

Abstract
After recanalization, cerebral blood flow (CBF) can increase above baseline in cerebral ischemia. However, the significance of post-ischemic hyperperfusion for tissue recovery remains unclear. To analyze the course of post-ischemic hyperperfusion and its impact on vascular function, we used magnetic resonance imaging (MRI) with pulsed arterial spin labeling (pASL) and measured CBF quantitatively during and after a 60 minute transient middle cerebral artery occlusion (MCAO) in adult rats. We added a 5% CO2 - challenge to analyze vasoreactivity in the same animals. Results from MRI were compared to histological correlates of angiogenesis. We found that CBF in the ischemic area recovered within one day and reached values significantly above contralateral thereafter. The extent of hyperperfusion changed over time, which was related to final infarct size: early (day 1) maximal hyperperfusion was associated with smaller lesions, whereas a later (day 4) maximum indicated large lesions. Furthermore, after initial vasoparalysis within the ischemic area, vasoreactivity on day 14 was above baseline in a fraction of animals, along with a higher density of blood vessels in the ischemic border zone. These data provide further evidence that late post-ischemic hyperperfusion is a sequel of ischemic damage in regions that are likely to undergo infarction. However, it is transient and its resolution coincides with re-gaining of vascular structure and function.
Introduction
“Luxury perfusion syndrome” is a term coined by Niels Lassen in 1966. It refers to a state of overabundant cerebral blood flow (CBF) relative to the metabolic needs of the brain tissue, as is observed within seconds after reopening of a formerly occluded artery in experimental stroke models [1]. Early post-ischemic hyperperfusion is usually abrupt, lasts only for minutes to a few hours and is closely related to severity and length of prior ischemia [2], [3]. High CBF in this early stage after ischemia has been correlated to more severe neuronal damage and worse outcome, mediated, in part, by the overproduction and release of toxic free radicals [4], [5].
Evidence for a similar phenomenon in humans comes from clinical studies, where CBF was increased above normal in some patients after vessel recanalization [6], [7], [8], [9]. Although there is consensus that at least partial recanalization is a prerequisite for hyperperfusion after stroke, the incidence as well the meaning of hyperperfusion for patient recovery have remained controversial [8], [10]. It may be assumed, however, that the mechanisms of early post-ischemic hyperperfusion in animals are distinct from late hyperperfusion observed in patients up to weeks after stroke [11]. Animal stroke models allow for the observation of blood flow over time following a controlled stroke and reperfusion paradigm. Perfusion-weighted MRI has been frequently applied as a non-invasive method to measure CBF [12], [13]. However, only very few data exist about CBF at more chronic time points after experimental ischemia, which would better resemble the clinical observation times of hyperperfusion in stroke patients [14], [15], [16]. We used a rat stroke model and quantified CBF with pulsed arterial spin labeling (pASL) MRI. Our goal was to find out how CBF is maintained at different time intervals after reperfusion and how the capacity for vasodilation recovers in the ischemic area.
Materials and Methods
Animals and experimental protocol
All experiments were performed in accordance with the National Institutes of Health animal protection guidelines and approved by the Institutional Animal Care and Use Committee, University of California, San Diego (Protocol number S04192).
In this study, 14 adult male Wistar rats (280–320 g body weight) were used. For MRI and surgery, animals were anesthetized using facemask inhalation of 1.5–2.5% isoflurane in a 2∶1 N2O∶O2 atmosphere. Temperature was maintained at 37°C by a feedback-controlled heating pad.
Six animals were subjected to a single MRI session for CBF quantification under the isoflurane anesthesia described above (referred to as “air”) and with an additional 5% CO2 challenge (referred to as 5% CO2). Using arterial blood gas analyses we found in a separate set of animals (using the identical set-up and performed within 6 months of the experiments presented here) that these anesthesia conditions resulted in normoxia (“air”) and hypercapnia (5% CO2) followed by a robust increase in CBF during hypercapnia [17]. In 8 rats, transient ischemia was induced for 60 minutes by MCAO using the intraluminal thread model [18]. Briefly, a 4-0 silicone-coated filament was introduced into the common carotid artery and advanced approximately 16 to 20 mm from the carotid bifurcation until a slight resistance was felt. The thread was left in place for 60 min. During that time and without discontinuation of anesthesia, the animal was placed in the custom-built cradle for the first (day0) MRI examination. After 60 minutes, the thread was withdrawn and the animal allowed to recover. Animals were re-anesthetized for repetitive MRI after MCAO: MRI assessment was performed at the following time points: day 0 (d0), d1, d4, and d14 after MCAO. For induction of hypercapnia, the isoflurane concentration was kept constant while the inhaled gas composition was changed to 5% CO2 in medical air. A 2 minute adjustment was allowed between switching the gas and initiating data acquisition. The overall duration of isoflurane anesthesia for the MRI scans on d1 to d14 was 45 minutes. Before MRI, stroke-induced functional deficits were assessed using an 18-point composite neurological score [19]. The test incorporated the observation of (1) spontaneous activity, (2) symmetry in limb movement, (3) forepaw outstretching, (4) climbing, (5) body proprioception, and (6) response to vibrissae touch, each scored with a maximum of three points, so that 18 points indicated no neurological deficit. After the last MRI, animals were killed by an overdose of pentobarbital and subjected to transcardial perfusion fixation with 0.1 mol/L phosphate-buffered saline (PBS), followed by 4% formalin in phosphate buffer. Brains were extracted for histological analysis.
MRI experiments
MRI experiments were carried out on a 3 T GE Signa Excite whole-body system with a body transmit coil and a custom-built passively decoupled single-loop receive-only head coil of 3 cm diameter [20]. For perfusion imaging, we used multislice flow-sensitive alternating inversion recovery (FAIR) pulsed arterial spin labeling (pASL) with the QUIPSS II modification to minimize the effects of transit delays [21]. Imaging parameters were as follows: multi-shot spiral; slice thickness: 2 mm (three slices); gap: 1 mm; field of view: 4×4 cm; matrix: 64×64; flip angle: 90°, number of interleaves: 8, number of repetitions: 12; TE: 4.3 ms; TI1/TI2: 700/1250 ms gap between tagging and imaging region: 5 mm. In-plane saturation was applied immediately after the preparatory inversion pulse for background suppression. Artefacts from pulsations of the carotid arteries below the brain were suppressed with a spatial saturation pulse. The image acquisition was immediately followed by a global saturation (PostSat) to reset the blood signal. For details of equilibrium magnetization reference scan and coil sensitivity profile estimation see [20].
CBF was calculated from the signal difference ΔS between tag and control ASL images according to:
| (1) |
where α is the tagging efficiency, M 0B is the MRI signal from a voxel full of arterial blood, τ is the temporal width of the blood bolus that reaches the region of interest, and T1B is the T1 of blood. 2αM 0B is the initial magnetization difference between tagged and control blood, the product τCBF is the amount of tagged blood that flows into the region of interest (ROI), and the exponential factor reflects T1relaxation of the tag.
In addition, anatomical images (T2-weighted, 256×256), as well as T1 and T2 maps were acquired with the same slice prescription as the ASL experiment.
MRI data processing and statistical analyses
For MRI data processing, images from all animals per group were first co-registered and then averaged using codes written in Matlab (Mathworks, Natick, MA, USA) software. Group values given in this study were obtained by ROI analysis on the co-registered and averaged MR images. Vasoreactivity (VR) maps were generated by subtracting the CBF map acquired in air from the one acquired in 5% CO2. To define voxels with hyperperfusion, the mean signal and standard deviation of the contralateral hemisphere (excluding the ventricles) were obtained on CBF maps. Voxels with CBF values above this mean plus two standard deviations were considered as “hyperperfused”. Similarly, voxels with T2 values above the mean and two standard deviations of the contralateral hemisphere were assigned to the final infarct. Statistical analyses were conducted using SPSS v12.0 for Windows. All values were reported as mean ± standard deviation. The area of RECA-positive signal within the ROIs was compared by independent t-test. CBF values between ischemic and contralateral voxels were compared with a paired t-test. The Pearson correlation coefficient was used for correlation analyses between MRI parameters. A Repeated Measures General Linear Model (GLM) Analysis was conducted for the comparison of ipsi-and contralateral CBF values. P values below 0.05 were considered significant.
Histology
After perfusion fixation, brains were removed, immersed in ice-cold 4% formalin in phosphate buffer overnight and transferred to 30% sucrose solution for at least 3 days. Coronal 40-µm-thick sections were cut on a freezing microtome (Leica, Nussloch, Germany). Premounted sections were stained with hematoxylin eosin (HE). Immunofluorescence staining of endothelial cells was performed using mouse anti-rat endothelial cell antigen (RECA-1; 1∶400; AbD Serotec) and fluorescein (FITC) -labeled donkey anti-mouse secondary antibody (1∶250; Dianova).
For analysis of RECA -staining, slices were acquired from a single staining session and photographed under a 20× objective of an epifluorescence microscope, while fluorescence gain parameters were kept constant. FITC - fluorescence signal was quantified using ImageJ software (NIH Bethesda, MD) within a 3×3 mm region of interest (ROI) positioned within the subcortical border zone of the lesion and the homologous contralateral area. The ipsilateral signal density was compared to the contralateral ROI.
Results
In a first step, CBF was determined in healthy adult Wistar rats (n = 6) under normal (breathing air) and high-flow (breathing 5% CO2) conditions. Hypercapnia resulted in a robust increase in CBF throughout the brain, with an average of 58.5% (Figure 1).
Figure 1. Assessment of vasoreactivity using 5% CO2.

CBF maps from six healthy animals were averaged. A: averaged CBF map during inhalation anaesthesia with air and B: with 5% CO2. C: Vasoreactivity map (VR) obtained by subtracting A from B. D: Anatomical location of brain slices analyzed in MRI with respect to intraaural (IA) line. The graph on the right side displays whole brain CBF values measured with air or CO2.
60 min of MCAO resulted in a profound drop in CBF within the ischemic hemisphere. We grouped voxels displaying a CBF of <30 ml/100 g/min on d0 to follow tissue with severely compromised perfusion over the course of reperfusion and beyond (Figure 2). On day 1 after MCAO, CBF within the d0-defined core voxels had recovered and was on average above contralateral CBF values. Towards d4, CBF rose further above values derived from the contralateral hemisphere. Voxels with an increase in CBF of more than two standard deviations above contralateral were classified as “hyperperfused”. Hyperperfusion evolved in different time courses after MCAO when individual animals were analyzed. Overall, we observed three patterns of hyperperfusion after MCAO (Table 1, Figures 3 and 4A). In large hemispheric cortico-subcortical strokes (type A, n = 2), post-stroke hyperperfusion was first visible at d4 after MCAO, further increasing towards d14. Animals with moderate cortico-subcortical strokes (type B, n = 2) showed a few voxels with hyperperfusion already on d1, with a maximum on d4. Hyperperfusion was clearly remittent towards d14. Predominantly subcortical infarctions (type C, n = 4) already reached the maximum of hyperperfusion on d1, remaining fairly stable or decreasing slightly until d14. To analyze vasodilation as an indicator of vasoreactivity, in addition to standard air conditions, CBF was measured during the application of a 5% CO2-challenge on d4 and 14 (Figure 3). In type A strokes, vasoreactivity was depressed on d4 and 14. In type B strokes, vasoreactivity was depressed on d4, but overshooting on d14. Vasoreactivity was either not affected at all or increased in punctate areas within the ischemic lesion on d4, more pronounced on d14, in type C strokes. The final infarct size - determined from T2 maps at day 14 - had a similar distribution as the region of hyperperfusion at its maximum (Figure 3: T2-ROI). The number of hyperperfused voxels on d4 was different between infarct types (Figure 4A) and correlated with final infarct size determined from T2w-MRI on d14 (r = 0.76; p = 0.028); the correlation was even stronger when the number of voxels at the time of maximal hyperperfusion (d1, d4 or d14) were correlated with T2-lesion size (r = 0. 88; p = 0.004). The absolute CBF values within the hyperperfused area could not be used to discriminate infarct type (A, B, C) at any observation time point (Figure 4B). The maximal number of voxels with hyperperfusion correlated with the number of voxels with a severe perfusion restriction (CBF<30 ml/100 g/min) on d0 (r = 0.72; p = 0.04), but not with functional outcome on d14 (r = −0.49). The number of hyperperfused voxels on d14 was a better indicator for functional outcome (r = −0.61), however, this did not reach significance (p = 0.1). In line with this finding, the increment in mean functional test score from type A and B to type C strokes (Figure 4C) was not strong enough to discriminate the three infarct types; whereas T2 lesion size was (Figure 4D).
Figure 2. CBF in ischemic voxels over time.

Black line: CBF changes within voxels with intra-ischemic CBF of <30 ml/100 g/min between d 0 (D0; during vascular occlusion) up to d14 after MCAO. Grey line: contralateral CBF values from the same animals. Mean ± SD values are shown. The GLM analysis for repeated measures demonstrated a significant effect of the within-subject factor “time” (p = 0.03), but not of the between-subject factor ipsi- or contralateral side (p = 0.7), but for the interception of the two factors (p<0.0001).
Table 1. Lesion characteristics and functional test scores in individual animals.
| CBF<30 d0 | HP d1 | HP d4 | HP d14 | T2 d14 | VR+ | functional | Stroke | |
| No1 | 187 | 21 | 112 | 218 | 398 | 0 | 10 | A |
| No2 | 119 | 67 | 66 | 68 | 83 | 1 | 14 | C |
| No3 | 55 | 37 | 5 | 17 | 32 | 1 | 18 | C |
| No4 | 120 | 37 | 253 | 22 | 278 | 1 | 14 | B |
| No5 | 150 | 20 | 172 | 204 | 308 | 0 | 15 | A |
| No6 | 145 | 87 | 54 | 52 | 50 | 0 | 16 | C |
| No7 | 185 | 100 | 239 | 74 | 208 | 1 | 15 | B |
| No8 | 92 | 73 | 35 | 39 | 28 | 0 | 14 | C |
Values from all 8 animals (No1 to No8) are shown. CBF<30 d0: Numbers of voxels with CBF below 30 ml/100 g/min on d0 (considered severely ischemic). HP: hyperperfusion on d 1, 4 and 14. T2 d14: number of voxels of the final infarct which was taken from T2 maps on d 14 as number of voxels with a T2 above the mean and 2 standard deviations of contralateral. VR+ refers to presence of increased vasoreactivity (overshooting CBF response to CO2-challenge) on d14. Functional score on d14 is shown (maximum 18 points, minimum 3 points). Stroke was categorized as large cortico-subcortical (A), moderate cortico-subcortical (B) or small, predominantly subcortical (C).
Figure 3. Patterns of hyperperfusion and changes in vasoreactivity.

Three representative image series (slice IA 7.5 mm) from animals with A: large cortico-subcortical., B: moderate cortico-subcortical and C: predominantly subcortical infarction. CBF maps on days 0, 4 and 14; along with vasoreactivity (VR) - maps and T2 maps (from d4 and 14/d14) are shown. On the right, the masked infarct area as extracted from D14 T2 maps is overlaid onto the CBF D4 map to show the spatial overlap between hyperperfusion and final infarct. White arrow: points to maximum of hyperperfusion, which occurs at different time points in A) and B). Red arrow in B): when the amount of hyperperfusion has declined, remaining voxels with hyperperfusion are localized more to the infarct border.
Figure 4. Hyperperfusion and CBF in individual animals.

4A: number of hyperperfused voxels on days 1, 4 and 14 (D1, D4, D14). The three infarct types (A, B, C) show differences in the number of hyperperfused voxels over time. 4B: absolute CBF values (given in mg/100 ml/min) within ischemic voxels (<30 mg/100 ml/min on d0) over time (D0, D1, D4, D14). 4C: Mean functional test scores on D 14 (higher scores indicate less functional impairment) show a gradual increase between infarct types (A, B, C); 4D: Infarct size determined from D14 T2-maps in the three different infarct types.
Voxels in which increased vasoreactivity was observed on d14 mapped to the region of infarct on T2-weighted images (Figure 5). At that time, the remaining voxels with hyperperfusion were localized more around the infarct border zone.
Figure 5. Distribution of voxels with increased d14 vasoreactivity.

(A) Infarct region of one animal on d14 - CBF map with hyperperfused voxels (B, shown in red) overlaid onto the d14 - T2 map (B and C). These voxels were localized to the infarct border. (D) Day 14 – vasoreactivity (VR) map. Voxels with increased vasoreactivity are shown in red and overlaid on the T2 map: E. Hematoxylin-eosin stained section of the corresponding region in F.
To test if increased vasoreactivity was correlated to vascular anatomy in the border zone of infarction on d14, we analyzed the expression of the endothelial marker RECA. Subcortical regions at the borderzone of infarction showed a higher density of RECA - positive staining compared to the contralateral side in animals with increased vasoreactivity (Figure 6). Absolute area of staining (in mm2) was similar in both groups on the contralateral side (3.6+/−0.8 mm2 vs. 3.5+/−0.7 mm2).
Figure 6. Increased density of blood vessels at perilesional regions with increased d14 vasoreactivity.
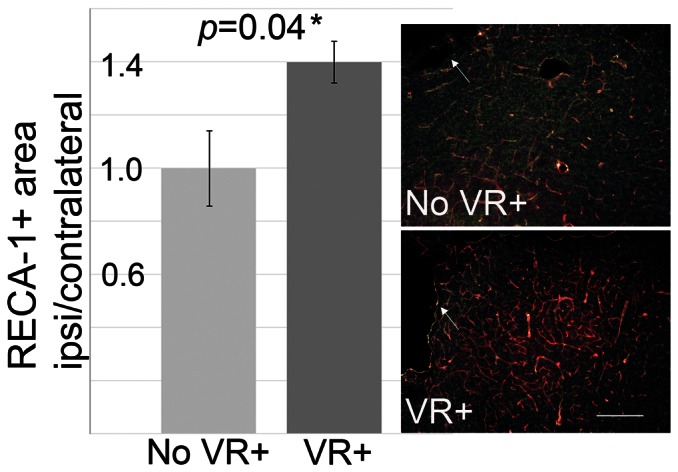
RECA-1 immunofluorescence staining (red) with DAPI nuclear staining (green) compared in animals without (No VR+) and with (VR+) increased vasoreactivity on d14 (n = 4 per group). Corresponding subcortical regions close to the infarct on d14 showed a higher density of RECA-1 signal. One example per group is shown on the right. White arrows point to the lesion. White line indicates 100 µm.
Discussion
Recanalization of the occluded vessel is highly correlated with a better functional outcome in patients affected by stroke [22], [23]. However, recanalization bears the risk of “reperfusion injury”, a phenomenon known and feared after surgical revascularization procedures, such as carotid endarterectomy [24], [25]. In early reports of post-stroke hyperperfusion, this phenomenon was suspected to be part of reperfusion injury; and a deleterious effect on stroke recovery was assumed [26]. In their studies of transient ischemia in cats, Heiss et al. found that the longer the duration of ischemia (30–120 minutes), the more severe and longer the immediate post-ischemic hyperperfusion, and the worse the chance of survival [5]. However, while these changes in CBF take place within the first minutes to hours after ischemia, not much is known about the late hyperperfusion phase observed here, occurring after days to weeks.
In clinical studies, depending on the time of analysis after stroke and the method used, between 10–50% of patients have areas of post-ischemic hyperperfusion [6], [7], [8], [10]. The fact that post-ischemic hyperperfusion could only be detected in some, but not all stroke patients, can be explained by recanalization that is partial or absent in some patients, as well as the transient nature of hyperperfusion which can be missed with a single examination. Kidwell et al. 2001 did not find a difference in clinical improvement in patients displaying hyperperfusion after stroke, but showed that tissue with post-ischemic hyperperfusion was very likely to become part of the final infarct.
In our study, late post-ischemic hyperperfusion between d1 and 14 was found in all rats, regardless of final lesion size. Similar to the findings of Kidwell et al., the area of hyperperfusion overlapped with the initial perfusion restriction, and even more, with final infarct volume.
The number of voxels with hyperperfusion was an indicator of infarct size. Small subcortical infarcts were characterized by an earlier onset of hyperperfusion (d1). In intermediate size cortico-subcortical infarcts, hyperperfusion was maximal on d4, but remittent on d14. Hyperperfusion was sustained and maximal on d14 in large hemispheric lesions. From such non-uniform temporal dynamics of blood flow adaptations it is difficult to extract a prognostic parameter at a single observation time point. However, an earlier occurrence and fewer affected voxels indicate a smaller final lesion. Although the number of hyperperfused voxels on d4 correlated to final infarct size, the correlation to functional test score on d14 was rather weak, which was most likely explained by our small sample size.
Based on our observations, we postulate that late post-ischemic hyperperfusion is a common part of the reperfusion cascade, and secondary to the initial ischemic impact.
Mechanistically, post-ischemic hyperperfusion may reflect a state of “vasoparalysis”, where endothelial cells within the affected region lose their autoregulatory capacity and remain in (sub)-maximal dilation. We tested this by analyzing the response to the vasodilatory stimulus CO2. We found that vasoreactivity within the ischemic area was depressed in most animals shortly after stroke, when hyperperfusion was maximal. Interestingly, we observed animals with an overshooting vasoreactivity on d14 after MCAO. These animals either had subcortical strokes with a rather low and constant number of hyperperfused voxels from d0 on, or intermediate cortico-subcortical lesions with a clear reduction of the hyperperfused area from d4 to d14. Such locally increased vasodilatory response to CO2 after ischemia-reperfusion has not been described before. In the series of events leading to CBF regulation after reperfusion, this probably reflects recovery of the vasculature from the vasoparalytic state. However, a limitation of our study is that arterial blood gases before and during the CO2 – challenge were not measured in the same set of animals subjected to longitudinal MRI, but in a separate group with identical settings used for MRI and anesthesia [17]. Our histological data, indicating a higher density of blood vessels in the vicinity of the infarct border in these animals, suggest that the high vasodilatory capacity might be due to young, still immature, blood vessels, which were formed in a compensatory reaction to facilitate supply to the peri-lesional tissue. Peri-lesional angiogenesis starts around 3 days post stroke; and blood vessel density has been shown to increase markedly after 7–15 days [27]. This has been associated with a higher peri-lesional CBF values [28].
Late post-ischemic hyperperfusion has been investigated after 30–90 minutes of MCAO in rats [29]. These authors also found a congruency between voxels with hyperperfusion and voxels that went on to be infarcted. The peak of hyperperfusion in their study was at 48 hours. Interestingly, they observed hyperperfusion in all animals after 30 minutes MCAO, but only in half of the 60 minute - and none of the 90 minute MCAO group. This is most likely due to the shorter observation span (only 24 hours in the 60 and 90 minute occlusion groups) in their study. As we show in our sample, larger infarcts have a later occurrence of hyperperfusion, so it was probably missed at 24 hours.
Summary
In conclusion, late post-ischemic hyperperfusion was detected between d1 and 14 after transient ischemia in our rat model. It is likely that hyperperfusion observed in stroke patients is similar to this late post-ischemic hyperperfusion (in contrast to immediate or early post-ischemic hyperperfusion). Although voxels included in late post-ischemic hyperperfusion areas are likely to undergo infarction, the occurrence of hyperperfusion per se is not related to an unfavorable functional outcome. An early occurrence and smaller area of maximal hyperperfusion after stroke are associated with smaller lesions, while a late occurrence and larger area of hyperperfusion indicate a large infarct. Vasodilatory capacity can recover in former areas of hyperperfusion and even be above baseline at later time points, indicating recovery of vascular function or integration of newly formed blood vessels into the infarct border zone.
Funding Statement
The work was supported by the Deutsche Forschungsgemeinschaft, the 6th Framework Program of the European Commission (Marie Curie Outgoing International Fellowship), the Swiss National Science Foundation (NCCR Neural Plasticity and Repair) (all to S.W.), and the National Institute of Health (R01 EB002096) (to E.C.W.). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Lassen NA (1966) The luxury-perfusion syndrome and its possible relation to acute metabolic acidosis localised within the brain. The Lancet 1113–1115. [DOI] [PubMed] [Google Scholar]
- 2. Gourley JK, Heistad DD (1984) Characteristics of reactive hyperemia in the cerebral circulation. Am J Physiol 246: H52–58. [DOI] [PubMed] [Google Scholar]
- 3. Marchal G, Young AR, Baron JC (1999) Early postischemic hyperperfusion: pathophysiologic insights from positron emission tomography. J Cereb Blood Flow Metab 19: 467–482. [DOI] [PubMed] [Google Scholar]
- 4. Soehle M, Heimann A, Kempski O (1998) Postischemic application of lipid peroxidation inhibitor U-101033E reduces neuronal damage after global cerebral ischemia in rats. Stroke 29: 1240–1246 discussion 1246–1247. [DOI] [PubMed] [Google Scholar]
- 5. Heiss WD, Graf R, Lottgen J, Ohta K, Fujita T, et al. (1997) Repeat positron emission tomographic studies in transient middle cerebral artery occlusion in cats: residual perfusion and efficacy of postischemic reperfusion. J Cereb Blood Flow Metab 17: 388–400. [DOI] [PubMed] [Google Scholar]
- 6. Olsen TS, Larsen B, Skriver EB, Herning M, Enevoldsen E, et al. (1981) Focal cerebral hyperemia in acute stroke. Incidence, pathophysiology and clinical significance. Stroke 12: 598–607. [DOI] [PubMed] [Google Scholar]
- 7. Tran Dinh YR, Ille O, Guichard JP, Haguenau M, Seylaz J (1997) Cerebral postischemic hyperperfusion assessed by Xenon-133 SPECT. J Nucl Med 38: 602–607. [PubMed] [Google Scholar]
- 8. Kidwell CS, Saver JL, Mattiello J, Starkman S, Vinuela F, et al. (2001) Diffusion-perfusion MRI characterization of post-recanalization hyperperfusion in humans. Neurology 57: 2015–2021. [DOI] [PubMed] [Google Scholar]
- 9. Heiss WD, Huber M, Fink GR, Herholz K, Pietrzyk U, et al. (1992) Progressive derangement of periinfarct viable tissue in ischemic stroke. J Cereb Blood Flow Metab 12: 193–203. [DOI] [PubMed] [Google Scholar]
- 10. Marchal G, Furlan M, Beaudouin V, Rioux P, Hauttement JL, et al. (1996) Early spontaneous hyperperfusion after stroke. A marker of favourable tissue outcome? Brain 119 (Pt 2) 409–419. [DOI] [PubMed] [Google Scholar]
- 11. Raynaud C, Rancurel G, Tzourio N, Soucy JP, Baron JC, et al. (1989) SPECT analysis of recent cerebral infarction. Stroke 20: 192–204. [DOI] [PubMed] [Google Scholar]
- 12. van Dorsten FA, Olah L, Schwindt W, Grune M, Uhlenkuken U, et al. (2002) Dynamic changes of ADC, perfusion, and NMR relaxation parameters in transient focal ischemia of rat brain. Magn Reson Med 47: 97–104. [DOI] [PubMed] [Google Scholar]
- 13. Lee SK, Kim DI, Jeong EK, Yoon PH, Cha SH, et al. (2002) Temporal changes in reversible cerebral ischemia on perfusion- and diffusion-weighted magnetic resonance imaging: the value of relative cerebral blood volume maps. Neuroradiology 44: 103–108. [DOI] [PubMed] [Google Scholar]
- 14. Li F, Silva MD, Sotak CH, Fisher M (2000) Temporal evolution of ischemic injury evaluated with diffusion-, perfusion-, and T2-weighted MRI. Neurology 54: 689–696. [DOI] [PubMed] [Google Scholar]
- 15. Lee SK, Kim DI, Kim SY, Kim DJ, Lee JE, et al. (2004) Reperfusion cellular injury in an animal model of transient ischemia. AJNR Am J Neuroradiol 25: 1342–1347. [PMC free article] [PubMed] [Google Scholar]
- 16. Wang L, Yushmanov VE, Liachenko SM, Tang P, Hamilton RL, et al. (2002) Late reversal of cerebral perfusion and water diffusion after transient focal ischemia in rats. J Cereb Blood Flow Metab 22: 253–261. [DOI] [PubMed] [Google Scholar]
- 17. Wegener S, Wong EC (2008) Longitudinal MRI studies in the isoflurane-anesthetized rat: long-term effects of a short hypoxic episode on regulation of cerebral blood flow as assessed by pulsed arterial spin labelling. NMR Biomed 21: 696–703. [DOI] [PubMed] [Google Scholar]
- 18. Koizumi J, Yoshida Y, Nakazawa T, Ohneda G (1986) Experimental studies of ischemic brain edema, I: a new experimental model of cerebral embolism in rats in which recirculation can be introduced in the ischemic area. Japanese Journal of Stroke 8: 1–8. [Google Scholar]
- 19. Garcia JH, Wagner S, Liu KF, Hu XJ (1995) Neurological deficit and extent of neuronal necrosis attributable to middle cerebral artery occlusion in rats. Statistical validation. Stroke 26: 627–634 discussion 635. [DOI] [PubMed] [Google Scholar]
- 20. Wegener S, Wu WC, Perthen JE, Wong EC (2007) Quantification of rodent cerebral blood flow (CBF) in normal and high flow states using pulsed arterial spin labeling magnetic resonance imaging. J Magn Reson Imaging 26: 855–862. [DOI] [PubMed] [Google Scholar]
- 21. Wong EC, Buxton RB, Frank LR (1998) Quantitative imaging of perfusion using a single subtraction (QUIPSS and QUIPSS II). Magn Reson Med 39: 702–708. [DOI] [PubMed] [Google Scholar]
- 22. Neumann-Haefelin T, du Mesnil de Rochemont R, Fiebach JB, Gass A, Nolte C, et al. (2004) Effect of incomplete (spontaneous and postthrombolytic) recanalization after middle cerebral artery occlusion: a magnetic resonance imaging study. Stroke 35: 109–114. [DOI] [PubMed] [Google Scholar]
- 23. Barber PA, Davis SM, Infeld B, Baird AE, Donnan GA, et al. (1998) Spontaneous reperfusion after ischemic stroke is associated with improved outcome. Stroke 29: 2522–2528. [DOI] [PubMed] [Google Scholar]
- 24. Kuroda S, Siesjo BK (1997) Reperfusion damage following focal ischemia: pathophysiology and therapeutic windows. Clin Neurosci 4: 199–212. [PubMed] [Google Scholar]
- 25. Aronowski J, Strong R, Grotta JC (1997) Reperfusion injury: demonstration of brain damage produced by reperfusion after transient focal ischemia in rats. J Cereb Blood Flow Metab 17: 1048–1056. [DOI] [PubMed] [Google Scholar]
- 26. Macfarlane R, Moskowitz MA, Sakas DE, Tasdemiroglu E, Wei EP, et al. (1991) The role of neuroeffector mechanisms in cerebral hyperperfusion syndromes. J Neurosurg 75: 845–855. [DOI] [PubMed] [Google Scholar]
- 27. Lee B, Clarke D, Al Ahmad A, Kahle M, Parham C, et al. (2011) Perlecan domain V is neuroprotective and proangiogenic following ischemic stroke in rodents. J Clin Invest 121: 3005–3023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Li L, Jiang Q, Zhang L, Ding G, Gang Zhang Z, et al. (2007) Angiogenesis and improved cerebral blood flow in the ischemic boundary area detected by MRI after administration of sildenafil to rats with embolic stroke. Brain Res 1132: 185–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Shen Q, Du F, Huang S, Duong TQ (2011) Spatiotemporal characteristics of postischemic hyperperfusion with respect to changes in T1, T2, diffusion, angiography, and blood-brain barrier permeability. J Cereb Blood Flow Metab [DOI] [PMC free article] [PubMed] [Google Scholar]