

Abstract
Gastrin-releasing peptide (GRP) is a pro-angiogenic ligand secreted by tumors and acts directly upon binding to GRP-receptor in endothelial cells. Angiogenesis plays a critical role in the pathology of various diseases, including cancer, as the formation of new blood vessels potentiates the rate of tumor growth and dissemination. GRP increases the migration of endothelial cells, but much is unknown about its role on endothelial cell proliferation and survival, as well as the signaling pathways involved. In the present study, we showed that GRP increases endothelial cell proliferation and tubule formation. There was a time-dependent increase in the levels of phosphorylated AKT, mTOR, and S6R in human umbilical vein endothelial cells treated with GRP. Interestingly, GRP treatment decreased the expression of pro-autophagic factors, ATG5, BECN1 and LC3 proteins. GRP also attenuated rapamycin-induced formation of autophagosomes. Moreover, overexpression of ATG5 or BECN1 significantly decreased tubule formation induced by exogenous GRP, whereas, siRNA against ATG5 or BECN1 resulted in increased tubule formation with GRP treatment. Our results show that GRP inhibits the process of autophagy in vascular endothelial cells, thereby, increasing endothelial cell proliferation and tubule formation. Here, we describe a novel role of GRP in the regulation of autophagy of endothelial cells, thereby, providing a potential new therapeutic strategy in targeting angiogenesis during cancer progression.
Keywords: AKT, autophagy, endothelial cells, GRP, mTOR
INTRODUCTION
Gastrin-releasing peptide (GRP) is both a gut and a neuropeptide that has been implicated in a number of pathophysiological conditions, including cancer. It has a mitogenic function on various cancer cell types including neuroblastoma in both autocrine as well as paracrine manner.1, 2 Moreover, GRP treatment activates intracellular signaling pathways such as the PI3K to stimulate angiogenesis and tumor growth.3–5 Though, GRP induces endothelial cell migration and enhances angiogenesis in an in vivo assay,6 much is unknown about its role in the regulation of vascular endothelial cell proliferation. Moreover, downstream signaling pathways mediating the angiogenic effects of GRP are yet to be elucidated.
Cancer progression requires formation of new blood vessels to suffice nutritional requirements of neoplastic cells.7 Anti-angiogenic therapies targeting tumor cell-derived angiogenic factors have been successfully introduced in clinical trials for metastatic cancer.8, 9 Tumor cell-derived vascular endothelial growth factor (VEGF) signaling promotes cancer progression by stimulating endothelial cell proliferation, migration and invasion, increased blood vessel permeability and by forming a microenvironment for the migration of endothelial cells.10 In turn, endothelial cells are recruited to tumors to provide “angiocrine” support for the dissemination of tumor cells during cancer progression.11 Moreover, endothelial cell-autonomous VEGF signaling pathway is critical for vascular homeostasis as genetic depletion of VEGF leads to progressive endothelial degeneration.12 However, the exact action of GRP, which is a known inducer of VEGF signaling, on vascular endothelial cells remains unclear.
Autophagy, an alternative mechanism for cell death, has been identified as a novel therapeutic strategy when there is a failure of the apoptotic machinery in cancer cells. Induction of pro-autophagic molecules requires a concomitant downregulation of the AKT/mTOR signal transduction pathway, as activated mTOR is a negative regulator of autophagy.13 Interestingly, GRP treatment enhances AKT signaling in cancer cells,14 which then phosphorylates mTOR.15 The role of GRP in the regulation of autophagy via the activation of AKT/mTOR signaling has not been studied yet. Moreover, how the induction of autophagy affects VEGF secretion and tubule formation by endothelial cells is yet to be determined. Targeting GRP-AKT-mTOR signaling axis might provide insights into using autophagy as a new tool to target tumor-associated vascular endothelial cells.
In this study, we demonstrate for the first time that GRP treatment enhances proliferation of human umbilical vein endothelial cells (HUVECs) via upregulation of the PI3K/AKT/mTOR signaling pathway. Moreover, GRP treatment enhanced tubule formation by HUVECs when compared to control. Interestingly, GRP treatment decreased the expression of key autophagic molecules in HUVECs in a time-dependent manner. Furthermore, overexpression or silencing autophagic molecules, ATG5 or BECN1, decreased or increased tubule formation by HUVECs, respectively. Our data indicates a proliferative role for GRP in vascular endothelial cells and identifies a novel role for this neuropeptide in the inhibition of autophagy, where, autophagy regulates tubule formation by vascular endothelial cells.
MATERIALS AND METHODS
Cell culture and reagents
HUVECs obtained from Dr. M. Freeman (Vanderbilt University Medical Center, Nashville, TN) were cultured in EMM-2 supplemented with growth factors (EGM-2 SingleQuot kit, Lonza, Walkersville, MD) at 37°C and humidified 5% CO2. GRP was obtained from TOCRIS Bioscience (Ellisville, MO). Matrigel was purchased from BD Bioscience (Bedford, MA).
Cell proliferation
HUVECs were seeded at 96-well plate at a density of 5000 cells/well. Next day, GRP (100 nM) was added to each well for up to 24 and 72 h. Twenty μl of the combined MTS/PMS solution were added into each well containing 100 μl of cells in culture media. The plate was incubated for 1 to 4 h at 37°C in a humidified, 5% CO2 atmosphere. Absorbance of the color was measured at a wavelength of 570 nm with Flexstation 3 Modecular devices (Bio-Tek Instruments Inc., Winooski, VT).
Endothelial cell tube formation assay
HUVECs grown to 70% confluence were trypsinized counted, and seeded with 48,000 cells per well in 24-well plates coated with 300 μl of Matrigel (BD Biosciences, Bedford, MA). These cells were periodically observed by microscope as they differentiated into capillary-like tubule structures. After 6 h, cells were stained with Hematoxylin & Eosin and photographs were taken via microscope, and representative photographs obtained. Tubules were developed from clear elongated cell bodies that connect to form a polygon network. The average number of tubules was quantified by randomly selecting three separate 200× fields and counting the number of tubules per field.
Co-culture assay
Six-well transwell cell culture chamber inserts without collagen and with 0.4 mm pores were used to plate HUVECs in the inserts and BE(2)-C cells in the chambers (in duplicate) at HUVEC:BE(2)-C ratio of 1:10. After 48 h, the insert was removed and stained with Hematoxylin & Eosin and photographs (200×) taken using a microscope.
Immunoblotting
Cells (5 × 105) were collected at various time points, and then washed with ice-cold PBS twice before adding lysis buffer (M-PER Mammalian Protein Extraction reagent, Thermo Scientific, Waltham, MA) and cocktail inhibitor (Sigma, 5 μg/ml). Equal amounts of protein were loaded onto each well and separated by NuPAGE 4–12% Bis-Tris gel, followed by transfer onto PVDF-membranes (Bio-Rad, Hercules, CA). Membranes were blocked with 5% non-fat milk in PBS-T for 1 h at room temperature. The blots were then incubated with antibodies against the following human proteins p-mTOR, mTOR, p-AKT, AKT, p-S6R, S6R, ATG5-ATG12, BECN1, β-actin and LC3I/II, for 1 h at 4°C. Goat anti-rabbit IgG secondary antibody (1:5000; Santa Cruz Biotechnology, Santa Cruz, CA) was incubated for 45 min at room temperature. Immunoblots were developed by using the chemiluminescence detection system (PerkinElmer, Waltham, MA) according to the manufacturer’s protocol and autoradiography was performed.
Cell transfection
siRNA ATG5, BECN1 and siRNA control were purchased from Santa Cruz (Santa Cruz, CA). Cells were transfected with 25 nM siRNAs, 3μg ATG5 plasmid or BECN1 plasmid using Lipofectamine 2000 (Invitrogen Life Technologies, Carlsbad, CA). Transfected cells were used for subsequent experiments 48 h later.
Autophagy assay
HUVECs were transfected with 3 μg of EGFP-LC3 expression plasmid (a gift from Dr. Norboru Mizushima) using Lipofectamine 2000 for 24 h. Cells with punctate GFP signaling were counted as autophagic cells based on characteristic lysosomal localization of LC3 protein during autophagy.16 Punctate GFP cells were quantified by randomly selecting three separate 100× fields and counting the number of punctate GFP cells per field. The percent of punctate GFP cells per total GFP transfected cells was calculated and experiments were conducted in triplicate. The fluorescence of EGFP-LC3 was observed using immunofluorescence microscopy.
Statistical analysis
All results are shown as mean value ± SEM from at least three independent experiments. Western blot scans are representative of three separate assays. Statistical analysis was performed using Student’s t-test. p values < 0.05 were considered to be statistically significant.
RESULTS
GRP increased HUVECs proliferation and tubule formation
GRP has been reported to induce migration of HUVECs.6 To examine whether GRP also induces proliferation of HUVECs, we treated HUVECs with GRP (100 nM) for 24 and 72 h. We found that GRP treatment significantly increased HUVEC proliferation (Fig. 1A). We next wanted to determine whether GRP treatment induces tubule formation in vitro. HUVECs plated on transwell plates coated with Matrigel were treated either with or without GRP (100 nM) for 6 h. GRP treatment significantly increased tubule formation by HUVECs compared to the control group (Fig. 1B). Taken together, these data suggest that exogenous GRP induces endothelial cell proliferation and tubule formation in vitro.
Figure 1. GRP increased HUVEC proliferation and tubule formation.

(A) HUVECs treated with GRP (100 nM) resulted in a significant increase in cell proliferation as measured by CellTiter 96® AQueous Non-Radioactive Cell Proliferation Assay when compared to control cells. (B) HUVECs were treated with GRP (100 nM) on 24-well plates coated with Matrigel for 6 h. Tubule staining was performed in triplicate (mean ± SEM; * = p < 0.05 vs. control).
GRP treatment downregulated key autophagic proteins in HUVECs
Activation of PI3K/AKT pathway is a crucial step in events leading to angiogenesis. GRP also stimulates cancer cell proliferation via activation of the PI3K/AKT pathway.3 The kinase mTOR is a critical negative regulator of autophagy, where, activated mTOR suppresses key molecules involved in the induction of autophagy.17 To validate whether the effect of GRP on HUVEC proliferation and tubule formation is mediated by PI3K/AKT/mTOR pathway, we treated HUVECs with GRP. As expected, we found that GRP treatment increased phosphorylation of AKT, mTOR and S6R in a time-dependent manner (Fig. 2A). Since, GRP treatment enhanced phosphorylation of mTOR in HUVECs, we investigated the role of GRP in the regulation of key autophagy proteins. When HUVECs were treated with GRP, key autophagy proteins, ATG5 and BECN1 were significantly downregulated in a time-dependent manner (Fig. 2B). Furthermore, expression of LC3I and LC3II was inhibited in a time-dependent manner, indicating decreased formation of autophagosomes upon GRP treatment.
Figure 2. GRP enhanced AKT/mTOR signaling and downregulated ATG5, BECN1.

(A) Phosphorylated and total AKT, mTOR and S6R protein levels in HUVECs over a time course after GRP treatment (100 nM) for 30 min by Western blotting (B) GRP (100 nM) inhibited expression of ATG5, BECN1 and LC3I/II in HUVECs. Data is representative of three separate experiments. Total proteins (35 μg per lane) were probed with the indicated antibodies in HUVECs. β-actin was probed to demonstrate equal protein loading. Numbers represent ratio of densitometric values of LC3I to β-actin levels. (C) HUVECs were transfected with EGFP-LC3 plasmid and exposed to 200 μM rapamycin for 24 h and assessed for autophagy after 48 h. Representative fluorescence microscopy photos are shown. Values are mean ± SEM of three separate experiments (* = p < 0.05 vs. control, † = p < 0.05 vs. rapamycin alone).
To further investigate whether GRP regulates autophagy via the mTOR pathway, we transfected EGFP-LC3 plasmid into HUVECs to visualize the formation of autophagosomes, a hallmark of autophagy. Diffuse cytoplasmic localization of EGFP-LC3 was observed in control cells, whereas rapamycin treated cells showed significantly increased punctate fluorescence of LC3 proteins at 48 h post-transfection. Moreover, GRP dramatically reduced the rapamycin-induced increased punctate fluorescence of EGFP-LC3 proteins (Fig. 2C). These data indicate that GRP regulates autophagy of HUVECs via activation of the AKT/mTOR/S6R signaling axis.
Overexpression of ATG5 and BECN1 decreased HUVECs tubule formation
Autophagy is an alternative cell death mechanism that can be activated in tumor cells when other modes of inducing cancer cell death fails. Since endothelial cells form new leaky vessels through autocrine and paracrine secretion of pro-angiogenic molecules during tumor progression, activation of autophagy in these cells might be critical to suppress tumorigenesis. To examine whether autophagy affects the production of tubular structures, HUVECs were transfected with plasmid ATG5 or BECN1 or control vector. A representative image and the average number of tubules counted in three separate (200×) fields are shown in Figures 3A and 3B, respectively. Tubules were developed from clear elongated cell bodies that connect to form a polygon network. Treatment of GRP increased the formation of such three dimensional capillary-like tubule structures when compared to controls (20±2 vs. 9±1). Overexpression of either ATG5 or BECN1 formed discontinued tubules and had significantly reduced formation of such polygon networks in spite of GRP treatment when compared to GRP treatment alone (20±2 vs. 3.3±0.58 and 20±2 vs. 6.7±1, respectively). Immunoblotting was used to confirm overexpression of either ATG5 or BECN1 by stable transfections with or without GRP treatment (Fig. 3C). These findings indicate that induction of the autophagy machinery may prevent GRP-induced angiogenesis by HUVECs.
Figure 3. Overexpression of ATG5 or BECN1 decreased HUVEC tubule formation.

(A) HUVECs (3×105 cells) were transfected with overexpression plasmids encoding ATG5 or BECN1. After 24 h, cells were trypsinized and replated on 24-well plates coated with Matrigel. After 6 h, cells were fixed and stained with Hematoxylin & Eosin. The slides were examined by microscope (200×), and representative fields are shown. (B) Stained tubules were based on three separate repeated experiments and shown as the mean ± SEM. (C) Overexpression of ATG5 and BECN1 in HUVECs were confirmed by immunoblotting. β-actin was probed to demonstrate equal loading (* = p < 0.05 vs. control, † = p < 0.05 vs. GRP alone).
Silencing ATG5 or BECN1 enhanced HUVECs tubule formation
To further characterize the role of autophagy in the regulation of angiogenesis, we downregulated the expression of the two essential autophagy proteins, ATG5 and BECN1. siRNA against either ATG5 or BECN1 increased formation of capillary-like tubule structures with GRP treatment in comparison to GRP treatment alone (27.6 ±1.5 vs. 8±1 and 23±1.5 vs. 8±1, respectively) (Fig. 4A). Moreover, the polygon networks formed upon silencing ATG5 or BECN1 were denser compared to the GRP treatment alone, indicating that targeting pro-autophagic genes enhanced tubule formation by HUVECs. The average number of tubules counted in three separate (200×) fields is shown in Figure 4B. Protein levels of ATG5 and BECN1 after siRNA-mediated silencing were confirmed by immunoblotting with or without GRP treatment (Fig. 4C). Taken together, these data further confirmed that GRP-mediated increase in angiogenesis by endothelial cells is further enhanced by the inhibition of autophagy.
Figure 4. Silencing ATG5 or BECN1 enhanced HUVEC tubule formation.

(A) HUVECs (3×105 cells) were transfected with 25 nM siRNA against ATG5 or BECN1 for 24 h. After 24 h, cells were trypsinized and treated with GRP on 24-well plates coated with Matrigel. After 6 h, cells were fixed and stained with Hematoxylin & Eosin. The slides were examined by microscopy (200×), and representative fields are shown. (B) Stained tubules were based on three separate repeated experiments and shown as the mean ± SEM. (C) Targeted silencing of ATG5 and BECN1 were confirmed by immunoblotting. β-actin was probed to demonstrate equal loading (* = p < 0.05 vs. control, † = p < 0.05 vs. GRP alone).
Inhibition of autophagy increased angiogenesis in co-culture of HUVECs with BE(2)-C cells
To investigate the angiogenic effects via paracrine mechanism, we co-cultured human neuroblastoma BE(2)-C cells and ATG5- or BECN1-silenced HUVECs using transwell inserts without collagen coating and a pore size of 0.4 μm. Interestingly, we found that in the presence of BE(2)-C cells, HUVECs transfected with siRNA against ATG5 or BECN1 exhibited increased formation of net-like structures in comparison to controls (Fig. 5; upper panels, arrows). In particular, GRP blocking antibody inhibited the net-like formation observed after silencing the autophagic machinery (Fig. 5; lower panels, arrows). Taken together, these data suggest a critical role for GRP-mediated inhibition of autophagy in the paracrine regulation of endothelial cells.
Figure 5. Net-like formation of co-culture with HUVECs transfected with siRNA against ATG5 or BECN1 in BE(2)-C cells.
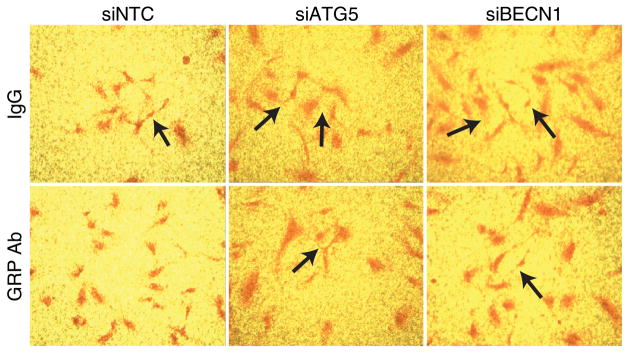
HUVECs were transfected with 25 nM siRNA against ATG5 or BECN1 for 24 h. After 24 h, HUVECs were trypsinized and co-cultured with BE(2)-C cells with or without GRP antibody (GRP Ab; 1μg/ mL) for 48 h. IgG was used as control. Arrows indicate increased net-like structures (magnification 200×).
DISCUSSION
GRP is a potent inducer of angiogenesis in various cancer cell types; however, its paracrine regulation of vascular endothelial cell proliferation and tubule formation has not been elucidated. In this report, we, for the first time, describe a novel mechanism of GRP regulation of autophagy in HUVEC proliferation and tubule formation via activation of the PI3K/AKT/mTOR signaling pathway. Further, GRP enhanced HUVECs tubule formation by downregulation of key autophagy proteins such as ATG5 and BECN1.
Angiogenesis, a process essential during normal development, requires an intricate and complex interplay of growth factors and inhibitors, endothelial cells and the extracellular matrix.18 Imbalance in any aspect of this critical process contributes to a wide variety of pathological conditions, including cancer. Major breakthroughs have identified signaling pathways and specific pro-angiogenic growth factors along with their receptors in cancer-derived neo-angiogenesis. Conversely, endothelial cells play a more critical role in the process of angiogenesis and cancer progression than acting as mere tubes for the circulation of blood.19 Cell-autonomous synthesis and secretion of pro-angiogenic factors are required for the proliferation and survival of endothelial cells, as pharmacological inhibition of pro-angiogenic receptors induced endothelial cell apoptosis.20 Moreover, targeting vascular endothelial cells using anticancer drugs have shown to induce apoptosis in these cells and inhibit tubule formation in vitro.21
GRP treatment has been reported to induce migration of endothelial cells in vitro and enhance angiogenesis in vivo.6 Our data indicates that GRP acts as a mitogen for endothelial cells and induces tubule formation in vitro. This observation further confirms the pro-angiogenic role of GRP during tumorigenesis, as endothelial cell survival, proliferation and vessel formation are critical steps in neovascularization. GRP and its receptor, GRP-R, utilize the PI3K/AKT signaling pathway as downstream effectors in cancer cells.14 Interestingly, GRP treatment activated the AKT/mTOR/S6 signaling axis in endothelial cells demonstrating the critical nature of this pathway downstream of GRP/GRP-R in different kinds of cells. Since, mTOR negatively regulates autophagy, we speculated a role for GRP in the inhibition of autophagy via the activation of AKT/mTOR pathway. As expected, GRP treatment decreased the expression of pro-autophagic proteins in a time-dependent manner, and also inhibited rapamycin-induced autophagy. This is a novel finding where a neuropeptide has been shown to suppress the process of autophagy in vascular endothelial cells providing a better understanding of how induction of autophagy might be a potential anti-vascular therapy. Interestingly, our results indicate that overexpression of pro-autophagic proteins inhibited the vasoactive role of GRP and suppressed tubule formation by endothelial cells. Also, silencing autophagic molecules synergistically enhanced tubule formation by GRP treatment. In particular, we found that co-culture system recapitulated component of the angiogenic phenotype, including formation of net-like structures. Targeting endothelial cells and the perivascular niche using next-generation combinational drug regimens is becoming increasingly important.20 Our co-culture data also highlights the importance of tumor-host interactions during tumor progression. It is plausible that tumor-cell secreted GRP enhances neovascularization in solid tumors by increasing endothelial cell proliferation, survival and motility. This area of tumor-host interaction becomes increasingly crucial in various cancers such as neuroblastoma, prostate and small-cell lung cancer, where GRP acts as a critical mitogen and has been implicated in disease progression.
In conclusion, the findings in this study identify a novel role for GRP in vascular endothelial cell proliferation and tubule formation. Our study has further ascertained a plausible mechanism by which tumor cells may enhance tubule formation by endothelial cells by increasing tumor cell-associated GRP secretion and subsequently suppressing endothelial cell autophagy via activation of the PI3K/AKT/mTOR pathway, highlighting the critical nature of host-tumor interactions. Moreover, activation of autophagy by genetic manipulation of the autophagic machinery inhibits the pro-angiogenic effect of GRP on endothelial cells, providing a potential new tool in targeting angiogenesis during cancer progression.
Acknowledgments
Grant support: R01 DK61470 from the National Institutes of Health
The authors thank Karen Martin for assistance with the manuscript preparation. This work was supported by a grant R01 DK61470 from the National Institutes of Health.
Abbreviations
- GRP
gastrin-releasing peptide
- GRP-R
GRP-receptor
- HUVECs
human umbilical vein endothelial cells
- mTOR
mammalian target of Rapamycin
- VEGF
vascular endothelial growth factor
References
- 1.Patel O, Shulkes A, Baldwin GS. Gastrin-releasing peptide and cancer. Biochim Biophys Acta. 2006;1766:23–41. doi: 10.1016/j.bbcan.2006.01.003. [DOI] [PubMed] [Google Scholar]
- 2.Paul P, Gillory LA, Kang J, Qiao J, Chung DH. Targeting gastrin-releasing peptide as a new approach to treat aggressive refractory neuroblastomas. Surgery. 2011;149:425–432. doi: 10.1016/j.surg.2010.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kang J, Ishola TA, Baregamian N, et al. Bombesin induces angiogenesis and neuroblastoma growth. Cancer Lett. 2007;253:273–281. doi: 10.1016/j.canlet.2007.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Qiao J, Kang JH, Cree J, Evers BM, Chung DH. Ets1 transcription factor mediates gastrin-releasing peptide-induced IL-8 regulation in neuroblastoma cells. Neoplasia. 2007;9:184–191. doi: 10.1593/neo.06841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Martinez A. A new family of angiogenic factors. Cancer Lett. 2006;236:157–163. doi: 10.1016/j.canlet.2005.04.008. [DOI] [PubMed] [Google Scholar]
- 6.Martinez A, Zudaire E, Julian M, Moody TW, Cuttitta F. Gastrin-releasing peptide (GRP) induces angiogenesis and the specific GRP blocker 77427 inhibits tumor growth in vitro and in vivo. Oncogene. 2005;24:4106–4113. doi: 10.1038/sj.onc.1208581. [DOI] [PubMed] [Google Scholar]
- 7.Hanahan D, Folkman J. Patterns and emerging mechanisms of the angiogenic switch during tumorigenesis. Cell. 1996;86:353–364. doi: 10.1016/s0092-8674(00)80108-7. [DOI] [PubMed] [Google Scholar]
- 8.Hurwitz H, Fehrenbacher L, Novotny W, et al. Bevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic colorectal cancer. N Engl J Med. 2004;350:2335–2342. doi: 10.1056/NEJMoa032691. [DOI] [PubMed] [Google Scholar]
- 9.Miller K, Wang M, Gralow J, et al. Paclitaxel plus bevacizumab versus paclitaxel alone for metastatic breast cancer. N Engl J Med. 2007;357:2666–2676. doi: 10.1056/NEJMoa072113. [DOI] [PubMed] [Google Scholar]
- 10.Ellis LM, Hicklin DJ. VEGF-targeted therapy: mechanisms of anti-tumour activity. Nat Rev Cancer. 2008;8:579–591. doi: 10.1038/nrc2403. [DOI] [PubMed] [Google Scholar]
- 11.Butler JM, Kobayashi H, Rafii S. Instructive role of the vascular niche in promoting tumour growth and tissue repair by angiocrine factors. Nat Rev Cancer. 2010;10:138–146. doi: 10.1038/nrc2791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lee S, Chen TT, Barber CL, et al. Autocrine VEGF signaling is required for vascular homeostasis. Cell. 2007;130:691–703. doi: 10.1016/j.cell.2007.06.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kim KW, Mutter RW, Cao C, et al. Autophagy for cancer therapy through inhibition of pro-apoptotic proteins and mammalian target of rapamycin signaling. J Biol Chem. 2006;281:36883–36890. doi: 10.1074/jbc.M607094200. [DOI] [PubMed] [Google Scholar]
- 14.Qiao J, Kang J, Ishola TA, Rychahou PG, Evers BM, Chung DH. Gastrin-releasing peptide receptor silencing suppresses the tumorigenesis and metastatic potential of neuroblastoma. Proc Natl Acad Sci U S A. 2008;105:12891–12896. doi: 10.1073/pnas.0711861105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hahn-Windgassen A, Nogueira V, Chen CC, Skeen JE, Sonenberg N, Hay N. Akt activates the mammalian target of rapamycin by regulating cellular ATP level and AMPK activity. J Biol Chem. 2005;280:32081–32089. doi: 10.1074/jbc.M502876200. [DOI] [PubMed] [Google Scholar]
- 16.Mizushima N, Yamamoto A, Hatano M, et al. Dissection of autophagosome formation using Apg5-deficient mouse embryonic stem cells. J Cell Biol. 2001;152:657–668. doi: 10.1083/jcb.152.4.657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jung CH, Ro SH, Cao J, Otto NM, Kim DH. mTOR regulation of autophagy. FEBS Lett. 2010;584:1287–1295. doi: 10.1016/j.febslet.2010.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Carmeliet P, Jain RK. Angiogenesis in cancer and other diseases. Nature. 2000;407:249–257. doi: 10.1038/35025220. [DOI] [PubMed] [Google Scholar]
- 19.Franses JW, Baker AB, Chitalia VC, Edelman ER. Stromal endothelial cells directly influence cancer progression. Sci Transl Med. 2011;3:66ra65. doi: 10.1126/scitranslmed.3001542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lu C, Kamat AA, Lin YG, et al. Dual targeting of endothelial cells and pericytes in antivascular therapy for ovarian carcinoma. Clin Cancer Res. 2007;13:4209–4217. doi: 10.1158/1078-0432.CCR-07-0197. [DOI] [PubMed] [Google Scholar]
- 21.Mailloux A, Grenet K, Bruneel A, Beneteau-Burnat B, Vaubourdolle M, Baudin B. Anticancer drugs induce necrosis of human endothelial cells involving both oncosis and apoptosis. Eur J Cell Biol. 2001;80:442–449. doi: 10.1078/0171-9335-00171. [DOI] [PubMed] [Google Scholar]