhibits translation. Mol Cell Biol. 2003;23:26–37. doi: 10.1128/MCB.23.1.26-37.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yang HS, Cho MH, Zakowicz H, Hegamyer G, Sonenberg N, Colburn NH. A novel function of the MA-3 domains in transformation and translation suppressor Pdcd4 is essential for its binding to eukaryotic translation initiation factor 4A. Mol Cell Biol. 2004;24:3894–906. doi: 10.1128/MCB.24.9.3894-3906.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Singh P, Wedeken L, Waters LC, Carr MD, Klempnauer KH. Pdcd4 directly binds the coding region of c-myb mRNA and suppresses its translation. Oncogene. 2011;30:4864–73. doi: 10.1038/onc.2011.202. [DOI] [PubMed] [Google Scholar]
- 6.Yang HS, Knies JL, Stark C, Colburn NH. Pdcd4 suppresses tumor phenotype in JB6 cells by inhibiting AP-1 transactivation. Oncogene. 2003;22:3712–20. doi: 10.1038/sj.onc.1206433. [DOI] [PubMed] [Google Scholar]
- 7.Yang HS, Matthews CP, Clair T, Wang Q, Baker AR, Li CC, et al. Tumorigenesis suppressor Pdcd4 down-regulates mitogen-activated protein kinase kinase kinase kinase 1 expression to suppress colon carcinoma cell invasion. Mol Cell Biol. 2006;26:1297–306. doi: 10.1128/MCB.26.4.1297-1306.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jansen AP, Camalier CE, Colburn NH. Epidermal expression of the translation inhibitor programmed cell death 4 suppresses tumorigenesis. Cancer Res. 2005;65:6034–41. doi: 10.1158/0008-5472.CAN-04-2119. [DOI] [PubMed] [Google Scholar]
- 9.Hilliard A, Hilliard B, Zheng SJ, Sun H, Miwa T, Song W, et al. Translational regulation of autoimmune inflammation and lymphoma genesis by programmed cell death 4. J Immunol. 2006;177:8095–102. doi: 10.4049/jimmunol.177.11.8095. [DOI] [PubMed] [Google Scholar]
- 10.Chen Y, Knösel T, Kristiansen G, Pietas A, Garber ME, Matsuhashi S, et al. Loss of PDCD4 expression in human lung cancer correlates with tumour progression and prognosis. J Pathol. 2003;200:640–6. doi: 10.1002/path.1378. [DOI] [PubMed] [Google Scholar]
- 11.Zhang Z, DuBois RN. Detection of differentially expressed genes in human colon carcinoma cells treated with a selective COX-2 inhibitor. Oncogene. 2001;20:4450–6. doi: 10.1038/sj.onc.1204588. [DOI] [PubMed] [Google Scholar]
- 12.Gao F, Zhang P, Zhou C, Li J, Wang Q, Zhu F, et al. Frequent loss of PDCD4 expression in human glioma: possible role in the tumorigenesis of glioma. Oncol Rep. 2007;17:123–8. [PubMed] [Google Scholar]
- 13.Wang X, Wei Z, Gao F, Zhang X, Zhou C, Zhu F, et al. Expression and prognostic significance of PDCD4 in human epithelial ovarian carcinoma. Anticancer Res. 2008;28(5B):2991–6. [PubMed] [Google Scholar]
- 14.Zhang H, Ozaki I, Mizuta T, Hamajima H, Yasutake T, Eguchi Y, et al. Involvement of programmed cell death 4 in transforming growth factor-beta1-induced apoptosis in human hepatocellular carcinoma. Oncogene. 2006;25:6101–12. doi: 10.1038/sj.onc.1209634. [DOI] [PubMed] [Google Scholar]
- 15.Wei ZT, Zhang X, Wang XY, Gao F, Zhou CJ, Zhu FL, et al. PDCD4 inhibits the malignant phenotype of ovarian cancer cells. Cancer Sci. 2009;100:1408–13. doi: 10.1111/j.1349-7006.2009.01210.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang X, Wang X, Song X, Liu C, Shi Y, Wang Y, et al. Programmed cell death 4 enhances chemosensitivity of ovarian cancer cells by activating death receptor pathway in vitro and in vivo. Cancer Sci. 2010;101:2163–70. doi: 10.1111/j.1349-7006.2010.01664.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shibahara K, Asano M, Ishida Y, Aoki T, Koike T, Honjo T. Isolation of a novel mouse gene MA-3 that is induced upon programmed cell death. Gene. 1995;166:297–301. doi: 10.1016/0378-1119(95)00607-9. [DOI] [PubMed] [Google Scholar]
- 18.Gao F, Wang X, Zhu F, Wang Q, Zhang X, Guo C, et al. PDCD4 gene silencing in gliomas is associated with 5’CpG island methylation and unfavourable prognosis. J Cell Mol Med. 2009;13:4257–67. doi: 10.1111/j.1582-4934.2008.00497.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Afonja O, Juste D, Das S, Matsuhashi S, Samuels HH. Induction of PDCD4 tumor suppressor gene expression by RAR agonists, antiestrogen and HER-2/neu antagonist in breast cancer cells. Evidence for a role in apoptosis. Oncogene. 2004;23:8135–45. doi: 10.1038/sj.onc.1207983. [DOI] [PubMed] [Google Scholar]
- 20.Wang WQ, Zhang H, Wang HB, Sun YG, Peng ZH, Zhou G, et al. Programmed cell death 4 (PDCD4) enhances the sensitivity of gastric cancer cells to TRAIL-induced apoptosis by inhibiting the PI3K/Akt signaling pathway. Mol Diagn Ther. 2010;14:155–61. doi: 10.1007/BF03256368. [DOI] [PubMed] [Google Scholar]
- 21.Eto K, Goto S, Nakashima W, Ura Y, Abe SI. Loss of programmed cell death 4 induces apoptosis by promoting the translation of procaspase-3 mRNA. Cell Death Differ. 2012;19:573–81. doi: 10.1038/cdd.2011.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Göke R, Barth P, Schmidt A, Samans B, Lankat-Buttgereit B. Programmed cell death protein 4 suppresses CDK1/cdc2 via induction of p21(Waf1/Cip1) Am J Physiol Cell Physiol. 2004;287:C1541–6. doi: 10.1152/ajpcell.00025.2004. [DOI] [PubMed] [Google Scholar]
- 23.Lankat-Buttgereit B, Lenschen B, Schmidt H, Göke R. The action of Pdcd4 may be cell type specific: evidence that reduction of dUTPase levels might contribute to its tumor suppressor activity in Bon-1 cells. Apoptosis. 2008;13:157–64. doi: 10.1007/s10495-007-0153-x. [DOI] [PubMed] [Google Scholar]
- 24.Choi KS. Autophagy and cancer. Exp Mol Med. 2012;44:109–20. doi: 10.3858/emm.2012.44.2.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Czyzyk-Krzeska MF, Meller J, Plas DR. Not all autophagy is equal. Autophagy. 2012;8:1155–6. doi: 10.4161/auto.20650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ogier-Denis E, Codogno P. Autophagy: a barrier or an adaptive response to cancer. Biochim Biophys Acta. 2003;1603:113–28. doi: 10.1016/s0304-419x(03)00004-0. [DOI] [PubMed] [Google Scholar]
- 27.Lin CI, Whang EE, Abramson MA, Jiang X, Price BD, Donner DB, et al. Autophagy: a new target for advanced papillary thyroid cancer therapy. Surgery. 2009;146:1208–14. doi: 10.1016/j.surg.2009.09.019. [DOI] [PubMed] [Google Scholar]
- 28.Yamaki M, Umehara T, Chimura T, Horikoshi M. Cell death with predominant apoptotic features in Saccharomyces cerevisiae mediated by deletion of the histone chaperone ASF1/CIA1. Genes Cells. 2001;6:1043–54. doi: 10.1046/j.1365-2443.2001.00487.x. [DOI] [PubMed] [Google Scholar]
- 29.Arico S, Petiot A, Bauvy C, Dubbelhuis PF, Meijer AJ, Codogno P, et al. The tumor suppressor PTEN positively regulates macroautophagy by inhibiting the phosphatidylinositol 3-kinase/protein kinase B pathway. J Biol Chem. 2001;276:35243–6. doi: 10.1074/jbc.C100319200. [DOI] [PubMed] [Google Scholar]
- 30.Inbal B, Bialik S, Sabanay I, Shani G, Kimchi A. DAP kinase and DRP-1 mediate membrane blebbing and the formation of autophagic vesicles during programmed cell death. J Cell Biol. 2002;157:455–68. doi: 10.1083/jcb.200109094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kvam E, Gable K, Dunn TM, Goldfarb DS. Targeting of Tsc13p to nucleus-vacuole junctions: a role for very-long-chain fatty acids in the biogenesis of microautophagic vesicles. Mol Biol Cell. 2005;16:3987–98. doi: 10.1091/mbc.E05-04-0290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hanada T, Noda NN, Satomi Y, Ichimura Y, Fujioka Y, Takao T, et al. The Atg12-Atg5 conjugate has a novel E3-like activity for protein lipidation in autophagy. J Biol Chem. 2007;282:37298–302. doi: 10.1074/jbc.C700195200. [DOI] [PubMed] [Google Scholar]
- 33.Fujita N, Itoh T, Omori H, Fukuda M, Noda T, Yoshimori T. The Atg16L complex specifies the site of LC3 lipidation for membrane biogenesis in autophagy. Mol Biol Cell. 2008;19:2092–100. doi: 10.1091/mbc.E07-12-1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tassa A, Roux MP, Attaix D, Bechet DM. Class III phosphoinositide 3-kinase--Beclin1 complex mediates the amino acid-dependent regulation of autophagy in C2C12 myotubes. Biochem J. 2003;376:577–86. doi: 10.1042/BJ20030826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Juhász G, Hill JH, Yan Y, Sass M, Baehrecke EH, Backer JM, et al. The class III PI(3)K Vps34 promotes autophagy and endocytosis but not TOR signaling in Drosophila. J Cell Biol. 2008;181:655–66. doi: 10.1083/jcb.200712051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Klionsky DJ, Abeliovich H, Agostinis P, Agrawal DK, Aliev G, Askew DS, et al. Guidelines for the use and interpretation of assays for monitoring autophagy in higher eukaryotes. Autophagy. 2008;4:151–75. doi: 10.4161/auto.5338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Vikhreva PN, Shepelev MV, Korobko EV, Korobko IV. [Pdcd4 tumor suppressor: properties, functions, and their application to oncology] Mol Gen Mikrobiol Virusol. 2010;2:3–11. [PubMed] [Google Scholar]
- 38.Wedeken L, Singh P, Klempnauer KH. Tumor suppressor protein Pdcd4 inhibits translation of p53 mRNA. J Biol Chem. 2011;286:42855–62. doi: 10.1074/jbc.M111.269456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mizushima N, Sugita H, Yoshimori T, Ohsumi Y. A new protein conjugation system in human. The counterpart of the yeast Apg12p conjugation system essential for autophagy. J Biol Chem. 1998;273:33889–92. doi: 10.1074/jbc.273.51.33889. [DOI] [PubMed] [Google Scholar]
- 40.Scherz-Shouval R, Weidberg H, Gonen C, Wilder S, Elazar Z, Oren M. p53-dependent regulation of autophagy protein LC3 supports cancer cell survival under prolonged starvation. Proc Natl Acad Sci U S A. 2010;107:18511–6. doi: 10.1073/pnas.1006124107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Stevens C, Lin Y, Harrison B, Burch L, Ridgway RA, Sansom O, et al. Peptide combinatorial libraries identify TSC2 as a death-associated protein kinase (DAPK) death domain-binding protein and reveal a stimulatory role for DAPK in mTORC1 signaling. J Biol Chem. 2009;284:334–44. doi: 10.1074/jbc.M805165200. [DOI] [PubMed] [Google Scholar]
- 42.Sherr CJ. Autophagy by ARF: a short story. Mol Cell. 2006;22:436–7. doi: 10.1016/j.molcel.2006.05.005. [Review] [DOI] [PubMed] [Google Scholar]
- 43.Takahashi Y, Coppola D, Matsushita N, Cualing HD, Sun M, Sato Y, et al. Bif-1 interacts with Beclin 1 through UVRAG and regulates autophagy and tumorigenesis. Nat Cell Biol. 2007;9:1142–51. doi: 10.1038/ncb1634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Moscat J, Diaz-Meco MT. p62: a versatile multitasker takes on cancer. Trends Biochem Sci. 2012;37:230–6. doi: 10.1016/j.tibs.2012.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Qu X, Zou Z, Sun Q, Luby-Phelps K, Cheng P, Hogan RN, et al. Autophagy gene-dependent clearance of apoptotic cells during embryonic development. Cell. 2007;128:931–46. doi: 10.1016/j.cell.2006.12.044. [DOI] [PubMed] [Google Scholar]
- 46.Pattingre S, Tassa A, Qu X, Garuti R, Liang XH, Mizushima N, et al. Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell. 2005;122:927–39. doi: 10.1016/j.cell.2005.07.002. [DOI] [PubMed] [Google Scholar]
- 47.Yousefi S, Perozzo R, Schmid I, Ziemiecki A, Schaffner T, Scapozza L, et al. Calpain-mediated cleavage of Atg5 switches autophagy to apoptosis. Nat Cell Biol. 2006;8:1124–32. doi: 10.1038/ncb1482. [DOI] [PubMed] [Google Scholar]
- 48.Hoppe-Seyler F, Butz K. Repression of endogenous p53 transactivation function in HeLa cervical carcinoma cells by human papillomavirus type 16 E6, human mdm-2, and mutant p53. J Virol. 1993;67:3111–7. doi: 10.1128/jvi.67.6.3111-3117.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Van Meir EG, Kikuchi T, Tada M, Li H, Diserens AC, Wojcik BE, et al. Analysis of the p53 gene and its expression in human glioblastoma cells. Cancer Res. 1994;54:649–52. [PubMed] [Google Scholar]
- 50.Khaoustov VI, Ozer A, Smith JR, Noda A, Mearns M, Krishnan B, et al. Induction of senescent cell-derived inhibitor of DNA synthesis gene, SDI1, in hepatoblastoma (HepG2) cells arrested in the G2-phase of the cell cycle by 9-nitrocamptothecin. Lab Invest. 1995;73:118–27. [PubMed] [Google Scholar]
- 51.Vikhanskaya F, Erba E, D’Incalci M, Broggini M. Introduction of wild-type p53 in a human ovarian cancer cell line not expressing endogenous p53. Nucleic Acids Res. 1994;22:1012–7. doi: 10.1093/nar/22.6.1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.