

Abstract
Necroptosis, a caspase-independent, receptor (TNFRSF)-interacting serine-threonine kinase 1 (RIPK1)/RIPK3-dependent necrotic cell death, occurs in cells when apoptosis is blocked. A high level of macroautophagy (herein referred to as autophagy) is usually detected in necroptotic cells, although it is still controversial as to whether excessive autophagy leads to cell death or is cytoprotective. In a recently published paper, we show that the anti-apoptotic protein CFLAR (CASP8 and FADD-like apoptosis regulator) long isoform (CFLARL) plays a critical role in all three fundamental intracellular processes: autophagy, necroptosis, and apoptosis in T lymphocytes. CFLARL-deficient T cells suffer from severe cell death upon T cell receptor stimulation, in which both apoptosis and necroptosis are involved. Autophagy is enhanced in both naïve and activated CFLARL-deficient T cells and plays a cytoprotective function. Here, we summarize our findings and discuss the future direction in the study of the interplay of autophagy, apoptosis and necroptosis in T lymphocytes.
Keywords: autophagy, necroptosis, apoptosis, c-FLIPL, T lymphocytes
Apoptosis can be triggered when death receptors on the cellular membrane meet their ligands. When sensing the death signals, death receptors activate CASP8, which in turn triggers the apoptotic cascade. However, blocking CASP8 activity does not protect cells from a fate of death. Loss of CASP8 activity leads to excessive cell death under certain conditions. This form of death, which is necrotic and depends on RIPK1 and RIPK3, is termed necroptosis. Using an isoform-specific conditional deletion mouse model, we found that CFLARL suppresses both apoptosis and necroptosis in T lymphocytes. CFLARL inhibits apoptosis by suppressing CASP8 activation. Interestingly, when T cells lack CFLARL, active CASP8 fails to control necroptosis, suggesting that CASP8-mediated cleavage of RIPK1 requires the presence of CFLARL.
A high level of autophagy is correlated with necroptosis induction in multiple cell types, suggesting some type of interplay between these two intracellular events. Interestingly, the anti-apoptotic and anti-necroptotic protein CFLARL is involved in autophagy regulation in primary T cells. Previous reports show that the various isoforms of CFLAR control the threshold of autophagy when overexpressed in cell lines. We detected elevated levels of autophagy in CFLARL-deficient T cells compared with wild-type lymphocytes. The LC3-II/LC3-I ratio in CFLARL-deficient T cells is higher than that in wild-type controls, under both resting and activated states. Consistent with the enhanced LC3 processing, the number of LC3-positive puncta is also increased in CFLARL-deficient T cells. Transmission electron microscopy analysis further confirms that CFLARL-deficient T cells contain more double-membrane vacuoles than wild-type T cells. Interestingly, autophagosomes in CFLARL-deficient T cells enclose multiple layers of membranes, structures that are absent in wild-type T cells.
When necroptosis was identified in L929 cells, the term “autophagic death” was used to describe this type of cell death, as inhibition of autophagy induction rescues the cell death. However, contrary results later appeared, arguing that the enhanced autophagy is protective, rather than harmful for cell survival. We examined whether the enhanced autophagy in CFLARL-deficient T cells is detrimental or protective to their survival by disabling the autophagy machinery in these cells. Induced deletion of Atg3 in CFLARL-deficient T cells further reduces the survival of activated CFLARL-deficient T lymphocytes. Meanwhile, acute deletion of Atg3 does not impair T lymphocyte survival. Our previous work showed that autophagy in peripheral T cells functions to remove excessive mitochondria and endoplasmic reticulum, and is essential for TCR-induced proliferation. Here we found that in stressed cells (in which both apoptosis and necroptosis are induced), autophagy plays a cytoprotective role.
Combined with published data from other groups, we present a model describing how CFLARL, along with CASP8, regulates the interplay of autophagy, apoptosis and necroptosis in T lymphocytes (Fig. 1). Still, several questions need to be answered to resolve the twisted relationships between these three fundamental intracellular events in T cells. First, it remains unclear as to the mechanism through which autophagy antagonizes apoptotic signals in CFLARL-deficient T cells. Second, little is known about whether and how autophagy protects the cells from necroptosis. Reactive oxygen species (ROS) contribute to the necroptosis in CFLARL-deficient T cells. Autophagy regulates the homeostasis of the major ROS resource, mitochondria. However, our results show that the mitochondrial levels are similar in cflarL−/− and wild-type T cells. It is possible that autophagy may remove damaged mitochondria in cflarL−/− T cells to promote their survival. Third, the mechanism of how necroptosis causes enhanced autophagy needs to be addressed. CFLAR proteins directly compete with LC3 for Atg3 through two death-effector domains. However, the high level of autophagy in zVAD-treated L929 cells and activated FADD-deficient T cells suggests that necroptotic conditions might induce autophagy that cannot be suppressed by endogenous CFLARL.
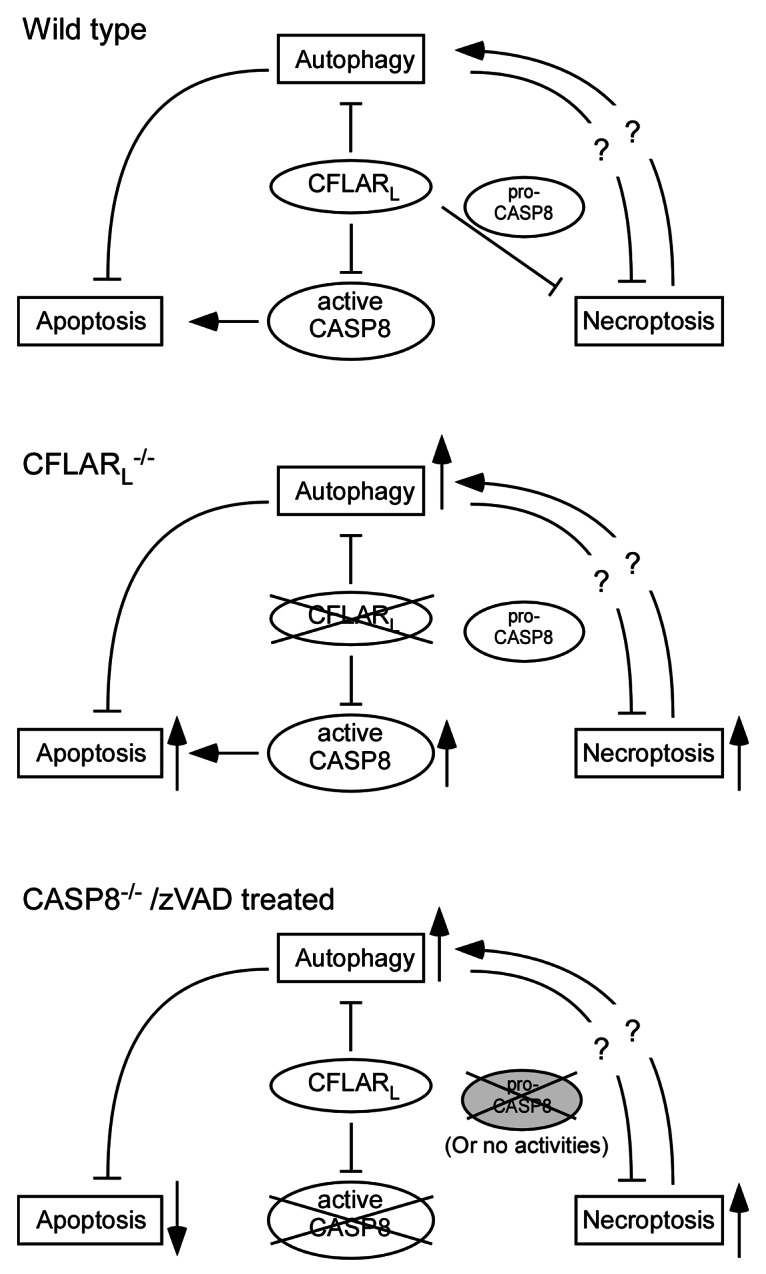
Figure 1. CFLARL-mediated interplay of autophagy, apoptosis and necroptosis in T lymphocytes. CFLARL controls apoptotic signaling (CASP8) activity and downregulates autophagy. Together with pro-CASP8 (with intact activity), CFLARL inhibits necroptosis induction in T cells. In the absence of CFLARL, autophagy, necroptosis and apoptosis are all enhanced. Active CASP8 fails to suppress necroptosis without CFLARL. In the absence of CASP8 or when caspase activities are inhibited, apoptosis is blocked. However, necroptosis is induced in the cells, as the catalytic activity of pro-CASP8 is required for RIPK1 inhibition. The autophagy level in casp8−/− T cells is yet to be determined.
Our study demonstrates that the three fundamental intracellular processes, autophagy, apoptosis and necroptosis, are coordinated by one factor, CFLARL. Under certain conditions, the three unique pathways may be aligned to ensure the best interest of the cells, either survival or death. Necroptosis is generally inhibited to avoid large-scale inflammation and tissue damage. However, it is possible that cells evolve a “backup” pathway to ensure death when cells fail to accomplish conventional apoptosis. Furthermore, inflammation caused by necrotic death may help initiate a robust immune response and promote the clearance of the pathogens. Autophagy induction has a close relationship with apoptosis and necroptosis. It is important in the future to determine whether the autophagy pathway is just recruited to the same field to battle with the other two pathways, or, whether autophagy balances the ratio of apoptotic and necroptotic signals and determines the final fate of the cell.
Acknowledgments
The work in the authors’ laboratory is supported by NIH grants AI073947, AI074754 and AI074944.
Glossary
Abbreviations:
- CASP8
caspase 8
- ROS
reactive oxygen species
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Footnotes
Previously published online: www.landesbioscience.com/journals/autophagy/article/23785