

Abstract
Cadmium (Cd) is an extremely toxic metal, capable of severely damaging several organs, including the brain. Studies have shown that Cd disrupts intracellular free calcium ([Ca2+]i) homeostasis, leading to apoptosis in a variety of cells including primary murine neurons. Calcium is a ubiquitous intracellular ion which acts as a signaling mediator in numerous cellular processes including cell proliferation, differentiation, and survival/death. However, little is known about the role of calcium signaling in Cd-induced apoptosis in neuronal cells. Thus we investigated the role of calcium signaling in Cd-induced apoptosis in primary rat cerebral cortical neurons. Consistent with known toxic properties of Cd, exposure of cerebral cortical neurons to Cd caused morphological changes indicative of apoptosis and cell death. It also induced elevation of [Ca2+]i and inhibition of Na+/K+-ATPase and Ca2+/Mg2+-ATPase activities. This Cd-induced elevation of [Ca2+]i was suppressed by an IP3R inhibitor, 2-APB, suggesting that ER-regulated Ca2+ is involved. In addition, we observed elevation of reactive oxygen species (ROS) levels, dysfunction of cytochrome oxidase subunits (COX-I/II/III), depletion of mitochondrial membrane potential (ΔΨm), and cleavage of caspase-9, caspase-3 and poly (ADP-ribose) polymerase (PARP) during Cd exposure. Z-VAD-fmk, a pan caspase inhibitor, partially prevented Cd-induced apoptosis and cell death. Interestingly, apoptosis, cell death and these cellular events induced by Cd were blocked by BAPTA-AM, a specific intracellular Ca2+ chelator. Furthermore, western blot analysis revealed an up-regulated expression of Bcl-2 and down-regulated expression of Bax. However, these were not blocked by BAPTA-AM. Thus Cd toxicity is in part due to its disruption of intracellular Ca2+ homeostasis, by compromising ATPases activities and ER-regulated Ca2+, and this elevation in Ca2+ triggers the activation of the Ca2+-mitochondria apoptotic signaling pathway. This study clarifies the signaling events underlying Cd neurotoxicity, and suggests that regulation of Cd-disrupted [Ca2+]i homeostasis may be a new strategy for prevention of Cd-induced neurodegenerative diseases.
Introduction
Cadmium (Cd) is an extremely toxic metal commonly found in industrial workplaces. It is also a food contaminant and a major component of cigarette smoke. It is toxic even at low doses since the metal accumulates and has a long biological half-life in humans (10–30 years) [1]. Cd is toxic to many organs, including liver, kidney, lung, testis and brain [2]–[7]. In addition, it can enter the brain parenchyma and neurons causing neurological alterations in humans and animal models, leading to lower attention, hypernociception, olfactory dysfunction and memory deficits [8], [9]. Increasing evidence has demonstrated that Cd is a possible etiological factor of neurodegenerative diseases, such as Alzheimer’s disease (AD) and Parkinson’s disease (PD) [10], [11].
Studies have shown that Cd disrupts calcium homeostasis, leading to apoptosis in a variety of cells [12]–[16]. Recently, Xu et al [17] have demonstrated that Cd-induced apoptosis in primary murine neurons occurs through a calcium-dependent pathway. Calcium is a universal messenger regulating many physiological and pathological functions, such as secretion, contraction, metabolism, gene transcription, and cell death [18], [19]. The cellular uptake of Cd occurs mainly through the Ca2+ channels (including both voltage-gated and receptor-operated Ca2+ channels) and Cd is a potent Ca2+ channel blocker and inhibits Ca2+ cellular uptake [20], [21]. The cellular toxicity of Cd is, in part, related to the alteration of intracellular calcium homeostasis, which can competitively reduce extracellular calcium influx or increase intracellular calcium concentration ([Ca2+]i) by inhibiting calcium-dependent ATPase or by stimulating the inositol triphosphate pathway [13], [22], [23]. Moreover, a number of studies have demonstrated that Cd interacts with the functions of many Ca2+-dependent enzymes such as endonuclease and regulatory proteins such as protein kinase C (PKC), mitogen-activated protein kinase, and phospholipase C, thus interfering with calcium homeostasis [21], [24]–[26]. Intracellular calcium homeostasis is very important in maintaining the normal function of the cell, in that variations in the concentration of calcium in cells can determine cell survival or death. For example, a high [Ca2+]i can cause disruption of mitochondrial Ca2+ equilibrium, which results in reactive oxygen species (ROS) formation due to the stimulation of electron flux along the electron transport chain (ETC) [27]. Under oxidative stress, mitochondrial Ca2+ accumulation can switch from a physiologically beneficial process to a cell death signal [28].
Cd can also induce apoptosis in vitro through direct targeting of mitochondria [29]. Recent studies have revealed the ability of Cd to compromise the mitochondrial membrane potential (ΔΨm) [16], [30]. ΔΨm triggers the release of proteins that are normally confined to the mitochondrial intermembrane space (IMS) into the cytosol. The proteins released include cytochrome c (which stimulates the cytosolic assembly of the apoptosome, the caspase-9 activation complex) and AIF (apoptosis-inducing factor) [31]. Finally, the activation of catabolic hydrolases, mainly caspases and nucleases, causes the cleavage of important cellular targets and leads to apoptotic cell death.
Moreover, it has been proven that Cd-induced neurotoxicity may be due to excessive ROS production in cerebral cortical neurons [4] and in various types of cells [17], [32], [33]. In addition, Cd-induced apoptosis of neuronal cells is triggered by elevated [Ca2+]i, leading to ROS induction and subsequent activation of caspase signaling pathway [17].
Thus Cd toxicity appears to involve Ca2+ signaling, mitochondrial-mediated apoptosis, and ROS production. However, it remains to be clarified how these events are inter-related and what the details are regarding the role of the mitochondrial in Cd toxicity. Therefore, in this study, we investigated the role of Ca2+ signaling pathway and downstream events in Cd-induced apoptosis in primary rat cerebral cortical neurons.
Materials and Methods
Reagents
Fetal calf serum (FCS) was obtained from Hyclone Laboratories (Logan, UT, USA). NEUROBASAL™ Medium, B27 Supplement, Trizol reagent, RNAase inhibitor, dNTPs, reverse primer and SuperScript™ III reverse transcriptase were purchased from Invitrogen (Grand Island, NY, USA). Dulbecco’s modified Eagle’s medium (DMEM)-F12 (1∶1), Fluo-4/AM, Rhodamine 123 (Rh123), cadmium acetate (CdAc2), 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT), 2-Aminoethoxydiphenyl borate (2-APB), 2′,7′-dichloro-dihydrofluorescein diacetate (H2-DCF-DA), antibody for β-actin, Hoechst 33258 staining, trypsin, penicillin/streptomycin were purchased from Sigma Chemical Co. (St. Louis, MO, USA). Antibodies against cleaved caspase-9, cleaved caspase-3, cleaved poly (ADP-ribose) polymerase (PARP), Bcl-2 and Bax were obtained from Cell Signaling Technology (Boston, MA, USA). N-benzyloxycarbonyl-Val-Ala-Asp-fluoromethylketone (Z-VAD-fmk) was supplied by Biovision (Mountain View, CA). The 1, 2-bis (2-amino-phenoxy) ethane-N,N,N′,N′-tetraacetic acid-tetraacetoxymethyl ester (BAPTA-AM) was from Alexis Biochemicals Corporation (San Diego, CA). Horseradish peroxidase (HRP)-conjugated goat anti-rabbit immunoglobulin G (IgG) was from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Na+/K+-APTase, and Ca2+/Mg2+-APTase analysis kits were obtained from Jiancheng Bioengineering Institute (Nanjing, China). Enhanced chemiluminescence solution was from Thermo Fisher Scientific (Waltham, MA). All other reagents were of analytical grade.
Cell Isolation and Culture
Fetal Sprague-Dawley rats of 18–19 days of gestation were obtained from Laboratory Animal Center in Yangzhou University (Yangzhou, China). This study was carried out in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Research Council. The animal care and use committee of Yangzhou University approved all experiments and procedures conducted on the animals (approval ID: SYXK (Su) 2007–0005). Primary rat cerebral cortical neurons were cultured from fetal Sprague-Dawley rats of 18–19 days of gestation, as described [34]. Isolated cells were seeded at a density of 1×106 cells/well in 6-well plates or 2×104 cells/well in 96-well plates coated with 100 mg/L poly-L-lysine in NEUROBASAL™ Medium supplemented with 2% B27 Supplement, 1 mM L-glutamine, 100 U/ml penicillin and 100 U/ml streptomycin. These cells were grown in a humid incubator (37°C, 5% CO2), with the media replaced every 3 days. The cells were used for experiments after 6 days of culture.
Cell Viability Assay and Morphology
Cells were seeded at a density of 2×104 cells/well in 96-well plates. Cells were treated with 0–25 µM Cd for 12 and 24 h, or with/without 10 µM Cd for 12 and 24 h following pre-incubation with/without BAPTA-AM (10 µM) for 30 min or Z-VAD-fmk (100 µM) for 1 h with 6 replicates of each treatment. At the designated time points, cell viability was measured by the MTT assay, which is based on the conversion of the tetrazolium salt to the colored product, formazan. In brief, 20 µl MTT solution (5.0 g/L in PBS) was added into each well of the 96-well plates (containing 100 µl medium and cells) 4 h before the end of incubation. The supernatant was then discarded, and 150 µl DMSO was added to dissolve the formazan. The absorbance was measured at 570/630 nm by the microplate reader (Sunrise, Austria).
For cell morphological analysis, cells were seeded at a density of 1×106 cells/well in six-well plates. At day 6, various Cd doses (0, 5, 10 and 20 µM) or Cd (10 µM) following 30 min of BAPTA-AM (10 µM) or 1 h of Z-VAD-fmk (100 µM) pre-incubation was added. After incubation for 24 h, the culture plates were examined and photographed by a DMI300 inverted phase microscopy (Leica, Germany) (400×) equipped with the Quick Imaging system.
Hoechst 33258 Staining
Apoptotic morphological changes in the nuclear chromatin were examined using Hoechst 33258 staining. The cells were fixed with 4% paraformaldehyde, stained with Hoechst 33258 (5 mg/L) for 10 min and examined by fluorescence microscopy to analyze cell chromatin condensation.
Analysis of Intracellular Free Ca2+concentration ([Ca2+]i)
Cells were cultured in six-well plates and pretreated with BAPTA-AM (10 µM), or 2-APB (50 µM) for 30 min, followed by treatment with various Cd concentrations (0, 5, 10 and 20 µM) for another 12 h. Fluo-4/AM was used as an intracellular free Ca2+ fluorescent probe to analyze [Ca2+]i in Cd-exposed cerebral cortical neurons. In short, the harvested cells were incubated with Fluo-4/AM (5 µmol/L final concentration) for 30 min at 37°C in the dark, washed with PBS, and analyzed on a BD-FACS Aria flow cytometry. Intracellular [Ca2+]i levels were represented by fluorescent intensity. Fluorescent intensity was recorded by excitation at 494 nm and emission at 516 nm. The data were analyzed by Cell Quest program (Becton Dickinson), and the mean fluorescence intensity was obtained by histogram statistics.
Measurement of Mitochondrial Membrane Potential (ΔΨm) and Reactive Oxygen Species (ROS) Production
Cells were cultured in six-well plates and pretreated with BAPTA-AM (10 µM) for 30 min, followed by treatment with various Cd concentrations (0, 5, 10 and 20 µM) for another 12 h. For the detection of mitochondrial ΔΨ, the harvested cells were incubated with Rh123 (10 mg/L final concentration) for 30 min in the dark at 37°C, harvested and re-suspended in PBS and analyzed on a BD-FACS Aria flow cytometry. The mitochondrial ΔΨ was based on Rh123 fluorescence intensity which was recorded by excitation at 488∼505 nm and emission at 530 nm. Generation of ROS was monitored by using H2-DCF-DA, a redox-sensitive fluorescent dye. Briefly, the harvested cells were incubated with H2-DCF-DA (50 µmol/L final concentration) for 30 min in the dark at 37°C. After treatment, cells were immediately washed twice, re-suspended in PBS, and analyzed on a BD-FACS Aria flow cytometry. ROS generation was based on fluorescent intensity which was recorded by excitation at 504 nm and emission at 529 nm. Each measurement was conducted on 10,000 events and analyzed on Cell Quest software (Becton Dickinson), and the mean fluorescence intensity was obtained by histogram statistics.
Activities of Na+/K+-ATPase, and Ca2+/Mg2+-ATPase
The harvested cells were homogenized in ice-cold physiological saline in an ultrasonic disintegrator. The cell homogenates were centrifuged at 1,000 g for 10 min, and supernatants were obtained. The concentration of protein in the supernatant was determined by the Folin phenol method, using bovine serum albumin as a standard. The ATPases activities were assayed by the quantization of phosphonium ions, which were performed in accordance with the ATPase detection protocol.
RNA Extraction and Real-time Quantitative PCR (qRT-PCR)
Total RNA was isolated from cerebral cortical neurons using Trizol reagent protocol (Invitrogen). A total of 5 µg of RNA was reverse transcribed into cDNA and amplified by qPCR using an ABI 7500 Real Time PCR System (Applied Biosystem, USA). Primer pairs specific for cytochrome oxidase subunits (COX-I/II/III) genes in rat, and β-actin were designed as shown in Table 1 and synthesized by Invitrogen (Shanghai, China). The calculations of the relative expression of COX-I, COX-II, and COX-III in the (normalized) experimental groups versus the (normalized) control group were compared using confidence intervals for ratios calculated by the comparative CT method (threshold cycle number at the cross-point between amplification plot and threshold) [35] and values were normalized to an internal β-actin control.
Table 1. Primers used for qRT-PCR.
| Gene Name | Primer (5'–3') | Product (base pairs) | Accession No. |
| COX-I | F: AGCTGGCTTCGTCCACTGAT | 223 bp | NM_AY172581 |
| R: GGCCGTAAGTGAGATGAATG | |||
| COX-II | F:AAGACGCCACATCACCTATCAT | 151 bp | NM_AY172581 |
| R: TCTTGGGCGTCTATTGTGCTT | |||
| COX-III | F: CAGGAGCCCTATCAGCTCTTC | 159 bp | NM_AY172581 |
| R: TGTGGTGGCCTTGGTATGTT | |||
| β-actin | F: CTCATGCCATCCTGCGTCT | 116 bp | NM_031144 |
| R: ACGCACGATTTCCCTCTCA |
Western Blotting Analysis
After treatment, cells were briefly washed twice with cold PBS. Cells were lysed in RIPA buffer (50 mM Tris, pH 7.4; 150 mM NaCl; 1% NP-40; 0.1% SDS) on ice. Lysates were sonicated for 10 sec and centrifuged at 12,000 g for 10 min at 4°C. Protein concentration was determined by bicinchoninic acid assay with bovine serum albumin as standard. Equivalent amounts of protein were separated on 10–15% SDS–polyacrylamide gels and transferred to nitrocellulose membranes. Membranes were incubated with TBS containing 0.05% Tween 20 and 5% nonfat dry milk to block nonspecific binding and were incubated overnight at 4°C with antibodies against cleaved caspase-3, cleaved caspase-9, cleaved PARP, Bcl-2 and Bax (1∶1,000 dilution) or β-actin (1∶2,000 dilution). Detection was performed with the appropriate horseradish peroxidase-conjugated secondary antibodies (1∶5,000 dilution) and enhanced chemiluminescence reagent. All assays were performed in duplicate.
Statistical Analysis
Values were expressed as the mean ± standard deviation (SD) and significance was calculated by Student’s t-test. A P<0.05 was considered statistically significant.
Results
Cd Triggers Apoptosis in Cerebral Cortical Neurons
We evaluated Cd toxicity on cerebral cortical neurons initially by phase-contrast microscopy. More round or shrunken cells appeared in cerebral cortical neurons, when exposed to increasing concentrations of Cd (5, 10, 20 µM) for 24 h (Fig. 1A). Moreover, cell density and neural network were decreased, with cells showing a great loss of neuronal integrity based on the disappearance of axons and dendrites caused by increasing concentrations of Cd. This is consistent with results from MTT assay which further demonstrate Cd-induced loss of cell viability in cerebral cortical neurons (Fig. 1B).
Figure 1. Cd induces apoptosis in cerebral cortical neurons.

(A) Morphological alterations in cerebral cortical neurons treated with different concentrations of Cd (0, 5, 10 and 20 µM) for 24 h. Cell morphology was assessed using a LEICA inverted phase-contrast microscope (400×) equipped with Quick Imaging system. All experiments were performed in duplicate. (B) Cell viability of cerebral cortical neurons, treated with 0–25 µM Cd for 12 and 24 h, was evaluated by MTT assay. Results are presented as mean ± SD (n = 6). *Statistical significance between control and Cd treatment (2.5, 5, 10, 20 and 25 µM); *P<0.05, **P<0.01 compared to respective control, using Student’s t-test. (C) Cd induced apoptotic morphological changes in cerebral cortical neurons. Cerebral cortical neurons were incubated with 0–20 µM Cd for 24 h, stained with Hoechst 33258. Cell morphology was analyzed by fluorescence microscopy (arrows, apoptotic cells). The original magnification is 400×. All experiments were performed in duplicate.
Apoptotic morphological changes induced by Cd in cerebral cortical neurons were assessed by fluorescence microscopy analysis of Hoechst 33258 staining (Fig.1C). In the control group, the majority of cells had uniformly stained nuclei, and the chromatin of normal nuclei was unaltered and spread uniformly throughout the entire nucleus. After exposure to CdAc2 (5, 10 and 20 µmol/L) for 24 h, the cells showed nuclear morphological changes typical of apoptosis, i.e., condensed nuclear chromatin and fragmented nuclei characterized by a scattered, drop-like structure. The nuclei of apoptotic cells were smaller than the nuclei of intact cells.
Cd Induces Calcium-dependent Apoptosis
To determine the cellular calcium level after treatment of cerebral cortical neurons with Cd, [Ca2+]i was measured by flow cytometry analysis of cells stained with Fluo-4 AM, a calcium indicator dye. We found that treatment with Cd (5, 10, 20 µM) resulted in a concentration-dependent increase of [Ca2+]i in cerebral cortical neurons (Fig. 2A). To verify the role of [Ca2+]i as a key second messenger, cells were pre-loaded with 10 µM BAPTA-AM for 30 min. BAPTA-AM is an effective membrane-permeable intracellular Ca2+ chelator, and becomes trapped in the cells after cytoplasmic hydrolysis. As shown in Fig. 2A, chelating intracellular Ca2+ with BAPTA-AM prevented the elevation of [Ca2+]i, demonstrating that the release of intracellular Ca2+ is essential for Cd-induced [Ca2+]i overloading. To explore other factors contributing to the calcium overload, we studied the effect of Cd on the activities of ATPases. As shown in Fig. 2B, treatment of cerebral cortical neurons with Cd resulted in a significant loss in the activities of ATPases (P<0.05 or P<0.01), which occurred in a dose-dependent manner. When exposed to 5, 10 and 20 µM of Cd for 12 h, the Na+/K+-ATPase activity decreased to 70.1%, 52.5% and 27.2% of the control value while the Ca2+/Mg+-ATPase activity decreased to 62.6%, 49.0% and 25.5% of the control value, respectively.
Figure 2. Cd-induced intracellular [Ca2+]i elevation in cerebral cortical neurons.
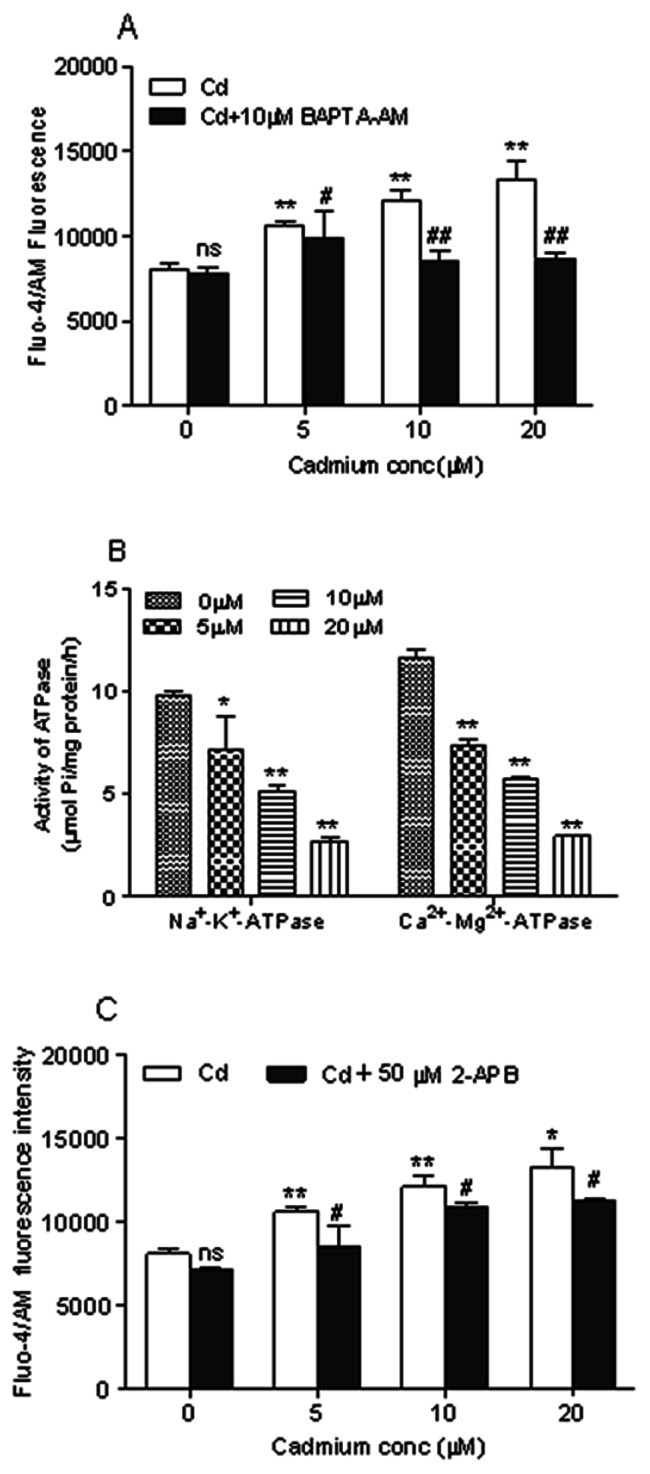
(A) Cd-induced elevation of [Ca2+]i was diminished by BAPTA-AM. Cells were pretreated with 10 µM BAPTA-AM for 30 min, followed by treatment with Cd for another 12 h and incubated with 5 µM Fluo-4 AM dye for a total of 30 min before analysis by flow cytometry. Results were expressed as mean fluorescence. Each bar represents mean ± SD (n = 6). *Statistical significance between control and Cd treatment (5, 10 and 20 µM); #Statistical significance between cells cultured in the absence and presence of BAPTA-AM. ns Not significant; **P<0.01; # P<0.05, ## P<0.01, using Student’s t-test. (B) Inhibition of Na+/K+-ATPase and Ca2+/Mg2+-ATPase activities by Cd in cerebral cortical neurons. The neurons were treated with Cd (0,5, 10 and 20 µM) for 12 h to measure the assays. Data are mean ± SD of three experiments from cells of different cultures, each one performed in triplicate. *P<0.05, **P<0.01 compared to control, using Student’s t-test. (C) Calcium released from ER through IP3R channels is a major cause of Cd-induced elevation of [Ca2+]i. Cells were pretreated with 50 µM 2-APB for 30 min, followed by treatment with Cd for another 12 h. The cytosolic calcium level was analyzed by Fluo-4 AM staining and flow cytometry. Results are expressed as mean fluorescence. Each bar represents mean ± SD (n = 6). *Statistical significance between control and Cd treatment (5, 10 and 20 µM); #Statistical significance between cells cultured in the absence and presence of 2-APB. ns Not significant; *P<0.05, **P<0.01; # P<0.05, using Student’s t-test.
The endoplasmic reticulum (ER) is one of the major calcium storage compartments in cells. To examine the role of the ER in Cd-induced elevation of [Ca2+]i, we incubated neurons with 2-APB, a blocker of the ER calcium channel (inositol-1, 4, 5-trisphosphate receptor, IP3R). As shown in Fig. 2C, we observed that the elevation of [Ca2+]i induced by Cd was suppressed by 2-APB after treatment with Cd for 12 h. Taken together, these results demonstrated that [Ca2+]i elevation induced by Cd in cerebral cortical neurons is linked to the release of calcium from the ER.
Next, to further determine the role of calcium in the regulation of Cd-induced apoptosis, cerebral cortical neurons were incubated with/without Cd (10 µM) in the absence or presence of BAPTA-AM (10 µM). As shown in Fig. 3A, Cd alone (10 µM) induced cell rounding and shrinkage, and BAPTA-AM itself did not alter cell shape. However, BAPTA-AM obviously blocked Cd-induced morphological changes. Furthermore, MTT assay results (Fig. 3B) further demonstrated that BAPTA-AM in part can suppress Cd-induced loss of cell viability in Cd-exposed cerebral cortical neurons. These results suggest that Cd-induced neuronal apoptosis might be associated with its induction of [Ca2+]i elevation (Fig. 3C).
Figure 3. Cd-induced apoptosis arises through calcium-mediated signaling.

(A) Morphological alterations in cerebral cortical neurons treated with/without Cd (10 µM) in the presence or absence of BAPTA-AM (10 µM) for 24 h. Morphology of cerebral cortical neurons was assessed using a Leica inverted phase-contrast microscope (400×) equipped with digital camera. All experiments were performed twice. (B) Cell viability of cerebral cortical neurons treated with/without Cd (10 µM) in the presence or absence of BAPTA-AM (10 µM) for 12 and 24 h. Results are presented as mean± SD (n = 6). *Statistical significance between control and Cd treatment (5, 10 and 20 µM); #Statistical significance between cells cultured in the absence and presence of BAPTA-AM. ns Not significant; *P<0.05, **P<0.01; # P<0.05, ## P<0.01, using Student’s t-test. (C) Cd-induced apoptosis were suppressed by BAPTA-AM. Cells were pretreated with BAPTA-AM (10 µM) for 30 min, followed by treatment with Cd (10 µM) for another 24 h to determine apoptosis by Hoechst 33258 staining under a fluorescence microscope (arrows, apoptotic cells). The original magnification is 400×. All experiments were performed twice.
Cd-elevated [Ca2+]i Induces ROS, Triggering Apoptosis of Cerebral Cortical Neurons
Studies have shown that Cd-induced neuronal apoptosis is attributed to the induction of ROS [36], [37]. In this study, we examined whether ROS production is involved in the Cd-induced apoptosis in cerebral cortical neurons. The expression levels of COX-I, II, and III are closely related to the generation of ROS [38]. COX is a terminal enzyme in the mitochondrial respiratory chain involved in aerobic ATP production, and has been used in several studies to assess the mitochondrial function and respiratory responses of organisms to toxic chemicals, especially Cd [39]–[41]. Thus we assessed the effects of Cd treatment on the mRNA expression levels of mitochondrial COX- I/II/III. As shown in Fig. 4, the relative expression levels of mitochondrial COX-I/II/III were all significantly (P<0.05 or P<0.01) decreased in the cerebral cortical neurons after 6 h of Cd exposure. In addition, production of ROS was detected in cells treated with Cd for 12 h (Fig. 5). As expected, BAPTA-AM significantly attenuated Cd-induced production of ROS, which is in agreement with our observation that BAPTA-AM prevented Cd-induced apoptosis and cell death in cerebral cortical neurons (Fig. 3). These data suggest that Cd elevates [Ca2+]i level, which induces ROS and in turn trigger apoptosis in cerebral cortical neurons.
Figure 4. Relative transcription levels of the genes in neurons exposed to Cd for 6 h.

The transcription levels were calculated relative to the control group, whose expression level was set to one. (A) Cytochrome oxidase subunit I (COX-I). (B) Cytochrome oxidase subunit II (COX-II). (C) Cytochrome oxidase subunit III (COX-III). Data represent mean ± SD determined by the comparative CT method (n = 6). ns Not significant; *P<0.05, **P<0.01; statistical significance between control and Cd treatment (5, 10 and 20 µM ).
Figure 5. Cd-elevated [Ca2+]i induces ROS in cerebral cortical neurons.

Cells were pretreated with BAPTA-AM (10 µM) for 30 min, followed by treatment with Cd for another 12 h. The generation of ROS was analyzed by H2-DCF-DA staining using flow cytometry. Results were expressed as mean fluorescence. Each bar represents mean ± SD (n = 6). *Statistical significance between control and Cd treatment (5, 10 and 20 µM); #Statistical significance between cells cultured in the absence and presence of BAPTA-AM. ns Not significant; *P<0.05, **P<0.01; # P<0.05, ## P<0.01 using Student’s t-test.
Cd-induced Apoptosis Occurs Through Ca2+-mitochondria Signaling
We examined the effect of Cd on the mitochondrial apoptosis pathway. A decline in Δψm is a morphological characteristic of mitochondrial function impairment. Rh123, a lipophilic cationic fluorescent dye, is selectively taken up by mitochondria and used to assess the mitochondrial ΔΨ of cerebral cortical neurons. As shown in Fig. 6, the mitochondrial ΔΨ decreased in cerebral cortical neurons after exposure to Cd (5, 10 and 20 µM) for 12 h. A significant reduction in mitochondrial ΔΨ was observed (P<0.05 or P<0.01).
Figure 6. Cd-induced dissipation of Δψm was inhibited by BAPTA-AM.

Cells were pretreated with BAPTA-AM (10 µM) for 30 min, followed by treatment with Cd for another 12 h. The Δψm was analyzed by Rh123 staining using flow cytometry. Results were expressed as mean fluorescence. Each bar represents mean ± SD (n = 6). *Statistical significance between control and Cd treatment (5, 10 and 20 µM); #Statistical significance between cells cultured in the absence and presence of BAPTA-AM. ns Not significant; *P<0.05, **P<0.01; # P<0.05, ## P<0.01 using Student’s t-test.
Activation of caspase-9 and -3 and cleavage of PARP are events downstream of the collapse of mitochondria. Our immunoblotting results showed that the levels of activated caspase-9 and -3 and cleaved PARP increased after treatment with 5, 10 and 20 µM Cd for 24 h or 10 µM Cd for 12, 24 and 48 h (Fig. 7A and B). To unveil whether there exists a caspase-dependent mechanism involved in Cd-induced neuronal apoptosis, cerebral cortical neurons were exposed to 10 µM Cd for 24 h after pretreatment with Z-VAD-fmk (100 µM), a pan caspase inhibitor, for 1 h. Cd-induced activation of caspase-9 and -3 and cleavage of PARP were obviously attenuated by Z-VAD-fmk (Fig. 7C). However, when cerebral cortical neurons were pretreated with this inhibitor for 1 h, followed by exposure to 10 µM Cd for 12 or 24 h, MTT assay and morphological analysis revealed that Z-VAD-fmk partially rescued cells from Cd-induced cell death and apoptosis (Fig. 7D–F). In addition, we noticed that Cd increased Bax and decreased Bcl-2 levels in a time- and concentration-dependent manner in cerebral cortical neurons (Fig. 8). Our data suggest that the mitochondrial apoptosis pathway may be involved in Cd-induced apoptosis of cerebral cortical neurons.
Figure 7. Cd induced neuronal apoptosis by caspase-dependent mechanism.

(A and B) Cd increased cleavage of caspase-9, caspase-3 and PARP. Cells were exposed to 0–20 µM Cd for 24 h, or 10 µM Cd for 0–48 h and then analyzed by Western blotting. All experiments were performed twice. (C) A broad caspase inhibitor, Z-VAD-fmk, reverses Cd-induced cleavage of caspase-9, caspase-3 and PARP. Cerebral cortical neurons, treated with 10 µM Cd for 24 h following pretreatment with a pan caspase inhibitor, Z-VAD-fmk (100 µM) for 1 h, were harvested. Cell lysates were analyzed by Western blotting. All experiments were performed twice. (D) Z-VAD-fmk prevented cell death induced by Cd. Cell viability of cerebral cortical neurons treated with/without Cd (10 µM) in the presence or absence of Z-VAD-fmk (100 µM) for 12 h. Results are presented as mean± SD (n = 6). *Statistical significance between control and Cd treatment (5, 10 and 20 µM); #Statistical significance between cells cultured in the absence and presence of Z-VAD-fmk. ns Not significant; **P<0.01; ##P<0.01, using Student’s t-test. (E) Z-VAD-fmk prevented morphological alterations induced by Cd. Morphology of cerebral cortical neurons, treated with/without 10 µM Cd in the presence or absence of 100 µM Z-VAD-fmk for 24 h, was assessed using a LEICA inverted phase-contrast microscope (200×) equipped with digital camera. All experiments were performed twice. (F) Cd-induced apoptosis were suppressed by Z-VAD-fmk. Cells were pretreated with Z-VAD-fmk (100 µM) for 1 h, followed by treatment with 10 µM Cd for another 24 h to determine apoptosis by Hoechst 33258 staining under a fluorescence microscope (arrows, apoptotic cells). The original magnification is 200×. All experiments were performed twice.
Figure 8. Cd regulated the expression levels of Bcl-2 and Bax.
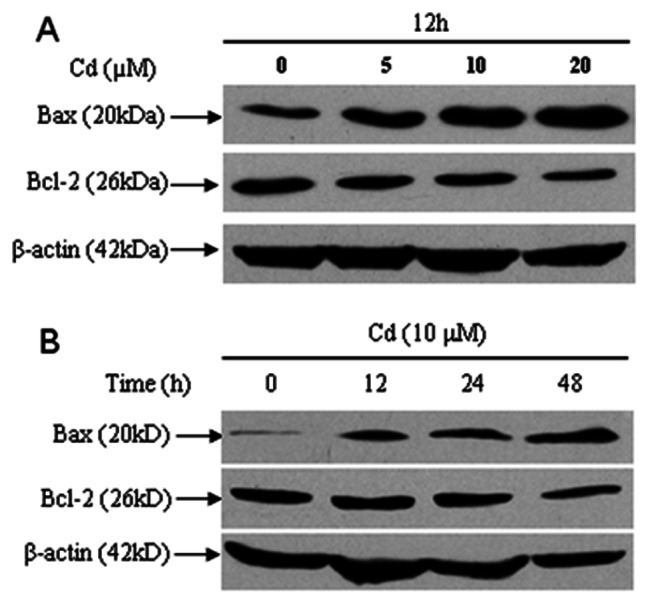
(A and B) Cerebral cortical neurons were treated with Cd (0, 5, 10, 20 µM) for 12 h, or 10 µM Cd for 0–48 h and then assessed by Western blotting. β-actin was used as an internal control. The expression level of Bcl-2 decreased, while that of Bax increased. All experiments were performed twice.
An increase in the cytoplasmic level of calcium is known to secondarily alter mitochondrial homeostasis. We found that BAPTA-AM significantly blocked disruption of Δψm in cells exposed to Cd (5, 10 and 20 µM) for 12 h (Fig. 6). Furthermore, cleavage of caspase-9, caspase-3 and PARP were significantly attenuated by BAPTA-AM (Fig. 9), which is in agreement with our observation that BAPTA-AM profoundly prevented Cd-induced apoptosis and cell death of cerebral cortical neurons (Fig. 3). However, increased Bax and decreased Bcl-2 levels were not blocked by BAPTA-AM (data not shown). These data suggest that calcium-mediated mitochondria-caspase b is involved in Cd-induced apoptosis. Moreover, our results collectively suggest that Cd-induced apoptosis of cerebral cortical neurons occurs through a calcium-mitochondria signaling pathway.
Figure 9. Activation of caspases induced by Cd occurs through calcium signaling.
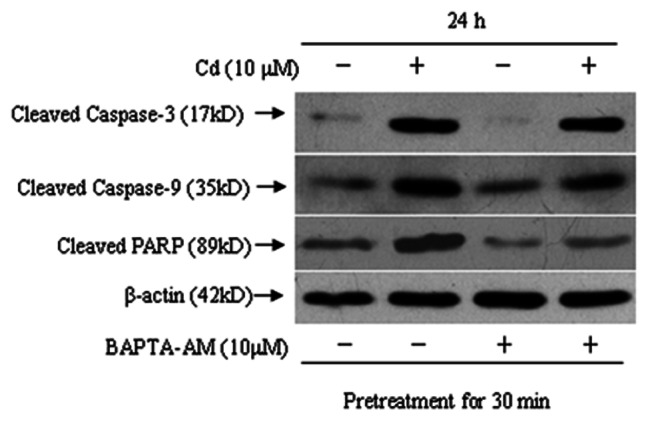
Cells were pretreated with BAPTA-AM (10 µM) for 30 min, followed by treatment with Cd (10 µM) for 24 h. Cell lysates were analyzed by Western blotting. All experiments were performed twice.
Discussion
Cerebral cortical neurons have been identified as targets of Cd-mediated toxicity [17]. Chen et al [42] have demonstrated that the effects of Cd-elevated [Ca2+]i on MAPK and mTOR network as well as apoptosis are through stimulating phosphorylation of calcium/calmodulin-dependent protein kinase II (CaMKII) in primary cortical neurons. In this study, we showed that Cd-induced apoptosis in cerebral cortical neurons is mediated through a calcium signaling pathway. Calcium has been recognized as a ubiquitous intracellular signal responsible for numerous cellular events, such as growth, proliferation, differentiation, and survival/apoptosis [19]. As a second messenger, Ca2+ mediates responses of neurons to neurotransmitters and neurotrophic factors, including cell survival or death signals [43]–[45]. Dysfunction of cellular Ca2+ homeostasis induces neuronal cell death, and this cellular event has been implicated in many neurodegenerative disorders, such as Parkinson’s disease and Alzheimer’s disease [46]–[48].
Cd has been previously demonstrated to interfere with calcium through different pathways, including inhibition of calcium-dependent ATPases or activation of the inositol triphosphate pathway [13], [22], [23]. Among these pathways, the Na+/K+-ATPase, and Ca2+/Mg2+-ATPase play critical roles in intracellular Ca2+ homeostasis, in that they pump Ca2+ in the cytosol across the plasma membrane, and disruption of this function can result in cytosolic calcium overload [49]. Cd may induce such inhibition of Na+/K+- and Ca2+/Mg2+-ATPases activities (Fig. 2B), compromising calcium homeostasis and markedly increasing [Ca2+]i.
The transition metals frequently cause elevation of [Ca2+]i which leads to a cytotoxicity effect [50]. We used BAPTA-AM to chelate intracellular calcium in order to demonstrate that elevation of [Ca2+]i plays a crucial role in Cd-induced apoptosis of cerebral cortical neurons (Fig. 3). Xie et al [51] have demonstrated that Cd-induced apoptosis was mediated by the release of Ca2+ from intracellular Ca storage. We also observed that pretreatment with 2-APB, a membrane-permeable inhibitor of IP3R [52], markedly attenuated Cd-induced [Ca2+]i elevation (Fig. 2C), suggesting that Cd-induced [Ca2+]i elevation may also involve induction of intracellular release of Ca2+ stores. It was reported that Cd could induce an increase of IP3, a ligand of IP3R, within 5 min after treatment with Cd [53], suggesting that Cd could activate the IP3R through increase of intracellular IP3 which then triggers the release of calcium from ER. ER is the major calcium storage compartment in the cell. Recent evidence has demonstrated that accumulation of calcium in the mitochondria and apoptosis induced by ER stress can be reduced by pretreatment with an inhibitor of the calcium channel of the ER (IP3R) [54], implicating the release of calcium from the ER in promoting apoptosis.
In addition, mitochondria play a role in stress responses and can produce ROS when damaged. Mitochondria are indeed a major source of ROS. ROS production is related to the level of ETC [55]; it is elevated when electron transport is reduced, which occurs in pathological situations [56]. The enzyme COX can serve as an indicator of mitochondrial function. This is because COX dysfunction increases ROS, reduces energy stores and impairs energy metabolism [57]. In addition, COX-I, II, and III levels are quite vulnerable to oxidative damage because their mRNA and protein are in close proximity to the sites of ROS generation [38]. Accordingly, COX-I, II, and III mRNA expression levels were measured using qRT-PCR to investigate the influence of Cd on COX damage. As shown in Fig. 4, the expression levels of COX-I, II, and III in the Cd treatment group were all significantly lower than that in the control group. These results suggest that a decrease in the abundance of specific COX subunits (I/II/III) may be responsible for the COX dysfunction induced by the Cd treatment, which may in turn affect the rates of mitochondrial respiration and ROS production. In metal-induced apoptosis, it is thought that the mitochondria are most pertinent in mediating apoptosis, putatively via metal-induced ROS production [58]. Cd is a well-known inducer of ROS generation in cells [16], [59]. Accumulating evidences indicate that Cd-induced neuronal toxicity is due to induction of ROS, which leads to oxidative stress [4], [36], [37]. Xu et al [17] demonstrated that Cd-elevated [Ca2+]i induces ROS, which then triggers apoptosis of neuronal cells. Our experiments also showed that Cd-induced [Ca2+]i elevation resulted in the induction of ROS in cerebral cortical neurons after a 12 h treatment with Cd (Fig. 5). BAPTA-AM attenuated Cd-induced ROS (Fig. 5) which suggests a mitochondrial origin for calcium-related ROS production. The elevated [Ca2+]i concentration and ROS may cause Ca2+ influx into mitochondria which would disrupt normal metabolism of mitochondria, leading to apoptosis and growth arrest.
Intracellular calcium overload may be related to the mitochondrial dysfunction. Mitochondria are vital organelles for cellular metabolism and bioenergetics, but they are also key regulators of cell death [60]. Since mitochondria are the major site of ATP production and mitochondrial ΔΨ is the driving force of ATP synthesis, a breakdown in the mitochondrial ΔΨ could lead to a fall in the ATP levels [61]. The resulting reduction in cellular ATP levels can disrupt ionic homeostasis which can cause an increase in [Ca2+]i and subsequent cellular apoptosis/necrosis [62]. Notably, in many (if not all) paradigms of apoptosis, ΔΨm represents the point of no return in the cascade of events that ultimately leads to cell death [63]. In cerebral cortical neurons, we have shown that Cd induces a loss in Δψm (Fig. 6), which precedes the observed apoptosis. In this study, BAPTA-AM was able to prevent this loss in Δψm (Fig. 6) and apoptosis (Fig. 3) from Cd toxicity, suggesting the collapse of mitochondria caused by the elevation of [Ca2+]i may play a role in Cd-induced apoptosis.
Members of the B-cell lymphoma (Bcl)-2 family proteins are thought to play regulatory roles in the apoptotic execution of the cells including regulation of the mitochondrial changes during apoptosis [64], [65]. Bcl-2 (an apoptosis-inhibiting gene) and Bcl-2-associated X protein (Bax) (an apoptosis-inducing gene) are two important members of the Bcl-2 family; the ratio between the two determines the survival of cells [66]. There is a negative correlation between the expression of Bcl-2 and Bax. Bcl-2 over-expression leads to cell proliferation, while Bax over-expression leads to cell death [67]. Cd exposure induces significant down-regulation of Bcl-2 expression and up-regulation of Bax [68]. In this study, we showed that Cd increased Bax and decreased Bcl-2 levels in a time- and concentration-dependent manner in cerebral cortical neurons (Fig. 8). These cellular events were not blocked by pre-treating cerebral cortical neurons with BAPTA-AM (data not shown). A number of studies have indicated that Bcl-2 prevents cytosolic calcium increase, while Bax may promote this increase during apoptosis [69], [70]. Cd reduces the expression of Bcl-2 and increases the expression of Bax, which results in an overload of Ca2+ in the mitochondria and promotes the opening of permeability transition pores. Mitochondria will then swell, with their outer membranes collapsing and exiting into the cytoplasm, which would consequently trigger apoptosis. In conclusion, Cd-induced apoptosis and [Ca2+]i elevation of cerebral cortical neurons may involve regulation of the expression of Bcl-2 family proteins.
Caspases, a family of cysteine-dependent aspartate-directed proteases, play critical roles in the initiation and execution of apoptosis [71]. Among various caspases, caspase-3 appears to be the main effector, due to (i) its critical role in inducing characteristic apoptotic changes, including chromatin condensation, DNA fragmentation, and formation of apoptotic bodies, and (ii) its close association with many other mediators in apoptosis such as caspase-9, cytochrome c, and PARP [72]. [Ca2+]i elevation may have mediated Cd-induced apoptosis through [Ca2+]i-ROS-JNK-caspase-3 and Ca2+-mitochondria-caspase signaling pathways [13], [73]. Recent studies have shown that Cd induces caspase-dependent and -independent apoptosis of PC12 and SH-SY5Y neuronal cells [36]. Furthermore, the caspase-3 pathway is involved in Cd-induced apoptosis in cortical neurons [74]. Our results demonstrated that Cd-induced apoptosis in cerebral cortical neurons involves the activation of caspase-3, caspase-9 and the cleavage of PARP. Pretreatment of the cells with a broad-spectrum caspase inhibitor, Z-VAD-fmk, did prevent cleavage of caspase-3, caspase-9 and PARP (Fig. 7A–C), but partially prevented Cd-induced cell death and apoptosis (Fig. 7D–F), revealing that Cd induces neuronal apoptosis through caspase-dependent mechanism. Similar findings have been reported in other neuronal cells, such as PC12 cells [36] and oligodendrocytes [29]. BAPTA-AM was able to reduce caspase activation and the cleavage of PARP (Fig. 9), suggesting apoptosis in Cd-treated cerebral cortical neurons is mediated by calcium-mitochondria-caspase signaling.
In the present study, we observed that Cd-induced apoptosis is associated with calcium-induced massive production of ROS, dissipation of ΔΨm, cleavage of caspase-9, caspase-3 and PARP. Collectively, our results demonstrate that Cd-induced apoptosis is mediated by calcium signaling pathway and calcium-mediated apoptosis occurs through the mitochondria-caspase signaling pathway. In conclusion, our results provide a molecular evidence to demonstrate a novel finding that Cd induces calcium-mitochondria-caspase-mediated apoptosis.
Acknowledgments
We are thankful to Dr. Cheng-ming Wang for the help with the English editing of the manuscript and Dr Mao-zhi Hu for technical assistance in flow cytometry analysis.
Funding Statement
This work was supported by the National Natural Science Foundation of China (No. 31101866, to Yan Yuan; No. 31172373, to Zong-ping Liu) (http://www.nsfc.gov.cn/Portal0/default152.htm) and a project Funded by Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD) (http://jsycw.ec.js.edu.cn/). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Wu X, Liang Y, Jin T, Ye T, Kong Q, et al. (2008) Renal effects evolution in a Chinese population after reduction of cadmium exposure in rice. Environ Res 108: 233–238. [DOI] [PubMed] [Google Scholar]
- 2. Oliveira H, Lopes T, Almeida T, Pereira Mde L, Santos C (2012) Cadmium-induced genetic instability in mice testis. Hum Exp Toxicol 31: 1228–1236. [DOI] [PubMed] [Google Scholar]
- 3. Baker JR, Edwards RJ, Lasker JM, Moore MR, Satarug S (2005) Renal and hepatic accumulation of cadmium and lead in the expression of CYP4F2 and CYP2E1. Toxicol Lett 159: 182–191. [DOI] [PubMed] [Google Scholar]
- 4. Lopez E, Arce C, Oset-Gasque MJ, Canadas S, Gonzalez MP (2006) Cadmium induces reactive oxygenspecies generation and lipid peroxidation in cortical neurons in culture. Free Radic Biol Med 40: 940–951. [DOI] [PubMed] [Google Scholar]
- 5. Coccini T, Barni S, Vaccarone R, Mustarelli P, Manzo L, et al. (2013) Pulmonary toxicity of instilled cadmium-doped silica nanoparticles during acute and subacute stages in rats. Histol Histopathol 28: 195–209. [DOI] [PubMed] [Google Scholar]
- 6. Prabu SM, Shagirtha K, Renugadevi J (2011) Naringenin in combination with vitamins C and E potentially protects oxidative stress-mediated hepatic injury in cadmium-intoxicated rats. J Nutr Sci Vitaminol (Tokyo) 57: 177–185. [DOI] [PubMed] [Google Scholar]
- 7. Napolitano JR, Liu MJ, Bao S, Crawford M, Nana-Sinkam P, et al. (2012) Cadmium-mediated toxicity of lung epithelia is enhanced through NF-kappaB-mediated transcriptional activation of the human zinc transporter ZIP8. Am J Physiol Lung Cell Mol Physiol 302: L909–918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lukawski K, Nieradko B, Sieklucka-Dziuba M (2005) Effects of cadmium on memory processes in mice exposed to transient cerebral oligemia. Neurotoxicol Teratol 27: 575–584. [DOI] [PubMed] [Google Scholar]
- 9. Nishimura Y, Yamaguchi JY, Kanada A, Horimoto K, Kanemaru K, et al. (2006) Increase in intracellular Cd2+ concentration of rat cerebellar granule neurons incubated with cadmium chloride: cadmium cytotoxicity under external Ca2+-free condition. Toxicol In Vitro 20: 211–216. [DOI] [PubMed] [Google Scholar]
- 10. Okuda B, Iwamoto Y, Tachibana H, Sugita M (1997) Parkinsonism after acute cadmium poisoning. Clin Neurol Neurosurg 99: 263–265. [DOI] [PubMed] [Google Scholar]
- 11. Jiang LF, Yao TM, Zhu ZL, Wang C, Ji LN (2007) Impacts of Cd(II) on the conformation and self-aggregation of Alzheimer's tau fragment corresponding to the third repeat of microtubule-binding domain. Biochim Biophys Acta 1774: 1414–1421. [DOI] [PubMed] [Google Scholar]
- 12. Son YO, Lee JC, Hitron JA, Pan J, Zhang Z, et al. (2010) Cadmium induces intracellular Ca2+- and H2O2-dependent apoptosis through JNK- and p53-mediated pathways in skin epidermal cell line. Toxicol Sci 113: 127–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Wang SH, Shih YL, Ko WC, Wei YH, Shih CM (2008) Cadmium-induced autophagy and apoptosis are mediated by a calcium signaling pathway. Cell Mol Life Sci 65: 3640–3652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Yeh JH, Huang CC, Yeh MY, Wang JS, Lee JK, et al. (2009) Cadmium-induced cytosolic Ca2+ elevation and subsequent apoptosis in renal tubular cells. Basic Clin Pharmacol Toxicol 104: 345–351. [DOI] [PubMed] [Google Scholar]
- 15. Biagioli M, Pifferi S, Ragghianti M, Bucci S, Rizzuto R, et al. (2008) Endoplasmic reticulum stress and alteration in calcium homeostasis are involved in cadmium-induced apoptosis. Cell Calcium 43: 184–195. [DOI] [PubMed] [Google Scholar]
- 16. Wang L, Cao J, Chen D, Liu X, Lu H, et al. (2009) Role of oxidative stress, apoptosis, and intracellular homeostasis in primary cultures of rat proximal tubular cells exposed to cadmium. Biol Trace Elem Res 127: 53–68. [DOI] [PubMed] [Google Scholar]
- 17. Xu B, Chen S, Luo Y, Chen Z, Liu L, et al. (2011) Calcium signaling is involved in cadmium-induced neuronal apoptosis via induction of reactive oxygen species and activation of MAPK/mTOR network. PLoS One 6: e19052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Berridge MJ, Bootman MD, Roderick HL (2003) Calcium signalling: dynamics, homeostasis and remodelling. Nat Rev Mol Cell Biol 4: 517–529. [DOI] [PubMed] [Google Scholar]
- 19. Clapham DE (2007) Calcium signaling. Cell 131: 1047–1058. [DOI] [PubMed] [Google Scholar]
- 20. Thevenod F, Jones SW (1992) Cadmium block of calcium current in frog sympathetic neurons. Biophys J 63: 162–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Beyersmann D, Hechtenberg S (1997) Cadmium, gene regulation, and cellular signalling in mammalian cells. Toxicol Appl Pharmacol 144: 247–261. [DOI] [PubMed] [Google Scholar]
- 22. Visser GJ, Peters PH, Theuvenet AP (1993) Cadmium ion is a non-competitive inhibitor of red cell Ca2+-ATPase activity. Biochim Biophys Acta 1152: 26–34. [DOI] [PubMed] [Google Scholar]
- 23. Wang SH, Shih YL, Lee CC, Chen WL, Lin CJ, et al. (2009) The role of endoplasmic reticulum in cadmium-induced mesangial cell apoptosis. Chem Biol Interact 181: 45–51. [DOI] [PubMed] [Google Scholar]
- 24. Lohmann RD, Beyersmann D (1993) Cadmium and zinc mediated changes of the Ca2+-dependent endonuclease in apoptosis. Biochem Biophys Res Commun 190: 1097–1103. [DOI] [PubMed] [Google Scholar]
- 25. Misra UK, Gawdi G, Akabani G, Pizzo SV (2002) Cadmium-induced DNA synthesis and cell proliferation in macrophages: the role of intracellular calcium and signal transduction mechanisms. Cell Signal 14: 327–340. [DOI] [PubMed] [Google Scholar]
- 26. Long GJ (1997) Cadmium perturbs calcium homeostasis in rat osteosarcoma (ROS 17/2.8) cells; a possible role for protein kinase C. Toxicol Lett. 91: 91–97. [DOI] [PubMed] [Google Scholar]
- 27. Chacon E, Acosta D (1991) Mitochondrial regulation of superoxide by Ca2+: an alternate mechanism for the cardiotoxicity of doxorubicin. Toxicol Appl Pharmacol 107: 117–128. [DOI] [PubMed] [Google Scholar]
- 28. Ermak G, Davies KJ (2002) Calcium and oxidative stress: from cell signaling to cell death. Mol Immunol 38: 713–721. [DOI] [PubMed] [Google Scholar]
- 29. Hossain S, Liu HN, Nguyen M, Shore G, Almazan G (2009) Cadmium exposure induces mitochondria-dependent apoptosis in oligodendrocytes. Neurotoxicology 30: 544–554. [DOI] [PubMed] [Google Scholar]
- 30. Krumschnabel G, Ebner HL, Hess MW, Villunger A (2010) Apoptosis and necroptosis are induced in rainbow trout cell lines exposed to cadmium. Aquat Toxicol 99: 73–85. [DOI] [PubMed] [Google Scholar]
- 31. Ferri KF, Kroemer G (2001) Organelle-specific initiation of cell death pathways. Nat Cell Biol 3: E255–263. [DOI] [PubMed] [Google Scholar]
- 32. Chatterjee S, Kundu S, Sengupta S, Bhattacharyya A (2009) Divergence to apoptosis from ROS induced cell cycle arrest: effect of cadmium. Mutat Res 663: 22–31. [DOI] [PubMed] [Google Scholar]
- 33. Cui ZG, Ogawa R, Piao JL, Hamazaki K, Feril LB Jr, et al. (2011) Molecular mechanisms involved in the adaptive response to cadmium-induced apoptosis in human myelomonocytic lymphoma U937 cells. Toxicol In Vitro 25: 1687–1693. [DOI] [PubMed] [Google Scholar]
- 34. Yan Y, Bian JC, Zhong LX, Zhang Y, Sun Y, et al. (2012) Oxidative stress and apoptotic changes of rat cerebral cortical neurons exposed to cadmium in vitro. Biomed Environ Sci 25: 172–181. [DOI] [PubMed] [Google Scholar]
- 35. Schmittgen TD, Livak KJ (2008) Analyzing real-time PCR data by the comparative C(T) method. Nat Protoc 3: 1101–1108. [DOI] [PubMed] [Google Scholar]
- 36. Chen L, Liu L, Huang S (2008) Cadmium activates the mitogen-activated protein kinase (MAPK) pathway via induction of reactive oxygen species and inhibition of protein phosphatases 2A and 5. Free Radic Biol Med 45: 1035–1044. [DOI] [PubMed] [Google Scholar]
- 37. Chen L, Xu B, Liu L, Luo Y, Zhou H, et al. (2011) Cadmium induction of reactive oxygen species activates the mTOR pathway, leading to neuronal cell death. Free Radic Biol Med 50: 624–632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Sohal RS, Toroser D, Bregere C, Mockett RJ, Orr WC (2008) Age-related decrease in expression of mitochondrial DNA encoded subunits of cytochrome c oxidase in Drosophila melanogaster. Mech Ageing Dev 129: 558–561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Garceau N, Pichaud N, Couture P (2010) Inhibition of goldfish mitochondrial metabolism by in vitro exposure to Cd, Cu and Ni. Aquat Toxicol 98: 107–112. [DOI] [PubMed] [Google Scholar]
- 41. Wang L, Li J, Li J, Liu Z (2010) Effects of lead and/or cadmium on the oxidative damage of rat kidney cortex mitochondria. Biol Trace Elem Res 137: 69–78. [DOI] [PubMed] [Google Scholar]
- 42. Chen S, Xu Y, Xu B, Guo M, Zhang Z, et al. (2011) CaMKII is involved in cadmium activation of MAPK and mTOR pathways leading to neuronal cell death. J Neurochem 119: 1108–1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Neher E, Sakaba T (2008) Multiple roles of calcium ions in the regulation of neurotransmitter release. Neuron 59: 861–872. [DOI] [PubMed] [Google Scholar]
- 44. Cheng A, Wang S, Yang D, Xiao R, Mattson MP (2003) Calmodulin mediates brain-derived neurotrophic factor cell survival signaling upstream of Akt kinase in embryonic neocortical neurons. J Biol Chem 278: 7591–7599. [DOI] [PubMed] [Google Scholar]
- 45. Surmeier DJ, Guzman JN, Sanchez-Padilla J (2010) Calcium, cellular aging, and selective neuronal vulnerability in Parkinson's disease. Cell Calcium 47: 175–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Kawahara M, Kuroda Y (2000) Molecular mechanism of neurodegeneration induced by Alzheimer's beta-amyloid protein: channel formation and disruption of calcium homeostasis. Brain Res Bull 53: 389–397. [DOI] [PubMed] [Google Scholar]
- 47. Mattson MP (2007) Calcium and neurodegeneration. Aging Cell 6: 337–350. [DOI] [PubMed] [Google Scholar]
- 48. Marambaud P, Dreses-Werringloer U, Vingtdeux V (2009) Calcium signaling in neurodegeneration. Mol Neurodegener 4: 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Fujita T, Inoue H, Kitamura T, Sato N, Shimosawa T, et al. (1998) Senescence marker protein-30 (SMP30) rescues cell death by enhancing plasma membrane Ca2+-pumping activity in Hep G2 cells. Biochem Biophys Res Commun 250: 374–380. [DOI] [PubMed] [Google Scholar]
- 50. Sabolic I (2006) Common mechanisms in nephropathy induced by toxic metals. Nephron Physiol 104: p107–114. [DOI] [PubMed] [Google Scholar]
- 51. Xie Z, Zhang Y, Li A, Li P, Ji W, et al. (2010) Cd-induced apoptosis was mediated by the release of Ca2+ from intracellular Ca storage. Toxicol Lett 192: 115–118. [DOI] [PubMed] [Google Scholar]
- 52. Ruiz A, Matute C, Alberdi E (2009) Endoplasmic reticulum Ca2+ release through ryanodine and IP3 receptors contributes to neuronal excitotoxicity. Cell Calcium 46: 273–281. [DOI] [PubMed] [Google Scholar]
- 53. Misra UK, Gawdi G, Pizzo SV (2003) Induction of mitogenic signalling in the 1LN prostate cell line on exposure to submicromolar concentrations of cadmium+. Cell Signal 15: 1059–1070. [DOI] [PubMed] [Google Scholar]
- 54. Deniaud A, Sharaf el dein O, Maillier E, Poncet D, Kroemer G, et al. (2008) Endoplasmic reticulum stress induces calcium-dependent permeability transition, mitochondrial outer membrane permeabilization and apoptosis. Oncogene 27: 285–299. [DOI] [PubMed] [Google Scholar]
- 55. Fleury C, Mignotte B, Vayssiere JL (2002) Mitochondrial reactive oxygen species in cell death signaling. Biochimie 84: 131–141. [DOI] [PubMed] [Google Scholar]
- 56. Wallace DC (2005) A mitochondrial paradigm of metabolic and degenerative diseases, aging, and cancer: a dawn for evolutionary medicine. Annu Rev Genet 39: 359–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Onyango IG, Bennett JP Jr, Tuttle JB (2005) Endogenous oxidative stress in sporadic Alzheimer's disease neuronal cybrids reduces viability by increasing apoptosis through pro-death signaling pathways and is mimicked by oxidant exposure of control cybrids. Neurobiol Dis 19: 312–322. [DOI] [PubMed] [Google Scholar]
- 58. Foster KA, Galeffi F, Gerich FJ, Turner DA, Muller M (2006) Optical and pharmacological tools to investigate the role of mitochondria during oxidative stress and neurodegeneration. Prog Neurobiol 79: 136–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Thevenod F (2009) Cadmium and cellular signaling cascades: to be or not to be? Toxicol Appl Pharmacol 238: 221–239. [DOI] [PubMed] [Google Scholar]
- 60. Fantin VR, Leder P (2006) Mitochondriotoxic compounds for cancer therapy. Oncogene 25: 4787–4797. [DOI] [PubMed] [Google Scholar]
- 61. Chakraborti T, Das S, Mondal M, Roychoudhury S, Chakraborti S (1999) Oxidant, mitochondria and calcium: an overview. Cell Signal 11: 77–85. [DOI] [PubMed] [Google Scholar]
- 62. Grammatopoulos TN, Johnson V, Moore SA, Andres R, Weyhenmeyer JA (2004) Angiotensin type 2 receptor neuroprotection against chemical hypoxia is dependent on the delayed rectifier K+ channel, Na+/Ca2+ exchanger and Na+/K+ ATPase in primary cortical cultures. Neurosci Res 50: 299–306. [DOI] [PubMed] [Google Scholar]
- 63. Kroemer G, Galluzzi L, Brenner C (2007) Mitochondrial membrane permeabilization in cell death. Physiol Rev 87: 99–163. [DOI] [PubMed] [Google Scholar]
- 64. Tsujimoto Y, Shimizu S (2000) Bcl-2 family: life-or-death switch. FEBS Lett 466: 6–10. [DOI] [PubMed] [Google Scholar]
- 65. Matsumoto A, Isomoto H, Nakayama M, Hisatsune J, Nishi Y, et al. (2011) Helicobacter pylori VacA reduces the cellular expression of STAT3 and pro-survival Bcl-2 family proteins, Bcl-2 and Bcl-XL, leading to apoptosis in gastric epithelial cells. Dig Dis Sci 56: 999–1006. [DOI] [PubMed] [Google Scholar]
- 66. Mertens HJ, Heineman MJ, Evers JL (2002) The expression of apoptosis-related proteins Bcl-2 and Ki67 in endometrium of ovulatory menstrual cycles. Gynecol Obstet Invest 53: 224–230. [DOI] [PubMed] [Google Scholar]
- 67. Cory S, Huang DC, Adams JM (2003) The Bcl-2 family: roles in cell survival and oncogenesis. Oncogene 22: 8590–8607. [DOI] [PubMed] [Google Scholar]
- 68. Zhang H, Cai C, Shi C, Cao H, Han Z, et al. (2012) Cadmium-induced oxidative stress and apoptosis in the testes of frog Rana limnocharis. Aquat Toxicol 122–123: 67–74. [DOI] [PubMed] [Google Scholar]
- 69. Lam M, Dubyak G, Chen L, Nunez G, Miesfeld RL, et al. (1994) Evidence that BCL-2 represses apoptosis by regulating endoplasmic reticulum-associated Ca2+ fluxes. Proc Natl Acad Sci U S A 91: 6569–6573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Foyouzi-Youssefi R, Arnaudeau S, Borner C, Kelley WL, Tschopp J, et al. (2000) Bcl-2 decreases the free Ca2+ concentration within the endoplasmic reticulum. Proc Natl Acad Sci U S A 97: 5723–5728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Budihardjo I, Oliver H, Lutter M, Luo X, Wang X (1999) Biochemical pathways of caspase activation during apoptosis. Annu Rev Cell Dev Biol 15: 269–290. [DOI] [PubMed] [Google Scholar]
- 72. Porter AG, Janicke RU (1999) Emerging roles of caspase-3 in apoptosis. Cell Death Differ 6: 99–104. [DOI] [PubMed] [Google Scholar]
- 73. Kim J, Sharma RP (2004) Calcium-mediated activation of c-Jun NH2-terminal kinase (JNK) and apoptosis in response to cadmium in murine macrophages. Toxicol Sci 81: 518–527. [DOI] [PubMed] [Google Scholar]
- 74. Lopez E, Figueroa S, Oset-Gasque MJ, Gonzalez MP (2003) Apoptosis and necrosis: two distinct events induced by cadmium in cortical neurons in culture. Br J Pharmacol 138: 901–911. [DOI] [PMC free article] [PubMed] [Google Scholar]