

Abstract
Several years ago, an explosion of research into pathogens and autophagy showed that viruses have a wide variety of relationships to this conserved homeostatic pathway. Often, autophagy acts as a host defense mechanism, degrading viruses before they can escape the host cell, and, as such, autophagy is suppressed or avoided by those viruses. A subset of viruses, however, induces and subverts the autophagic machinery to promote their own replication. Many of these viruses inhibit the degradative step in the autophagic pathway, presumably to prevent degradation of cytosolic virions before they exit the cell. Recently, we published a study showing that poliovirus (PV), a well-studied model virus, induces true autophagic degradation. The remainder of our study provided surprising clues about the role of autophagy in promoting virus production. The purpose of this punctum is to discuss the significance of our findings to a general understanding of the autophagic pathway and its relationship to a common class of cellular pathogens.
Keywords: virus, amphisome, acidification, degradation, secretion
Picornaviruses are efficient cytosolic pathogens, which are physically made up of two parts: a small plus-strand RNA genome and an inert capsid. With no enzymes in the virion, the RNA has evolved to directly bind cellular ribosomes, initiating immediate translation of a single large polyprotein from the genome. The polyprotein auto-cleaves to produce the enzymes, structural proteins and other peptide products required to replicate the genome and generate progeny viruses. Picornaviruses are famous for being rapid to replicate and ruthless to the infected cell. In tissue culture models, within 2 h of PV infection essentially all protein translation is viral, within 4–5 h the cytosol has been rearranged beyond recognition, and within 8–10 h the cell lyses.
There is considerable variety in cellular pathway regulation, tropism, and disease outcome among the picornaviruses. However, one feature common to picornaviruses, and positive-strand RNA viruses in general, is that RNA replication complexes associate with the cytosolic face of cellular membranes. The reasons for this are unclear. It has been speculated that membrane association eases complex assembly, or reduces the physical search space for templates, nucleotides, or other replication complexes. Several papers from the Kirkegaard lab, including some by the senior author of this punctum, showed that PV replication proteins can be found in association with double-membraned, autophagosome-like, cytosolic vesicles. We hypothesized that the autophagic pathway was being induced by the virus to generate replication membranes.
A competing model developed in the laboratories of Kurt Beinz, Ellie Ehrenfeld, and others, as they demonstrated that PV replication proteins could be found on single-membraned vesicles, colocalizing with proteins of the secretory apparatus (Fig. 1). Both groups proposed that PV subverts normal cellular secretion to generate replication membranes. Recently, the Ehrenfeld lab published EM tomography data showing the presence of single-membraned vesicles at 3 h post-infection (hpi), convoluted, invaginated membranes at 4 hpi, and double-membraned vesicles at 7 hpi. Since 3–4 hpi is the peak of viral RNA replication, they have modified their model to suggest that the single-membraned vesicles act as the primary sites of RNA replication, but that these vesicles later morph into double-membraned vesicles.
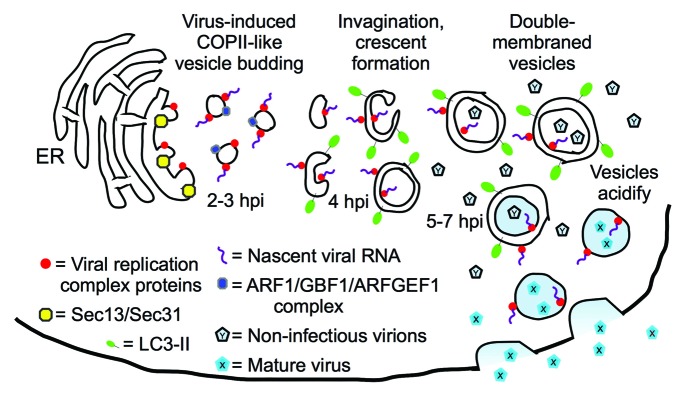
Figure 1. Model for vesicle morphogenesis throughout poliovirus infection. Single-membraned vesicles predominate at 3–4 hpi, the peak of viral RNA production, suggesting that those vesicles are the primary sites of genomic replication. Imaging showed Sec13 and Sec31 to be involved in vesicle formation at the ER, and ARF1 and GEFs co-localize with PV replication proteins. Since inhibiting vesicle acidification blocks a late step in maturation of the fully formed virus, we proposed that double-membraned, amphisome-like acidic vesicles observed later in infection are harbors for virus maturation. The virus would then exit cells through an autophagy-dependent, noncanonical secretory pathway. However, the autophagic pathway inhibitor 3-MA blocks viral RNA replication, which is a much earlier step in virus production. Therefore, we speculate that 3-MA blocks the ER budding of single-membraned autophagosome precursors thought to act as substrates for the RNA replication complex. This model suggests that, at least during PV infection, autophagosome-like vesicles are derived from secretory vesicles that contain Golgi-resident markers.
Meanwhile, a crucial question remained: what purpose does the autophagic pathway serve for poliovirus? The closely related picornavirus coxsackievirus B3 was shown to induce the autophagic pathway while inhibiting degradation. This finding makes sense for a cytosolic virus, so that the pathogen can avoid degradation. These data are also in line with models of bacterial pathogenesis: Legionella pneumophila, for example, delays maturation of the autophagosomes it occupies to generate a replication niche, and successful progeny bacteria escape the vesicles before they fuse with lysosomes. To our surprise, however, we found that poliovirus allows autophagic degradation to occur during infection, indicating a very different relationship between PV and autophagy.
We hypothesized that autophagy might degrade an antiviral factor, but protease inhibitors do not affect virus production at all. We next tested inhibitors of vesicle acidification, in order to reduce amphisome-lysosome fusion. We found a 6- to 10-fold drop in virus when we treated with NH4Cl or bafilomycin A1. This was surprising to us, as picornavirologists, since the entire life cycle of these viruses is thought to be cytosolic. Therefore, altering the internal pH of a vesicle would not be expected to affect virus production. We investigated the virus life cycle in the presence of NH4Cl and found that virus entry, translation, polyprotein processing, RNA replication and virion assembly all appeared normal. The only change was a reduction in the final step of producing an infectious virus, the cleavage of an internal capsid protein. This poorly understood event is required for infectivity, occurs only after assembly of the virus capsid around an RNA genome, and is thought to be autocatalytic.
We want to highlight two implications of these findings, which are modeled in Figure 1. First, our data show that PV replication, long thought to be exclusively cytosolic, is affected by the internal pH of vesicles. Since we think that the predominant vesicle in the cell late in infection is amphisome-like, we suspect that the pH of these vesicles affects PV infectivity. While it is possible that amphisomes act as “ion sinks” to modulate the pH of the cytosol, EM images showing virus in the interior of double-membraned vesicles led us to model that virions are maturing inside these vesicles. This leads to a topology problem, because unlike some viruses, picornaviruses do not exit cells by budding, and therefore do not take an enveloping membrane from the host cell. A picornavirus inside a double-membraned vesicle has three lipid bilayers to pass before it can exit the cell. In 2005, we suggested PV could exit cells through an autophagy-dependent secretory pathway, and recent work in Pichia pastoris and Dictyostelium discoideum has demonstrated that such a pathway exists. Therefore, our model shows virus exiting cells directly from autophagosomes, although there are other possibilities.
The second interesting facet of our data, when considered in light of the recent work from the Ehrenfeld lab, is the possible link between canonical secretory vesicles and autophagosomes. In the “unified model,” viral RNA replication complexes are initially found on single-membraned secretory vesicles. RNA replication and encapsidation are tightly coupled, so virions would form nearby. If the model is correct, and those vesicles morph into autophagosomes, many of these viruses would be captured and directed into an acidic amphisome (Fig. 1). The viruses would likely survive autophagosome maturation: poliovirus is a virus of the gut, after all, and adapted to acidic, degradative environments. This would mean that, at least in the context of PV, the autophagosome-like vesicles are derived from secretory vesicles. This hypothesis could provide a clue about the origin of normal cellular autophagosomes. Studies of cellular autophagy will show if the virus model can resolve long-standing controversies about the origin of autophagic membranes.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Footnotes
Previously published online: www.landesbioscience.com/journals/autophagy/article/23962