

Abstract
Non-transfusion-dependent thalassemias include a variety of phenotypes that, unlike patients with beta (β)-thalassemia major, do not require regular transfusion therapy for survival. The most commonly investigated forms are β-thalassemia intermedia, hemoglobin E/β-thalassemia, and α-thalassemia intermedia (hemoglobin H disease). However, transfusion-independence in such patients is not without side effects. Ineffective erythropoiesis and peripheral hemolysis, the hallmarks of disease process, lead to a variety of subsequent pathophysiologies including iron overload and hypercoagulability that ultimately lead to a number of serious clinical morbidities. Thus, prompt and accurate diagnosis of non-transfusion-dependent thalassemia is essential to ensure early intervention. Although several management options are currently available, the need to develop more novel therapeutics is justified by recent advances in our understanding of the mechanisms of disease. Such efforts require wide international collaboration, especially since non-transfusion-dependent thalassemias are no longer bound to low- and middle-income countries but have spread to large multiethnic cities in Europe and the Americas due to continued migration.
Introduction
Inherited hemoglobin disorders can be divided into two main groups. The first group includes structural hemoglobin variants, such as hemoglobin S, C, and E. The second group includes the alpha (α)- and beta (β)-thalassemias which result from the defective synthesis of the α- or β-globin chains of adult hemoglobin A. Inheritance of such disorders follows a typical Mendelian-recessive manner whereby asymptomatic heterozygous parents, or carriers, pass on one copy of a defective gene to their children. The high prevalence of hemoglobin mutations in particular parts of the world often leads to simultaneous inheritance of two different thalassemia mutations from each parent or co-inheritance of thalassemia together with structural hemoglobin variants. Thus there are a wide variety of clinically distinct thalassemia syndromes.1 Since the hallmark of disease in these syndromes is ineffective erythropoiesis, peripheral hemolysis, and subsequent anemia, transfusion-dependence has been an essential factor in characterizing the various thalassemia phenotypes and their severity. For instance, a diagnosis of β-thalassemia major entails lifelong regular transfusion requirement for survival. The main concern with transfusion-dependence is secondary iron overload, which if left untreated leads to target-organ toxicity and death.2 However, considerable advances have been made, in iron overload assessment and management strategies for transfusion-dependent patients, especially in the last decade, and these have translated into improved patient survival.2 Non-transfusion-dependent thalassemias (NTDT) is a term used to label patients who do not require lifelong regular transfusions for survival, although they may require occasional or even frequent transfusions in certain clinical settings and usually for defined periods of time (Figure 1). NTDT encompasses three clinically distinct forms: β-thalassemia intermedia, hemoglobin E/β-thalassemia (mild and moderate forms), and α-thalassemia intermedia (hemoglobin H disease).3 Although patients with hemoglobin S/β-thalassemia and hemoglobin C/β-thalassemia may have transfusion requirements similar to NTDT patients, these forms have other specific characteristics and management peculiarities and are better considered as separate entities. NTDT are primarily to be found in the low- or middle-income countries of the tropical belt stretching from sub-Saharan Africa, through the Mediterranean region and the Middle East, to South and Southeast Asia.3–4 This is primarily attributed to the high frequency of consanguineous marriages in these regions, as well as to a conferred resistance of carriers to severe forms of malaria in regions where the infection has been, or is still, prevalent.3–4 Improvements in public health standards in these regions have also helped to improve survival and the number of affected patients. Increasing incidences of these disorders in other areas of the world, such as North Europe and North America, previously relatively unaffected by these conditions, have also been reported.3–5
Figure 1.

Transfusion requirement in various thalassemia forms.
The aims of this review are 3-fold. First, to highlight those genetic and environmental factors that explain the milder disease form in NTDT compared with transfusion-dependent patients. Second, to overview prominent pathophysiological mechanisms, especially in the absence of transfusions, and illustrate how these translate into clinical morbidity. Third, to outline current knowledge on the role of available management options and summarize novel advances in therapeutic strategies. Curative therapy including bone marrow transplantation and gene therapy will not be covered as these have been recently reviewed elsewhere.6
Genetic and environmental modifiers of phenotype
β-thalassemia
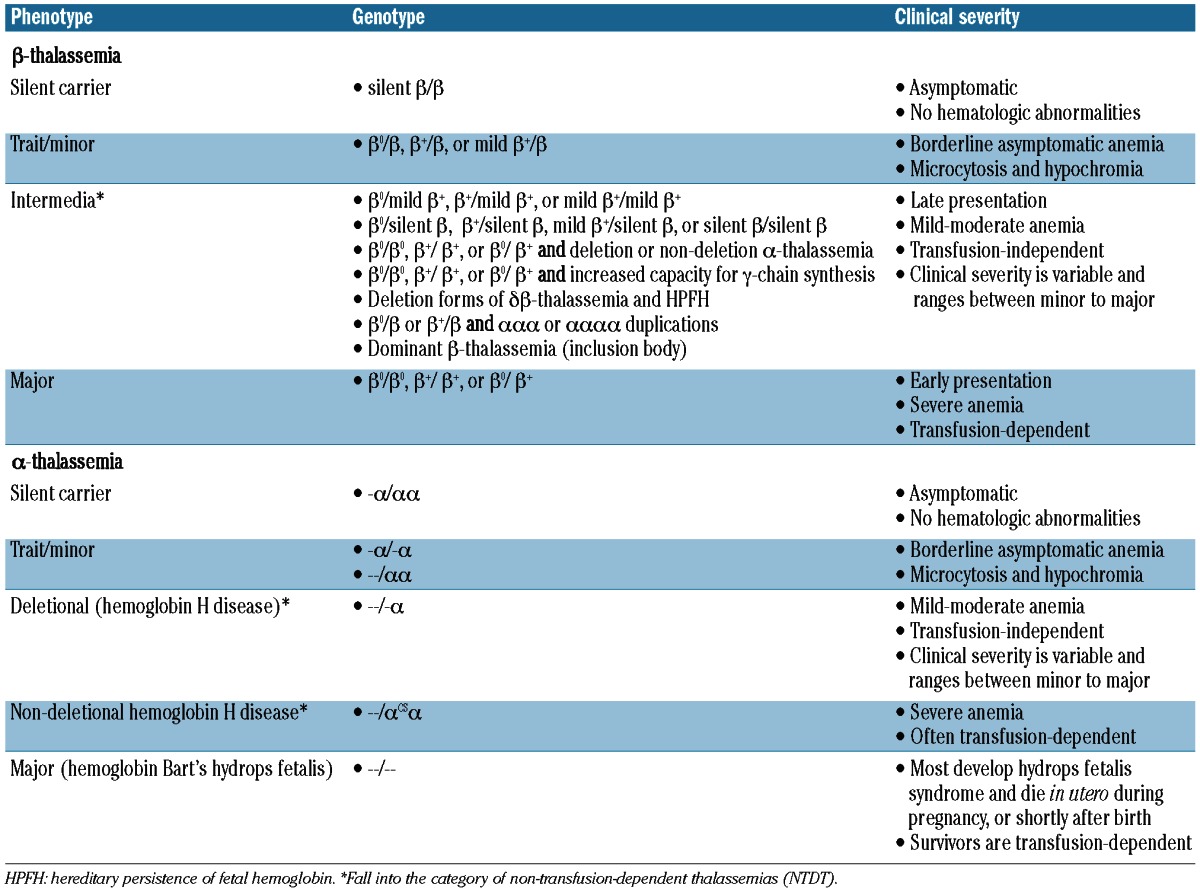
Distinction of the various phenotypes of thalassemia is mostly based on clinical parameters, although a genotype-phenotype association is established in both α- and β-thalassemia syndromes (Table 1). In patients with β-thalassemia intermedia, the primary modifier of phenotype is the broad diversity of mutations that affect the β-globin gene in the homozygous or compound heterozygous state (>200 disease-causing mutations. Up-dated list available from: http://globin.cse.psu.edu).7 These range from mild promoter mutations that cause a slight reduction in β-globin chain synthesis to the many different mutations that result in the β0-thalassemias; that is, a complete absence of β-globin. Deletions of the β-globin gene are uncommon. The diversity of mutations and the consequent variable degree of α/β-globin chain imbalance are the main determinants for milder anemia and phenotype. Secondary modifiers are those that are involved directly in modifying the degree of α/β-globin chain imbalance including co-inheritance of different molecular forms of α-thalassemia and effective synthesis of γ-chains in adult life.8 Several genes have been uncovered which could modify γ-chain production and ameliorate phenotype; some of them are encoded in the β-globin gene cluster (δβ0-thalassemia or point mutations at A-γ or G-γ promoters), while others are on different chromosomes (BCL11A, KLF1, HBS1L-MYB).7 Tertiary modifiers include polymorphisms that are not related to globin chain production but may have an ameliorating effect on specific complications of the disease such as iron absorption, bilirubin metabolism, bone metabolism, cardiovascular disease, and susceptibility to infection.9–10 β-thalassemia intermedia may also result from the increased production of α-globin chains by a triplicated or quadruplicated α-genotype associated with β-heterozygosity.11
Table 1.
Genotype-phenotype associations in β- and α-thalassemia.
Hemoglobin E/β-thalassemia
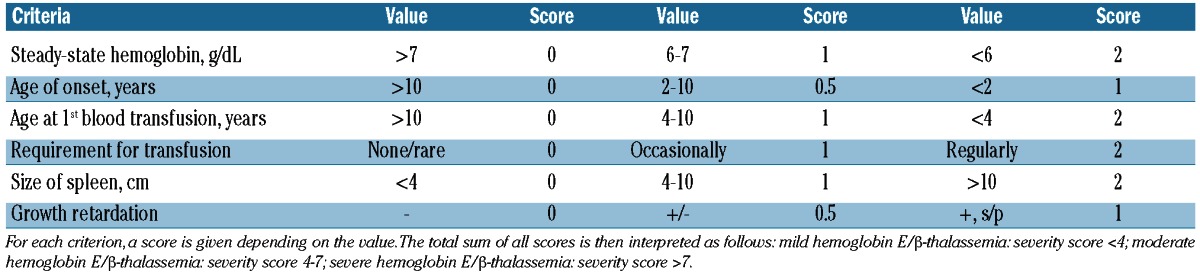
Hemoglobin E is caused by a G-to-A substitution in codon number 26 of the β-globin gene, which produces a structurally abnormal hemoglobin and an abnormally spliced non-functional mRNA. Hemoglobin E is synthesized at a reduced rate and behaves like a mild β-thalassemia. Patients with hemoglobin E/β-thalassemia co-inherit a β-thalassemia allele from one parent, and the structural variant, hemoglobin E, from the other.8,12 Hemoglobin E/β-thalassemia is further classified into severe (hemoglobin level as low as 4–5 g/dL, transfusion-dependent, clinical symptoms similar to β-thalassemia major), moderate (hemoglobin levels between 6 and 7 g/dL, transfusion-independent, clinical symptoms similar to β-thalassemia intermedia), and mild (hemoglobin levels between 9 and 12 g/dL, transfusion-independent, usually do not develop clinically significant problems) clinical forms; with the latter two falling into the category of NTDT.13–14 A disease scoring system that helps classify patients into mild, moderate, and severe has been proposed (Table 2).15 Similar to patients with β-thalassemia intermedia, modifiers of disease severity in hemoglobin E/β-thalassemia include the type of β-thalassemia mutation, co-inheritance of α-thalassemia and determinants that increase fetal hemoglobin production, as well as tertiary modifiers of complications such as the inherited variability in the function of the gene for UDP-glu-curonosyltransferase-1 underlying the more severe chronic hyperbilirubinemia and an increased occurrence of gallstones observed in some patients.13,14 It should be noted that patients with hemoglobin E/β-thalassemia also show different phenotypic severity at particular stages of development. Advancing age has an independent and direct effect on the background level of erythropoietin production in response to anemia.16–18 The most notable environmental factor influencing phenotype in patients with hemoglobin E/β-thalassemia is infection with malaria, particularly Plasmodium vivax.19
Table 2.
Mahidol score for hemoglobin E/β-thalassemia severity classification.15
α-thalassemia
Unlike β-thalassemia, deficient synthesis of α-globin chains in α-thalassemia is typically caused by deletions within the α-globin gene cluster on chromosome 16. Approximately 128 different molecular defects are known to cause α-thalassemia.20,21 There are many different sized deletions of the α-globin genes. Southeast Asian deletion (SEA) is the most common and involves both α-genes, but not embryonic globin genes. Larger deletions such as (-THAI) affect embryonic genes and may be more severe. The different phenotypes in α-thalassemia are primarily attributed to whether one (α+) or both (α0) α-globin genes are deleted in each of the two loci (Table 1). Hemoglobin Bart’s hydrops fetalis (α-thalassemia major) is caused by deletion of all four α-globin genes, and patients often die in utero. Deletion of three α-globin genes results in hemoglobin H disease (α-thalassemia intermedia).20,21 In addition to deletional forms, there are at least 7 forms of non-deletional hemoglobin H disease which are typically associated with a more severe phenotype, the most commonly described non-deletional hemoglobin H disease forms are hemoglobin H Constant Spring but also including hemoglobin H Paksé, Quong Sze, and Suan Dok.22–25 These deletional and non-deletional hemoglobin H diseases are the NTDT forms covered in this review. It should be noted that the most severe forms of non-deletional hemoglobin H disease may become completely transfusion-dependent (hemoglobin H hydrops) in which case they are considered similar to {β}-thalassemia major patients. Studies on the role of modifiers of disease severity in α-thalassemia are limited. Genetic modification may occur with co-inheritance of mutations in β-globin genes resulting in β-thalassemia, also referred to as hemoglobin H/β-thalassemia trait.22 In non-deletional hemoglobin H disease there may be a role for the α-hemoglobin stabilizing protein in ameliorating disease severity, although this warrants further study.8
Pathophysiology and clinical manifestations
In the absence of transfusion therapy, the plethora of underlying pathophysiological mechanisms emanating from ineffective erythropoiesis and peripheral hemolysis lead to a multitude of clinical complications in patients with NTDT (Figure 2). In cross-sectional surveys, these complications are often noted at a higher rate than in transfusion-dependent patients26 (Figure 3). This section describes key pathological processes and associated clinical complications in NTDT.
Figure 2.

Pathophysiological mechanisms and clinical complications in non-transfusion dependent thalassemias.
Figure 3.

Differences in common clinical complications profile between non-transfusion-dependent thalassemias compared to regularly transfused β-thalassemia major patients. The figure illustrates those complications that are often observed at a higher prevalence in one group over the other in available studies and clinical settings, although all mentioned complications can exist in both entities at varying rates.
Ineffective erythropoiesis and peripheral hemolysis
In NTDT, erythropoiesis is ineffective due to the imbalance in the production of α- and β-globin chains. Unstable globin chain tetramers precipitate and undergo oxidation into methemoglobin and hemichromes with eventual separation of heme from globin. The free iron released from heme disintegration in thalassemia erythroid cells eventually catalyzes the formation of reactive oxygen species which leads to oxidation of membrane proteins, structural membrane defects, and exposure of red-cell senescence antigens such as phosphatidylserine causing premature cell death within the bone marrow (ineffective erythropoiesis) or peripheral circulation (hemolysis).27,28 In states of ineffective erythropoiesis, erythroid precursors proliferate in great numbers, but a larger fraction fails to mature. Therefore, ineffective erythropoiesis is characterized by expansion, limited differentiation, and premature death of erythroid precursors.27,28 Expansion of the erythron in the bone marrow is not only associated with osteoporosis and bone deformities but is also associated with homing and proliferation of erythroid precursors in the spleen and liver (extramedullary hematopoiesis) leading to hepatosplenomegaly. The expansion of the erythron, observed both in stress erythropoiesis and ineffective erythropoiesis, suggests that the activity of factors that control steady-state erythropoiesis are enhanced to increase the production of erythroid cells. In fact, it has been shown that increased activation of Janus Kinase 2 (JAK2)/STAT5 pathway is associated with an excessive proliferation of mouse and human erythroid β-thalassemic progenitors.29
Ineffective erythropoiesis in NTDT patients also forces expansion of the hematopoietic tissue in areas other than the liver and spleen, mostly in the form of masses termed extramedullary hematopoietic pseudotumors (prevalence of approx. 20% compared to <1% in transfusion-dependent patients26,30). Almost all body sites may be involved including the lymph nodes, thymus, heart, breasts, prostate, broad ligaments, kidneys, adrenal glands, pleura, retroperitoneal tissue, skin, peripheral and cranial nerves, and the spinal canal. Paraspinal involvement (11–15% of cases) receives most attention due to the debilitating clinical consequences secondary to spinal cord compression.31
Morbidity in patients with NTDT is directly proportional to the severity of ineffective erythropoiesis and peripheral hemolysis32 as these remain the hallmark of subsequent pathophysiological mechanisms including iron overload and hypercoagulability (Figure 2).
Dysregulated iron homeostasis and clinical iron overload
In NTDT, ineffective erythropoiesis is the central process that leads to inappropriately low hepcidin levels and increased intestinal iron absorption.33–35 Proposed regulators of hepcidin production include growth differentiation factor-15, twisted gastrulation factor-1, hypoxia inducible transcription factors, and transmembrane protease serine-6 (TMPRSS6).36,37 However, more recently, growth differentiation factor-15 was shown to be not essential for systemic iron homeostasis in phlebotomized mice and humans.38–39 Therefore, additional erythropoietic factors are likely to regulate hepcidin expression in NTDT patients. Regardless of the signaling mechanism, the end result is suppression of hepcidin levels, increased intestinal iron absorption, and increased release of recycled iron from the reticuloendothelial system.27 This in turn leads to depletion of macrophage iron, relatively lower levels of serum ferritin, and preferential portal and hepatocyte iron loading (increased liver iron concentration),40 with subsequent release into the circulation of free iron species (labile plasma iron and non-transferrin bound iron with a consequent increase in intracellular labile iron pool) that can cause target-organ damage.41 By contrast, regularly transfused patients do not have low hepcidin levels, and iron is preferentially distributed to the reticuloendothelial system, thereby stimulating ferritin synthesis and its release into circulation, resulting in high serum ferritin levels. It should be noted that apart from this primary source of iron overload in NTDT patients, they can eventually accumulate iron from occasional or more frequent transfusions which may be indicated as illustrated in the management section.37
The accumulation of iron from intestinal absorption in NTDT patients is slower than that observed in transfusional siderosis and may reach 3–4 mg/day or as much as 1,000 mg/year.37 A mean annual increase in liver iron concentration of 0.38±0.49 mg Fe/g dry weight was observed in a recent trial including NTDT patients.42 Nonetheless, iron overload in NTDT patients is a cumulative process as evident from studies documenting positive correlations between iron overload indices and advancing age.25,41,43–45 Thus, a considerable proportion of NTDT patients eventually accumulate iron to liver iron concentration thresholds of clinical significance.25,40,42,46,47 An association between iron loading evident from longitudinal elevations in serum ferritin level and worsening of hepatic fibrosis in patients with β-thalassemia intermedia has been established.48 Several case reports and case series also suggest an association between iron overload and hepatocellular carcinoma in this patient population.49 Interestingly, cardiac siderosis and subsequent cardiac disease do not seem to be a major concern in NTDT, even in patients with considerably elevated liver iron concentration.50–53 In a recent cross-sectional study of 168 patients with β-thalassemia intermedia, higher liver iron concentration values on magnetic resonance imaging were associated with a significantly increased risk of developing thrombosis, pulmonary hypertension, hypothyroidism, hypogonadism, and osteoporosis.46 Levels of 5 mg Fe/g or over were associated with a considerable morbidity risk increase.54 An association between iron overload and renal tubular dysfunction, as evident from proteinuria, has also been recently reported in β-thalassemia intermedia patients.55
Hypercoagulability and vascular disease
A hypercoagulable state has been identified in children and adults with thalassemia and remains an active area of investigation, particularly in patients with β-thalassemia.56,57 Abnormalities of platelets and pathological red blood cells are believed to be the key factors causing hypercoagulability and subsequent thrombotic events, especially in splenectomized and transfusion-independent patients, making this pathophysiology highly relevant to patients with NTDT.58 Several other factors have also been identified and it is often a combination of these factors that leads to a hypercoagulable state resulting in clinical thrombosis (Figure 4).59–61
Figure 4.
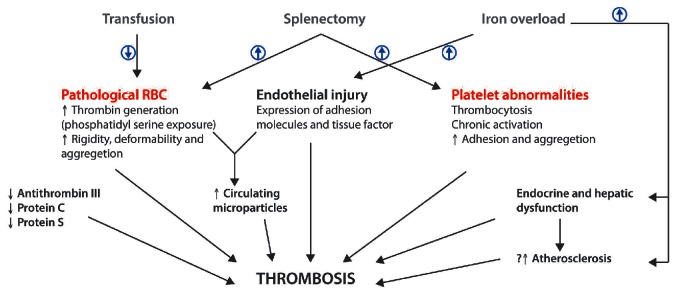
Factors contributing to a hypercoagulable state and subsequent thrombotic events in non-transfusion dependent thalassemias. RBC: red blood cells.
Clinically, the prevalence of thrombotic events in patients with β-thalassemia intermedia can reach up to 20%, compared to less than 1% in patients with β-thalassemia major.30,62–64 These events are mostly venous and primarily occur in splenectomized patients (22.5% vs. 3.5% prevalence rate in splenectomized vs. non-splenectomized patients; P<0.001).30,64 Other risk factors for thrombotic events include advancing age, total hemoglobin level less than 9 g/dL, history of thrombotic or other vascular events, elevated platelet (>500×109/L) and nucleated red blood cell counts (>300×106/L),30,43,64,65 while other conventional risk factors are often absent in such patients. The prevalence of overt strokes in β-thalassemia intermedia patients with a history of thrombosis ranges between 5% to 9%.66 However, a higher prevalence of silent cerebral ischemia (up to 60%) has been consistently documented, especially in splenectomized adults with elevated platelet counts.66 Such lesions were usually small (<0.5 cm), multiple, and involved the frontal and parietal lobes. Recent studies have also documented a high prevalence of large cerebral vessel disease (magnetic resonance angiography) and decreased neuronal function (positron emission tomography-computed tomography) primarily in the temporal and parietal lobes in similar patient cohorts.66–68 A significant association between the occurrence of these abnormalities and elevated iron overload indices was noted.67,68 In the general population, and in patients with sickle cell disease, these silent cerebrovascular abnormalities are associated with subsequent risk of overt stroke and neurocognitive decline, further highlighting the importance of these findings.66
Another vascular complication of NTDT (primarily β-thalassemia intermedia and hemoglobin E/β-thalassemia) that was found to occur at a relatively high frequency compared to patients with β-thalassemia major is pulmonary hypertension.69 However, the diagnosis of pulmonary hypertension in most available studies was performed by echocardiography and not cardiac catheterization and this may increase the rate of false positive results.70 Chronic anemia and hypoxia, iron overload, splenectomy, hypercoagulability, and microthrombotic disease of the pulmonary circulation have all been implicated in the pathophysiology of pulmonary hypertension in NTDT.71,72 Recently, decreased arginine bioavailability and nitric oxide depletion secondary to hemolysis have also been associated with pulmonary hypertension in patients with thalassemia.73–75 Although pulmonary hypertension is neither associated with myocardial siderosis nor with left ventricular dysfunction in NTDT patients, it is a leading cause of right-sided heart failure.69
The risk of leg ulcers in NTDT patients increases with advancing age.43 The skin at the extremities of elderly patients can be thin due to reduced tissue oxygenation making the subcutaneous tissue fragile and increasing the risk of ulceration after minimal trauma. Transfusion-naïvety, splenectomy and hypercoagulability, low fetal hemoglobin levels, and iron overload are all risk factors for the development of leg ulcers.30,46,76 Local iron overload is also thought to be a perpetuating factor causing chronicity of lesions especially when the heme from the degraded red blood cells accumulates locally and gives a dark hue.77
Management
The clinical morbidities observed in patients with NTDT may involve several organs and organ systems.71 Without appropriate treatment, the incidence of these morbidities increases with advancing age.43 Moreover, the multiplicity of morbidity in NTDT patients has a direct effect on patients’ quality of life.78–79 This observation highlights the importance of timely management and prevention in this patient population. There are currently no available guidelines for the management of patients with NTDT; however, emerging data from recent studies alongside expert opinion usually help put forward a management framework for this group of patients.
Splenectomy
Splenectomy in NTDT patients can increase the total hemoglobin level by 1–2 g/dL and avoid blood transfusion therapy.14,24 However, in view of the multiplicity of adverse events associated with splenectomy, it is suggested that splenectomy should be reserved for cases of:71,80 1) worsening anemia leading to poor growth and development when transfusion therapy is not possible or iron chelation therapy is unavailable; 2) hypersplensim leading to worsening anemia, leukopenia, or thrombocytopenia and resulting in recurrent bacterial infection or bleeding; and 3) splenomegaly accompanied by symptoms such as left upper quadrant pain or early satiety or massive splenomegaly (largest dimension >20 cm) with concern about possible splenic rupture.71,80
As described above, abnormalities of platelets and pathological red blood cells are believed to be the key factors causing a hypercoagulable state in patients with NTDT.58 These abnormalities become more prominent following splenectomy considering the beneficial role of the spleen in scavenging these procoagulant platelets and red blood cells. This puts this subgroup of patients at a higher risk of thrombotic and vascular events.60,81 For instance, about 80% of pathological red blood cells are removed extravascularly by macrophages present mainly in the spleen.82 Several clinical studies in patients with β-thalassemia intermedia confirm that splenectomized NTDT patients have a higher risk of venous thromboembolism (approx. 5-fold), pulmonary hypertension (approx. 4-fold), leg ulcers (approx. 4-fold), and silent cerebral infarction than non-splenectomized patients.24,30,66 The median time to thrombosis following splenectomy is approximately eight years.65 This delay indicates that thrombosis in splenectomized NTDT patients is not an acute complication, but a manifestation of a chronic underlying process, further emphasizing the need for long-term treatment modalities for prevention.
It has also been suggested that the spleen may be a reservoir of excess iron and may have a possible scavenging effect on iron free species such as non-transferrin bound iron, which may explain the higher serum level of this free iron species in splenectomized NTDT patients41 and the observation that splenectomized patients have a higher rate of iron-related organ morbidity than their non-splenectomized peers.30
Splenectomy also places NTDT patients of all ages at risk of morbidity and mortality due to infection. These infections could have an overwhelming, fatal course such as in meningitis and sepsis. Appropriate vaccinations and antibiotic prophylaxis are critical steps in preventing overwhelming infection after splenectomy.80
Transfusion therapy
Patients with NTDT may still require occasional blood transfusions during infection, pregnancy, surgery, or any setting with anticipated acute blood loss. They may also require more frequent, yet temporary, transfusions in the case of poor growth or development during childhood, or for the management of specific complications in adulthood, a setting in which the benefit of transfusion therapy has been established.24,83,84 Observational studies continue to confirm that NTDT patients who receive transfusions experience fewer leg ulcers, thrombotic events, pulmonary hypertension, and silent brain infarcts compared with transfusion-naïve patients.30,64,80,85,86 Successful management of the hematopoietic compensatory extramedullary pseudotumors has also been reported using transfusion therapy with and without radiation or surgery, especially in the most debilitating cases with paraspinal involvement.31
It is absolutely essential to assess the patient carefully over the first few months after the diagnosis is established and not to embark on any treatment modality, especially transfusion therapy, too hastily. Many patients with NTDT, who may not need regular transfusion, embark on a life of unnecessary treatment of this kind, particularly if they present with an unusually low hemoglobin level during a period of intercurrent infection. Even if a few transfusions have been administered in the acute situation, immediate commitment to a transfusion program is not recommended. It is worthwhile trying to evaluate the patient in the non-emergency situation from the untransfused baseline: i.e. to withdraw transfusions and observe the situation carefully. In fact, some children with NTDT, specifically with hemoglobin E/β-thalassemia, have a remarkable ability to adapt to low hemoglobin levels.16–17 Instead, the patient’s wellbeing, particularly with respect to activity, growth, development, and the early appearance of skeletal changes or other disease-complications are the factors to be taken into consideration.80
The main concern with transfusion therapy is the risk of iron overload, especially in NTDT patients who have already accumulated considerable amounts of iron due to increased intestinal absorption. The risk of alloimmunization should also be considered in minimally transfused and newly transfused patients, those at an old age at first transfusion, and in splenectomized patients. The risk of alloimunization is 1–1.6% after transfusion of one blood unit.87 This is of particular importance during pregnancy when fully phenotyped matched blood or transfusion alternatives should be considered.88
Iron chelation therapy
The same measures available for the assessment of iron overload in patients with transfusion-dependent β-thalassemia major can be used in NTDT. Whenever available, measurement of liver iron concentration by non-invasive means (R2 or R2* magnetic resonance imaging) is recommended.37 Considering the slow kinetics of iron loading in NTDT, there is no need to start assessment of liver iron concentration before patients reach ten years of age,36 particularly since there is a low prevalence of iron-related morbidities in patients under ten years of age.43 These can be measured at one- or even 2-year intervals; however, closer monitoring may be needed to tailor therapy in patients eligible for iron chelation.36 In resource-poor countries where measurement of liver iron concentration may not be available, serial measurements of serum ferritin level every three months are recommended. However, serum ferritin levels should be interpreted with caution.37 Although there is a positive correlation between serum ferritin level and liver iron concentration in NTDT patients,25,42,44,47 the ratio of serum ferritin to liver iron concentration is lower than in patients with β-thalassemia major.40,44,47,51 Therefore, spot measurements of serum ferritin level may underestimate iron overload and delay therapy in patients with NTDT if they are to be interpreted in the same way as β-thalassemia major patients. Although current evidence suggests that patients with NTDT are less likely to develop cardiac siderosis,50–53 cardiac magnetic resonance T2* assessment may still be warranted in older patients with high iron burden.37 Data on the use of other iron overload indices, such as transferrin saturation or non-transferrin bound iron, in NTDT patients are still limited; therefore, no recommendations regarding their use in clinical practice can be made at this time.37
Iron chelation therapy is indicated in NTDT patients aged ten years or over, (or fifteen years and over in deletional hemoglobin H disease) and having liver iron concentration levels 5 mg Fe/g or over dry weight (or serum ferritin level ≥800 ng/mL when liver iron concentration measurement is unavailable) as these thresholds indicate increased iron-related morbidity risk.36,43,54,89,90 Deferasirox is the only iron chelator to have been evaluated in a randomized clinical trial in patients with NTDT.42,91 The drug showed efficacy in reducing liver iron concentration in NTDT patients aged ten years or over and with a liver iron concentration of 5 mg Fe/g or over dry weight at starting doses of 5 or 10 mg/kg/day. It recently received approval by the US Food and Drug Administration (FDA) and the European Medicines Agency (EMA) for NTDT. The drug showed acceptable safety down to liver iron concentration values of 3 mg Fe/g dry weight (serum ferritin level of 300 ng/mL).
Available reports on the efficacy and safety of other iron chelators (deferoxamine and deferiprone) in reducing iron burden in patients with NTDT are limited to case reports and small case series, although benefits have been observed and warrant further consideration, especially in resource-poor countries.92–94 A beneficial effect of iron chelation in reducing clinical morbidity risk in patients with NTDT is suggested by observational studies and further long-term studies in this direction are needed.30,48
Fetal hemoglobin induction
As discussed above, increased production of γ-globin, which, similar to β-globin, combines with α-globin chain (fetal hemoglobin), results in improvement in α/β-globin chain imbalance and more effective erythropoiesis. This partly explains the more favorable phenotype in some patients with β-thalassemia intermedia and hemoglobin E/β-thalassemia compared with transfusion-dependent β-thalassemia major. Recent clinical studies have substantiated the quantitative ameliorating effect of increased fetal hemoglobin production on the clinical course in a variety of patients with NTDT.76,95–100 The earliest attempts to induce fetal hemoglobin through DNA methylation inhibition with 5-azacytidine were encouraging, although these were later hampered by concerns about the safety of this agent.101 More recently, this approach has been revisited with the use of a safer demethylating agent, decitabine. A pilot study on 5 patients with β-thalassemia intermedia showed that subcutaneous decitabine given at 0.2 mg/kg two times per week for 12 weeks increased total hemoglobin level by an average of 1 g/dL. Favorable changes in red blood cell indices were also noted and a call for larger studies was made.102 After being identified as a potent inducer of fetal hemoglobin, hydroxyurea became one of the key therapeutic agents for the management of patients with sickle cell disease. In studies including NTDT patients, mean increases in total hemoglobin level average approximately 1.5 g/dL, although results in these studies are highly variable.101 This increase remains essential since, for example, a difference between a severe and mild hemoglobin E/β-thalassemia patient is only 1–2 g/dL.15 Improvement in anemia is usually associated with better exercise tolerance, appetite, and sense of general wellbeing. Favorable effects on certain morbidities such as pulmonary hypertension, leg ulcer, and extramedullary hematopoietic pseudotumors have also been observed.101 However, available evidence on hydroxyurea comes from small single-arm trials or retrospective cohort studies, and it has been difficult to determine predictors of response or the optimal dose and duration of therapy.101 Randomized clinical trials with hydroxyurea in patients with NTDT are called for, especially given that the effects of hydroxyurea seem to extend beyond fetal hemoglobin induction and may improve the hypercoagulable state of the disease through effects on phosphatidylserine externalization in the red cell.103 Favorable responses to short-chain fatty acid (butyrate derivatives) inducers of fetal hemoglobin in small studies involving NTDT patients have also been documented, although effects were less notable in long-term therapy.101
The use of recombinant human erythropoietin or the newer erythropoietic stimulating agent darbepoetin alfa in patients with NTDT is associated with increases in total hemoglobin level.104 When such agents were combined with fetal hemoglobin inducers in NTDT patients, an additive effect on total hemoglobin augmentation was noted, although mostly at high doses.101 However, so far, such treatment options remain investigational and should be performed under carefully controlled trials.
Management of specific complications
The Online Supplementary Appendix highlights management options for specific clinical complications in NTDT patients.
Novel therapeutic approaches
In this section, we discuss the rationale and progress with several key strategies that are being or deserve to be developed to provide novel management options for patients with NTDT.
Modulators of erythropoiesis and iron metabolism in NTDT
Mouse models mimicking human NTDT (exhibiting non-transfusion dependent anemia, aberrant erythrocyte morphology, hepatosplenomegaly, and iron overload) have been generated and allowed several potential therapeutic targets to be evaluated. This is done by heterozygous deletion of both the β-minor and β-major or deletion of both β-major genes, respectively indicated as th3/+ and th1/th1.
JAK2 inhibitors
As indicated previously, JAK2 is a signaling molecule that regulates proliferation, differentiation, and survival of erythroid progenitors in response to erythropoietin. In murine models and patients with β-thalassemia, erythroid precursors express elevated levels of phosphorylated active JAK2 (pJAK2) and other downstream signaling molecules that promote proliferation and inhibit differentiation of erythroid progenitors. In th3/+ mice, erythroid hyperplasia and massive extramedullary hematopoiesis were associated with high erythropoietin levels and persistent JAK2 phosphorylation. Furthermore, early erythroid progenitors fail to differentiate, and hyperproliferate in the bone marrow, spleen and liver, thus contributing to hepatosplenomegaly.27,29 Given the role of JAK2 in the pathophysiology of ineffective erythropoiesis, it has been hypothesized that JAK2 inhibitors (JAK2is) may be effective in preventing the severe complications associated with β-thalassemia.105 Studies in th3/+ mice have shown that a short treatment with a JAK2i can affect ineffective erythropoiesis and decrease spleen size. In particular, in untransfused th3/+ mice, a JAK2i reduces both ineffective erythropoiesis (fewer bone marrow erythroid progenitors) and splenomegaly with minimal effect on red blood cell synthesis.29,106,107
If data from th3/+ mice can be translated to humans, JAK2i may allow NTDT patients to avoid splenectomy, thereby preventing additional pathophysiological sequelae, such as increased iron absorption, potential infections and thrombosis. Results from clinical studies in patients with myeloproliferative disorders, characterized by activating JAK2 mutations, suggest that the JAK2i ruxolitinib is an effective treatment option with a tolerable safety profile.108 Concerns were raised in patients treated chronically with ruxolitinib for more than eight months.108 However, NTDT differ considerably from JAK2-related neoplasms in that the activity of JAK2 is mediated by relatively high erythropoietin levels (and not a mutation in JAK2) and the progression of splenomegaly and extramedullary hematopoiesis occur more slowly in NTDT than in polycythemia vera/myelofibrosis patients. Thus, it is possible that the beneficial effects of JAK2is in NTDT will be achieved with reduced doses, shorter intermittent courses, and relatively fewer complications.105
Hepcidin modulation
Because of hepcidin deficiency, NTDT patients develop iron overload in a manner similar to hereditary hemochromatosis. Thus, hepcidin therapy curbing hyperabsorption of dietary iron may be beneficial in these patients. Furthermore, high hepcidin levels may lead to redistribution of iron from parenchyma to macrophages that could potentially limit target-organ toxicity. It is also possible that in NTDT patients, excessive iron supply to developing erythrocytes may contribute to the hyperproliferation of erythroid precursors and stimulate ineffective and extramedullary erythropoiesis. Finally, it might also decrease heme synthesis, limiting the formation of hemichromes. Therefore, administration of hepcidin could not only help manage iron loading but could also diminish the severity of erythroid pathologies.
Since in th3/+ mice endogenous hepcidin is inappropriately low and the iron absorbed by th3/+ mice is excessive in relation to the amount of iron needed to maintain their hemoglobin levels, it has been hypothesized that moderate hepcidin supplementation could limit iron absorption without interfering with the need of iron for erythropoiesis.34 To test this hypothesis, th3/+ mice that over-express hepcidin in the liver were generated.109 In fact, moderate hepcidin overexpression in th3/+ mice reduced iron content in the liver and spleen. Furthermore, these mice exhibited higher hemoglobin levels, decreased reticulocyte counts, reduction in splenomegaly and liver extramedullary hematopoiesis, and more normal spleen architecture.109 Due to the important role of TMPRSS6 in suppressing hepcidin, studies have evaluated whether the increased expression of hepcidin observed in Tmprss6−/− mice could be beneficial in animals affected by β-thalassemia. Lack of Tmprss6 in mice affected by β-thalassemia significantly improved iron overload and anemia in Tmprss6−/− th3/+ mice.110 Overall, these observations suggest that development of new pharmacological agents to increase hepcidin expression could be extremely valuable in the treatment of NTDT.111 Hepcidin agonists (minihepcidins) have been recently developed and showed benefit in mouse models with severe hemochromatosis.112–113
Apo-transferrin
Iron is transported between sites of acquisition, storage, and utilization by transferrin. The main role of this liver-synthesized molecule is to deliver iron to cells by receptor-mediated endocytosis. Low hepcidin expression causes excess circulatory iron, saturation of transferrin, and accumulation of toxic non-transferrin bound iron. Likewise, hypotransferrinemia, an inherited defect in transferrin expression, is associated with increased plasma non-transferrin bound iron and low hepcidin expression.34 Transferrin circulates in three forms: diferric-transferrin, monoferric-transferrin, and apo-transferrin, depending on the iron available. In th1/th1 mice, daily apo-transferrin injections resulted in increased hemoglobin, reduced reticulocytosis, smaller red blood cells with lower mean corpuscular hemoglobin, normalized red blood cell survival (likely as a consequence of reduced hemichromes precipitation on red blood cell membranes), decreased erythropoietin, improved maturation and decreased apoptosis of erythroid precursors, reversed splenomegaly, improved extramedullary hematopoiesis, and increased hepcidin expression.114
These observations demonstrate that apo-transferrin therapy is useful not only in the management of iron overload, but could also diminish the severity of ineffective erythropoiesis and anemia and indicate the importance of transferrin as a regulator of liver hepcidin expression. Taken together, apo-transferrin therapy would simultaneously reduce circulating non-transferrin bound iron and aberrant parenchymal iron deposition, improve anemia and ineffective erythropoiesis, and reduce further iron overload. If administration of apo-transferrin proves to be relatively free of side effects, it could provide an additional major improvement over existing therapy in patients with NTDT.
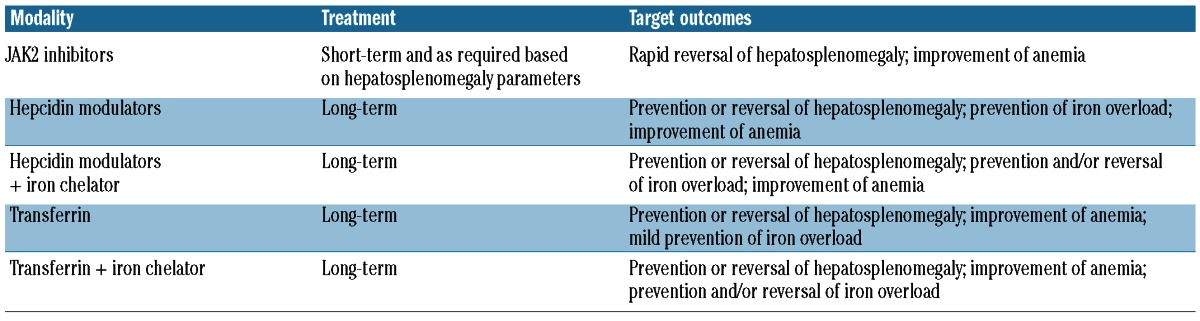
Table 3 illustrates how such novel therapies may potentially be used in the management of NTDT patients. These new approaches, especially hepcidin modulators and transferrin, may ameliorate ineffective erythropoiesis and prevent iron overload in the early stage of the disease. However, they might also be beneficial if used in later stages of the disease (after iron overload is already established) in combination with iron chelators to slow down the iron absorption while iron is being removed and hence to accelerate the detoxification.
Table 3.
Potential new treatment modalities for ineffective erythropoiesis and iron overload in non-transfusion dependent patients.
Targeted fetal hemoglobin induction
Although several molecular determinants of fetal hemoglobin expression have been previously identified, only recently were the details of the developmental switching of fetal to adult hemoglobin uncovered. Importantly, recent molecular investigations of this process have provided promising targets for therapeutic purposes to induce fetal hemoglobin. Such molecules include BCL11A, MYB, and KLF1 that have largely been identified as a result of human genetic studies.115 Correction of sickle cell disease in adult mice by interference with fetal hemoglobin silencing (inactivation of BCL11A) has been recently documented.116 Moreover, epigenetic partners of these factors have been identified for which small molecule inhibitors already exist. The clinical development of therapeutics for these targets could be the future path for NTDT and other hemoglobinopathies.101,115
Conclusions
It has now been established that morbidity in NTDT patients is more common and serious than previously recognized, and these patients should be carefully followed for early diagnosis and management of complications. This is essential given that the prevalence of such thalassemia forms is shifting towards a global distribution that could have serious implications for public health. It should be noted, however, that several of these complications still lack sufficient data to allow us to recommend specific guidelines for treatment. Therefore, recommendations about specific treatments should be made separately for each patient on an individualized basis until more data are available. Moreover, better understanding of the underlying pathophysiological mechanisms should allow novel therapeutics to be developed in clinical trial programs.
Footnotes
The online version of this article has a Supplementary Appendix.
Authorship and Disclosures
Information on authorship, contributions, and financial & other disclosures was provided by the authors and is available with the online version of this article at www.haematologica.org.
References
- 1.Weatherall DJ, Clegg JB. The thalassaemia syndromes. 4th ed Oxford: Blackwell Science; 2001 [Google Scholar]
- 2.Rachmilewitz EA, Giardina PJ. How I treat thalassemia. Blood. 2011;118(13):3479–88 [DOI] [PubMed] [Google Scholar]
- 3.Weatherall DJ. The definition and epidemiology of non-transfusion-dependent thalassemia. Blood Rev. 2012;(26 Suppl 1):S3–6 [DOI] [PubMed] [Google Scholar]
- 4.Weatherall DJ. The inherited diseases of hemoglobin are an emerging global health burden. Blood. 2010;115(22):4331–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Michlitsch J, Azimi M, Hoppe C, Walters MC, Lubin B, Lorey F, et al. Newborn screening for hemoglobinopathies in California. Pediatr Blood Cancer. 2009;52(4): 486–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Higgs DR, Engel JD, Stamatoyannopoulos G. Thalassaemia. Lancet. 2012;379(9813): 373–83 [DOI] [PubMed] [Google Scholar]
- 7.Danjou F, Anni F, Galanello R. Beta-thalassemia: from genotype to phenotype. Haematologica. 2011;96(11):1573–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Galanello R. Recent advances in the molecular understanding of non-transfusion-dependent thalassemia. Blood Rev. 2012;26 (Suppl 1):S7–S11 [DOI] [PubMed] [Google Scholar]
- 9.Weatherall D. 2003 William Allan Award address. The Thalassemias: the role of molecular genetics in an evolving global health problem. Am J Hum Genet. 2004; 74(3):385–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Galanello R, Piras S, Barella S, Leoni GB, Cipollina MD, Perseu L, et al. Cholelithiasis and Gilbert’s syndrome in homozygous beta-thalassaemia. Br J Haematol. 2001;115 (4):926–8 [DOI] [PubMed] [Google Scholar]
- 11.Graziadei G, Refaldi C, Barcellini W, Cesaretti C, Cassinero E, Musallam KM, et al. Does absolute excess of alpha chains compromise the benefit of splenectomy in patients with thalassemia intermedia?. Haematologica. 2012;97(1):151–3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fucharoen S, Weatherall DJ. The hemoglobin e thalassemias. Cold Spring Harb Perspect Med. 2012;2(8) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Olivieri NF, Pakbaz Z, Vichinsky E. HbE/beta-thalassemia: basis of marked clinical diversity. Hematol Oncol Clin North Am. 2010;24(6):1055–70 [DOI] [PubMed] [Google Scholar]
- 14.Olivieri NF, Muraca GM, O’Donnell A, Premawardhena A, Fisher C, Weatherall DJ. Studies in haemoglobin E beta-thalassaemia. Br J Haematol. 2008;141(3):388–97 [DOI] [PubMed] [Google Scholar]
- 15.Sripichai O, Makarasara W, Munkongdee T, Kumkhaek C, Nuchprayoon I, Chuansumrit A, et al. A scoring system for the classification of beta-thalassemia/Hb E disease severity. Am J Hematol. 2008;83(6):482–4 [DOI] [PubMed] [Google Scholar]
- 16.O’Donnell A, Premawardhena A, Arambepola M, Allen SJ, Peto TE, Fisher CA, et al. Age-related changes in adaptation to severe anemia in childhood in developing countries. Proc Natl Acad Sci USA. 2007;104(22):9440–4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Allen A, Fisher C, Premawardhena A, Peto T, Allen S, Arambepola M, et al. Adaptation to anemia in hemoglobin E-ss thalassemia. Blood. 2010;116(24):5368–70 [DOI] [PubMed] [Google Scholar]
- 18.Premawardhena A, Fisher CA, Olivieri NF, de Silva S, Arambepola M, Perera W, et al. Haemoglobin E beta thalassaemia in Sri Lanka. Lancet. 2005;366(9495):1467–70 [DOI] [PubMed] [Google Scholar]
- 19.O’Donnell A, Premawardhena A, Arambepola M, Samaranayake R, Allen SJ, Peto TE, et al. Interaction of malaria with a common form of severe thalassemia in an Asian population. Proc Natl Acad Sci USA. 2009;106(44):18716–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Harteveld CL, Higgs DR. Alpha-thalassaemia. Orphanet J Rare Dis. 2010;5:13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Higgs DR, Weatherall DJ. The alpha thalassaemias. Cell Mol Life Sci. 2009;66(7):1154–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vichinsky E. Complexity of alpha thalassemia: growing health problem with new approaches to screening, diagnosis, and therapy. Ann NY Acad Sci. 2010;1202:180–7 [DOI] [PubMed] [Google Scholar]
- 23.Singer ST, Kim HY, Olivieri NF, Kwiatkowski JL, Coates TD, Carson S, et al. Hemoglobin H-constant spring in North America: an alpha thalassemia with frequent complications. Am J Hematol. 2009; 84(11):759–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vichinsky E. Advances in the treatment of alpha-thalassemia. Blood Rev. 2012;26 (Suppl 1):S31–4 [DOI] [PubMed] [Google Scholar]
- 25.Lal A, Goldrich ML, Haines DA, Azimi M, Singer ST, Vichinsky EP. Heterogeneity of hemoglobin H disease in childhood. N Engl J Med. 2011;364(8):710–8 [DOI] [PubMed] [Google Scholar]
- 26.Taher A, Isma’eel H, Cappellini MD. Thalassemia intermedia: revisited. Blood Cells Mol Dis. 2006;37(1):12–20 [DOI] [PubMed] [Google Scholar]
- 27.Rivella S. The role of ineffective erythropoiesis in non-transfusion-dependent thalassemia. Blood Rev. 2012;26 (1 Suppl):S12–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rivella S. Ineffective erythropoiesis and thalassemias. Curr Opin Hematol. 2009;16(3): 187–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Melchiori L, Gardenghi S, Rivella S. beta-Thalassemia: HiJAKing Ineffective Erythropoiesis and Iron Overload. Adv Hematol. 2010;2010:938640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Taher AT, Musallam KM, Karimi M, El-Beshlawy A, Belhoul K, Daar S, et al. Overview on practices in thalassemia intermedia management aiming for lowering complication rates across a region of endemicity: the OPTIMAL CARE study. Blood. 2010;115(10):1886–92 [DOI] [PubMed] [Google Scholar]
- 31.Haidar R, Mhaidli H, Taher AT. Paraspinal extramedullary hematopoiesis in patients with thalassemia intermedia. Eur Spine J. 2010;19(6):871–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Musallam KM, Taher AT, Duca L, Cesaretti C, Halawi R, Cappellini MD. Levels of growth differentiation factor-15 are high and correlate with clinical severity in transfusion-independent patients with beta thalassemia intermedia. Blood Cells Mol Dis. 2011;47(4):232–4 [DOI] [PubMed] [Google Scholar]
- 33.Gardenghi S, Marongiu MF, Ramos P, Guy E, Breda L, Chadburn A, et al. Ineffective erythropoiesis in beta-thalassemia is characterized by increased iron absorption mediated by down-regulation of hepcidin and up-regulation of ferroportin. Blood. 2007;109(11): 5027–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ginzburg Y, Rivella S. beta-thalassemia: a model for elucidating the dynamic regulation of ineffective erythropoiesis and iron metabolism. Blood. 2011;118(16):4321–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rivella S, Nemeth E, Miller JL. Crosstalk between Erythropoiesis and Iron Metabolism. Adv Hematol. 2010;2010. pii: 317095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Musallam KM, Cappellini MD, Taher AT. Iron overload in beta-thalassemia intermedia: an emerging concern. Curr Opin Hematol. 2013. February 18 [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 37.Musallam KM, Cappellini MD, Wood JC, Taher AT. Iron overload in non-transfusion-dependent thalassemia: a clinical perspective. Blood Rev. 2012;26 (Suppl 1):S16–9 [DOI] [PubMed] [Google Scholar]
- 38.Casanovas G, Vujic Spasic M, Casu C, Rivella S, Strelau J, Unsicker K, et al. The murine growth differentiation factor 15 is not essential for systemic iron homeostasis in phlebotomized mice. Haematologica. 2013;98(3):444–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tanno T, Rabel A, Lee YT, Yau YY, Leitman SF, Miller JL. Expression of growth differentiation factor 15 is not elevated in individuals with iron deficiency secondary to volunteer blood donation. Transfusion. 2010;50(7): 1532–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Origa R, Galanello R, Ganz T, Giagu N, Maccioni L, Faa G, et al. Liver iron concentrations and urinary hepcidin in beta-thalassemia. Haematologica. 2007;92(5):583–8 [DOI] [PubMed] [Google Scholar]
- 41.Taher A, Musallam KM, El Rassi F, Duca L, Inati A, Koussa S, et al. Levels of non-trans-ferrin-bound iron as an index of iron overload in patients with thalassaemia intermedia. Br J Haematol. 2009;146(5):569–72 [DOI] [PubMed] [Google Scholar]
- 42.Taher AT, Porter J, Viprakasit V, Kattamis A, Chuncharunee S, Sutcharitchan P, et al. Deferasirox reduces iron overload significantly in nontransfusion-dependent thalassemia: 1-year results from a prospective, randomized, double-blind, placebo-controlled study. Blood. 2012;120(5):970–7 [DOI] [PubMed] [Google Scholar]
- 43.Taher AT, Musallam KM, El-Beshlawy A, Karimi M, Daar S, Belhoul K, et al. Age-related complications in treatment-naive patients with thalassaemia intermedia. Br J Haematol. 2010;150(4):486–9 [DOI] [PubMed] [Google Scholar]
- 44.Taher A, El Rassi F, Isma’eel H, Koussa S, Inati A, Cappellini MD. Correlation of liver iron concentration determined by R2 magnetic resonance imaging with serum ferritin in patients with thalassemia intermedia. Haematologica. 2008;93(10):1584–6 [DOI] [PubMed] [Google Scholar]
- 45.Chen FE, Ooi C, Ha SY, Cheung BM, Todd D, Liang R, et al. Genetic and clinical features of hemoglobin H disease in Chinese patients. N Engl J Med. 2000;343(8):544–50 [DOI] [PubMed] [Google Scholar]
- 46.Musallam KM, Cappellini MD, Wood JC, Motta I, Graziadei G, Tamim H, et al. Elevated liver iron concentration is a marker of increased morbidity in patients with beta thalassemia intermedia. Haematologica. 2011;96(11):1605–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pakbaz Z, Fischer R, Fung E, Nielsen P, Harmatz P, Vichinsky E. Serum ferritin underestimates liver iron concentration in transfusion independent thalassemia patients as compared to regularly transfused thalassemia and sickle cell patients. Pediatr Blood Cancer. 2007;49(3):329–32 [DOI] [PubMed] [Google Scholar]
- 48.Musallam KM, Motta I, Salvatori M, Fraquelli M, Marcon A, Taher AT, et al. Longitudinal changes in serum ferritin levels correlate with measures of hepatic stiffness in transfusion-independent patients with beta-thalassemia intermedia. Blood Cells Mol Dis. 2012;49(3–4):136–9 [DOI] [PubMed] [Google Scholar]
- 49.Maakaron JE, Cappellini MD, Graziadei G, Ayache JB, Taher AT. Hepatocellular carcinoma in hepatitis-negative patients with thalassemia intermedia: a closer look at the role of siderosis. Ann Hepatol. 2013;12(1):142–6 [PubMed] [Google Scholar]
- 50.Origa R, Barella S, Argiolas GM, Bina P, Agus A, Galanello R. No evidence of cardiac iron in 20 never- or minimally-transfused patients with thalassemia intermedia. Haematologica. 2008;93(7):1095–6 [DOI] [PubMed] [Google Scholar]
- 51.Taher AT, Musallam KM, Wood JC, Cappellini MD. Magnetic resonance evaluation of hepatic and myocardial iron deposition in transfusion-independent thalassemia intermedia compared to regularly transfused thalassemia major patients. Am J Hematol. 2010;85(4):288–90 [DOI] [PubMed] [Google Scholar]
- 52.Roghi A, Cappellini MD, Wood JC, Musallam KM, Patrizia P, Fasulo MR, et al. Absence of cardiac siderosis despite hepatic iron overload in Italian patients with thalassemia intermedia: an MRI T2* study. Ann Hematol. 2010;89(6):585–9 [DOI] [PubMed] [Google Scholar]
- 53.Mavrogeni S, Gotsis E, Ladis V, Berdousis E, Verganelakis D, Toulas P, et al. Magnetic resonance evaluation of liver and myocardial iron deposition in thalassemia intermedia and b-thalassemia major. Int J Cardiovasc Imaging. 2008;24(8):849–54 [DOI] [PubMed] [Google Scholar]
- 54.Musallam KM, Cappellini MD, Taher AT. Evaluation of the 5mg/g liver iron concentration threshold and its association with morbidity in patients with beta-thalassemia intermedia. Blood Cells Mol Dis. 2013;51 (1):35–8 [DOI] [PubMed] [Google Scholar]
- 55.Ziyadeh FN, Musallam KM, Mallat NS, Mallat S, Jaber F, Mohamed AA, et al. Glomerular Hyperfiltration and Proteinuria in Transfusion-Independent Patients with beta-Thalassemia Intermedia. Nephron Clin Pract. 2012;121(3–4):c136–c43 [DOI] [PubMed] [Google Scholar]
- 56.Eldor A, Durst R, Hy-Am E, Goldfarb A, Gillis S, Rachmilewitz EA, et al. A chronic hypercoagulable state in patients with beta-thalassaemia major is already present in childhood. Br J Haematol. 1999;107(4):739–46 [DOI] [PubMed] [Google Scholar]
- 57.Eldor A, Rachmilewitz EA. The hypercoagulable state in thalassemia. Blood. 2002;99(1):36–43 [DOI] [PubMed] [Google Scholar]
- 58.Cappellini MD, Musallam KM, Poggiali E, Taher AT. Hypercoagulability in non-transfusion-dependent thalassemia. Blood Rev. 2012;26 (1 Suppl):S20–3 [DOI] [PubMed] [Google Scholar]
- 59.Ataga KI, Cappellini MD, Rachmilewitz EA. Beta-thalassaemia and sickle cell anaemia as paradigms of hypercoagulability. Br J Haematol. 2007;139(1):3–13 [DOI] [PubMed] [Google Scholar]
- 60.Musallam KM, Taher AT. Thrombosis in thalassemia: why are we so concerned?. Hemoglobin. 2011;35(5–6):503–10 [DOI] [PubMed] [Google Scholar]
- 61.Cappellini MD, Motta I, Musallam KM, Taher AT. Redefining thalassemia as a hypercoagulable state. Ann NY Acad Sci. 2010;1202:231–6 [DOI] [PubMed] [Google Scholar]
- 62.Borgna Pignatti C, Carnelli V, Caruso V, Dore F, De Mattia D, Di Palma A, et al. Thromboembolic events in beta thalassemia major: an Italian multicenter study. Acta Haematol. 1998;99(2):76–9 [DOI] [PubMed] [Google Scholar]
- 63.Cappellini MD, Robbiolo L, Bottasso BM, Coppola R, Fiorelli G, Mannucci AP. Venous thromboembolism and hypercoagulability in splenectomized patients with thalassaemia intermedia. Br J Haematol. 2000;111 (2):467–73 [DOI] [PubMed] [Google Scholar]
- 64.Taher A, Isma’eel H, Mehio G, Bignamini D, Kattamis A, Rachmilewitz EA, et al. Prevalence of thromboembolic events among 8,860 patients with thalassaemia major and intermedia in the Mediterranean area and Iran. Thromb Haemost. 2006;96 (4):488–91 [PubMed] [Google Scholar]
- 65.Taher AT, Musallam KM, Karimi M, El-Beshlawy A, Belhoul K, Daar S, et al. Splenectomy and thrombosis: the case of thalassemia intermedia. J Thromb Haemost. 2010;8(10):2152–8 [DOI] [PubMed] [Google Scholar]
- 66.Musallam KM, Taher AT, Karimi M, Rachmilewitz EA. Cerebral infarction in beta-thalassemia intermedia: Breaking the silence. Thromb Res. 2012;130(5):695–702 [DOI] [PubMed] [Google Scholar]
- 67.Musallam KM, Nasreddine W, Beydoun A, Hourani R, Hankir A, Koussa S, et al. Brain positron emission tomography in splenectomized adults with beta-thalassemia intermedia: uncovering yet another covert abnormality. Ann Hematol. 2012;91(2):235–41 [DOI] [PubMed] [Google Scholar]
- 68.Musallam KM, Beydoun A, Hourani R, Nasreddine W, Raad R, Koussa S, et al. Brain magnetic resonance angiography in splenectomized adults with beta-thalassemia intermedia. Eur J Haematol. 2011;87(6):539–46 [DOI] [PubMed] [Google Scholar]
- 69.Farmakis D, Aessopos A. Pulmonary hypertension associated with hemoglobinopathies: prevalent but overlooked. Circulation. 2011;123(11):1227–32 [DOI] [PubMed] [Google Scholar]
- 70.Parent F, Bachir D, Inamo J, Lionnet F, Driss F, Loko G, et al. A hemodynamic study of pulmonary hypertension in sickle cell disease. N Engl J Med. 2011;365(1):44–53 [DOI] [PubMed] [Google Scholar]
- 71.Musallam KM, Taher AT, Rachmilewitz EA. beta-Thalassemia Intermedia: A Clinical Perspective. Cold Spring Harb Perspect Med. 2012;2(7):a013482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Morris CR, Vichinsky EP. Pulmonary hypertension in thalassemia. Ann NY Acad Sci. 2010;1202:205–13 [DOI] [PubMed] [Google Scholar]
- 73.Morris CR, Kim H-Y, Wood JC, Trachtenberg F, Klings ES, Porter JB, et al. Sildenafil therapy in patients with thalassemia and an elevated tricuspid regurgitant jet velocity (TRV) on doppler echocardiography at risk for pulmonary hypertension: report from the thalassemia clinical research network. Blood. 2012;120 (21):1023 [Google Scholar]
- 74.Morris CR, Kim H-Y, Wood JC, Trachtenberg F, Klings ES, Porter JB, et al. Cardiopulmonary and laboratory profiling of patients with thalassemia at risk for pulmonary hypertension: report from the thalassemia clinical research network. Blood. 2012;120(21):2122 [Google Scholar]
- 75.Morris CR, Kuypers FA, Kato GJ, Lavrisha L, Larkin S, Singer T, et al. Hemolysis-associated pulmonary hypertension in thalassemia. Ann NY Acad Sci. 2005;1054:481–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Musallam KM, Sankaran VG, Cappellini MD, Duca L, Nathan DG, Taher AT. Fetal hemoglobin levels and morbidity in untransfused patients with beta-thalassemia intermedia. Blood. 2012;119(2):364–7 [DOI] [PubMed] [Google Scholar]
- 77.Levin C, Koren A. Healing of refractory leg ulcer in a patient with thalassemia intermedia and hypercoagulability after 14 years of unresponsive therapy. Isr Med Assoc J. 2011;13(5):316–8 [PubMed] [Google Scholar]
- 78.Musallam KM, Khoury B, Abi-Habib R, Bazzi L, Succar J, Halawi R, et al. Health-related quality of life in adults with transfusion-independent thalassaemia intermedia compared to regularly transfused thalassaemia major: new insights. Eur J Haematol. 2011;87(1):73–9 [DOI] [PubMed] [Google Scholar]
- 79.Pakbaz Z, Treadwell M, Yamashita R, Quirolo K, Foote D, Quill L, et al. Quality of life in patients with thalassemia intermedia compared to thalassemia major. Ann N Y Acad Sci. 2005;1054:457–61 [DOI] [PubMed] [Google Scholar]
- 80.Taher AT, Musallam KM, Cappellini MD, Weatherall DJ. Optimal management of beta thalassaemia intermedia. Br J Haematol. 2011;152(5):512–23 [DOI] [PubMed] [Google Scholar]
- 81.Tso SC, Chan TK, Todd D. Venous thrombosis in haemoglobin H disease after splenectomy. Aust NZ J Med. 1982;12(6): 635–8 [DOI] [PubMed] [Google Scholar]
- 82.Mannu F, Arese P, Cappellini MD, Fiorelli G, Cappadoro M, Giribaldi G, et al. Role of hemichrome binding to erythrocyte membrane in the generation of band-3 alterations in beta-thalassemia intermedia erythrocytes. Blood. 1995;86(5):2014–20 [PubMed] [Google Scholar]
- 83.Taher AT, Musallam KM, Karimi M, Cappellini MD. Contemporary approaches to treatment of beta-thalassemia intermedia. Blood Rev. 2012;26 (Suppl 1):S24–7 [DOI] [PubMed] [Google Scholar]
- 84.Olivieri NF. Treatment strategies for hemoglobin E beta-thalassemia. Blood Rev. 2012;26 (Suppl 1):S28–30 [DOI] [PubMed] [Google Scholar]
- 85.Karimi M, Musallam KM, Cappellini MD, Daar S, El-Beshlawy A, Belhoul K, et al. Risk factors for pulmonary hypertension in patients with beta thalassemia intermedia. Eur J Intern Med. 2011;22(6):607–10 [DOI] [PubMed] [Google Scholar]
- 86.Taher AT, Musallam KM, Nasreddine W, Hourani R, Inati A, Beydoun A. Asymptomatic brain magnetic resonance imaging abnormalities in splenectomized adults with thalassemia intermedia. J Thromb Haemost. 2010;8(1):54–9 [DOI] [PubMed] [Google Scholar]
- 87.Kosaryan M, Mahdavi MR, Roshan P, Hojjati MT. Prevalence of alloimmunisation in patients with beta thalassaemia major. Blood Transfus. 2012;10(3):396–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Chou ST, Liem RI, Thompson AA. Challenges of alloimmunization in patients with haemoglobinopathies. Br J Haematol. 2012;159(4):394–404 [DOI] [PubMed] [Google Scholar]
- 89.Musallam KM, Cappellini MD, Daar S, Karimi M, El-Beshlawy A, Taher AT. Serum ferritin levels and morbidity in β-thalassemia intermedia: a 10-year cohort study. Blood. 2012;120(21):1021 [Google Scholar]
- 90.Taher A, Porter J, Viprakasit V, Kattamis A, Chuncharunee S, Sutcharitchan P, et al. Estimation of liver iron concentration by serum ferritin measurement in non-transfusion-dependent thalassemia patients: analysis from the 1-year THALASSA study. Haematologica. 2012;96(S1):0927 [Google Scholar]
- 91.Taher AT, Porter JB, Viprakasit V, Kattamis A, Chuncharunee S, Sutcharitchan P, et al. Deferasirox continues to reduce iron overload in non-transfusion-dependent thalassemia: a one-year, open-label extension to a one-year, randomized, double-blind, placebo-controlled study (THALASSA). Blood. 2012;120(21):3258. [DOI] [PubMed] [Google Scholar]
- 92.Cossu P, Toccafondi C, Vardeu F, Sanna G, Frau F, Lobrano R, et al. Iron overload and desferrioxamine chelation therapy in beta-thalassemia intermedia. Eur J Pediatr. 1981;137(3):267–71 [DOI] [PubMed] [Google Scholar]
- 93.Olivieri NF, Koren G, Matsui D, Liu PP, Blendis L, Cameron R, et al. Reduction of tissue iron stores and normalization of serum ferritin during treatment with the oral iron chelator L1 in thalassemia intermedia. Blood. 1992;79(10):2741–8 [PubMed] [Google Scholar]
- 94.Pippard MJ, Weatherall DJ. Iron balance and the management of iron overload in beta-thalassemia intermedia. Birth Defects Orig Artic Ser. 1988;23(5B):29–33 [PubMed] [Google Scholar]
- 95.Nuinoon M, Makarasara W, Mushiroda T, Setianingsih I, Wahidiyat PA, Sripichai O, et al. A genome-wide association identified the common genetic variants influence disease severity in beta0-thalassemia/hemoglobin E. Hum Genet. 2010;127(3):303–14 [DOI] [PubMed] [Google Scholar]
- 96.Galanello R, Sanna S, Perseu L, Sollaino MC, Satta S, Lai ME, et al. Amelioration of Sardinian beta0 thalassemia by genetic modifiers. Blood. 2009;114(18):3935–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Danjou F, Anni F, Perseu L, Satta S, Dessi C, Lai ME, et al. Genetic modifiers of beta-thalassemia and clinical severity as assessed by age at first transfusion. Haematologica. 2012;97(7):989–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Badens C, Joly P, Agouti I, Thuret I, Gonnet K, Fattoum S, et al. Variants in genetic modifiers of beta-thalassemia can help to predict the major or intermedia type of the disease. Haematologica. 2011;96(11):1712–4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Uda M, Galanello R, Sanna S, Lettre G, Sankaran VG, Chen W, et al. Genome-wide association study shows BCL11A associated with persistent fetal hemoglobin and amelioration of the phenotype of beta-thalassemia. Proc Natl Acad Sci USA. 2008;105(5):1620–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Ma Q, Abel K, Sripichai O, Whitacre J, Angkachatchai V, Makarasara W, et al. Beta-globin gene cluster polymorphisms are strongly associated with severity of HbE/beta(0)-thalassemia. Clin Genet. 2007; 72(6):497–505 [DOI] [PubMed] [Google Scholar]
- 101.Musallam KM, Taher AT, Cappellini MD, Sankaran VG. Clinical experience with fetal hemoglobin induction therapy in patients with beta-thalassemia. Blood. 2013. March 21;121(12):2199–212; quiz 2372 [DOI] [PubMed] [Google Scholar]
- 102.Olivieri NF, Saunthararajah Y, Thayalasuthan V, Kwiatkowski J, Ware RE, Kuypers FA, et al. A pilot study of subcutaneous decitabine in beta-thalassemia intermedia. Blood. 2011;118 (10):2708–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Singer ST, Vichinsky EP, Larkin S, Olivieri N, Sweeters N, Kuypers FA. Hydroxycarbamide-induced changes in E/beta thalassemia red blood cells. Am J Hematol. 2008;83(11):842–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Singer ST, Vichinsky EP, Sweeters N, Rachmilewitz E. Darbepoetin alfa for the treatment of anaemia in alpha- or beta- thalassaemia intermedia syndromes. Br J Haematol. 2011;154(2):281–4 [DOI] [PubMed] [Google Scholar]
- 105.Rivella S, Rachmilewitz E. Future alternative therapies for beta-thalassemia. Expert Rev Hematol. 2009;2(6):685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Libani IV, Guy EC, Melchiori L, Schiro R, Ramos P, Breda L, et al. Decreased differentiation of erythroid cells exacerbates ineffective erythropoiesis in beta-thalassemia. Blood. 2008;112(3):875–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Melchiori L, Gardenghi S, Guy EG, Rachmilewitz E, Giardina PJ, Grady RW, et al. Use of JAK2 inhibitors to limit ineffective erythropoiesis and iron absorption in mice affected by β-thalassemia and other disorders of red cell production [abstract]. Blood. 2009;114(22):202019587374 [Google Scholar]
- 108.Tefferi A. JAK inhibitors for myeloproliferative neoplasms: clarifying facts from myths. Blood. 2012;119(12):2721–30 [DOI] [PubMed] [Google Scholar]
- 109.Gardenghi S, Ramos P, Marongiu MF, Melchiori L, Breda L, Guy E, et al. Hepcidin as a therapeutic tool to limit iron overload and improve anemia in beta-thalassemic mice. J Clin Invest. 2010;120(12):4466–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Nai A, Pagani A, Mandelli G, Lidonnici MR, Silvestri L, Ferrari G, et al. Deletion of TMPRSS6 attenuates the phenotype in a mouse model of beta-thalassemia. Blood. 2012;119(21):5021–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Parrow NL, Gardenghi S, Rivella S. Prospects for a hepcidin mimic to treat beta-thalassemia and hemochromatosis. Expert Rev Hematol. 2011;4(3):233–5 [DOI] [PubMed] [Google Scholar]
- 112.Ramos E, Ruchala P, Goodnough JB, Kautz L, Preza GC, Nemeth E, et al. Minihepcidins prevent iron overload in a hepcidin-deficient mouse model of severe hemochromatosis. Blood. 2012;120(18):3829–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Preza GC, Ruchala P, Pinon R, Ramos E, Qiao B, Peralta MA, et al. Minihepcidins are rationally designed small peptides that mimic hepcidin activity in mice and may be useful for the treatment of iron overload. J Clin Invest. 2011;121(12):4880–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Li H, Rybicki AC, Suzuka SM, von Bonsdorff L, Breuer W, Hall CB, et al. Transferrin therapy ameliorates disease in beta-thalassemic mice. Nat Med. 2010;16(2): 177–82 [DOI] [PubMed] [Google Scholar]
- 115.Sankaran VG. Targeted therapeutic strategies for fetal hemoglobin induction. Hematology Am Soc Hematol Educ Program. 2011;2011:459–65 [DOI] [PubMed] [Google Scholar]
- 116.Xu J, Peng C, Sankaran VG, Shao Z, Esrick EB, Chong BG, et al. Correction of sickle cell disease in adult mice by interference with fetal hemoglobin silencing. Science. 2011;334(6058):993–6 [DOI] [PMC free article] [PubMed] [Google Scholar]