

Abstract
Therapy-related myelodysplastic syndromes and acute myelogenous leukemia comprise a poor-risk subset of myelodysplastic syndromes and acute myelogenous leukemia. Large-scale mutation profiling efforts in de novo myelodysplastic syndromes have identified mutations that correlate with clinical features, but such mutations have not been investigated in therapy-related myelodysplastic syndromes and acute myelogenous leukemia. Genomic DNA from 38 patient samples were subjected to high throughput polymerase chain reaction and sequenced for TP53, TET2, DNMT3A, ASXL1, IDH1, IDH2, EZH2, EED, SUZ12, RBBP4, SRSF2, U2AF35, and SF3B1. We identified somatic mutations in 16 of 38 (42%) patients. TP53 mutations were the most common lesion, detected in 8 of 38 (21%) patients, followed by TET2 in 4 of 38 (10.5%). Cases with a TP53 mutation or loss of the TP53 locus had a worse overall survival compared to those with wild-type TP53 (8.8 vs. 37.4 months; P=0.0035).
Introduction
Therapy-related myelodysplastic syndromes (t-MDS) and acute myeloid leukemia (t-AML) are known complications of therapy given to treat a spectrum of hematologic and epithelial malignancies, with incidence rates varying between 0.2–1% after adjuvant breast cancer therapy to as high as 10% after autologous stem cell transplant for lymphoma.1–3 The unique etiology of t-MDS/AML prompted the creation of a distinct ‘therapy-related myeloid neoplasms’ category within the World Health Organization (WHO) classification.4 The development of t-MDS/AML has been linked to exposure to specific chemotherapy agents and ionizing radiation. Deletion of chromosomes 5 and/or 7 commonly occurs in the context of prior exposure to alkylating agents with disease usually arising 5–7 years after exposure.5 Topoisomerase II inhibitor exposure has been associated with balanced translocations involving MLL at chromosome 11q23, a shorter latency period (2–3 years), and abrupt onset of AML.6
Although t-MDS/AML is classified as a separate entity and is associated with overall poorer prognosis, patients with t-MDS/AML receive standard induction and consolidation therapy followed by allogeneic transplantation whenever possible, since there is no known unique biological feature that can be targeted with existing therapies.7 In addition, clinical trials frequently exclude t-MDS/AML patients, resulting in fewer treatment options and missed opportunity to gain insight into disease biology. Outcomes with standard therapy are generally poor, with an estimated median survival of between 8 and 16 months.8,9 Allogeneic stem cell transplantation remains the only potentially curative option, but the overall cure rate is lower than for de novo MDS/AML, perhaps due to the higher incidence of poor risk cytogenetics in t-MDS/AML.10–12 Using targeted sequencing, a recent study by Bejar et al. identified mutations in five genes which predicted inferior clinical outcomes in de novo MDS.13 These mutations were found to confer significant prognostic value independent of the International Prognostic Scoring System (IPSS). In AML, the negative impact of FLT3 mutations on prognosis is widely recognized, and preliminary data has also shown that specific somatic mutations may correlate with response to therapy in MDS and in AML.13–16 However, the frequency and impact of known somatic mutations on clinical outcomes and response to therapy in t-MDS/AML have not been the subject of systematic study. To generate a preliminary assessment of the frequency and impact of such mutations, we studied samples from t-MDS/AML patients and correlated our findings with clinical features and outcome.
Design and Methods
Patient selection
Eligible patients had a documented history of a benign or malignant condition for which they had received chemotherapy and/or radiation therapy prior to the diagnosis of MDS or AML. Study cases included those with untreated or previously treated active t-MDS or t-AML with available viably cryopreserved mononuclear cells. All patients provided informed consent for research sample procurement using Memorial Sloan-Kettering Cancer Center (MSKCC) institutional review board approved protocols.
Cell separation
Mononuclear cells were separated using fluorescence activated cell sorting (FACS) for bone marrow samples or affinity column immunomagnetic separation for peripheral blood samples. Lymphocytes were separated from the mononuclear cell fraction by labeling for CD19, B220, CD3, CD4, and or CD8 in both methods. FACS was performed on a BD Biosciences FACS Aria cell sorter. Affinity column selection was performed using a Miltenyi magnetic cell separation device according to the manufacturer’s instructions.
Exon sequencing
DNA was prepared from the myeloid cell fractions using the Qiagen DNA Mini Kit or Puregene Kit. Genomic DNA was then subjected to genome wide amplification using phi29 polymerase. Exons were sequenced following polymerase chain reaction (PCR) amplification and bi-directional Sanger sequencing as previously described.16 Target genes included TP53, TET2, DNMT3A, ASXL1, IDH1, IDH2, EZH2, EED, SUZ12, RBBP4, SRSF2, U2AF35, and SF3B1. Traces were reviewed using Mutation Surveyor (Soft Genetics). Variants not in COSMIC were validated by repeat PCR and Sanger sequencing of the primary unamplified sample using lymphocyte DNA as a matched normal control.
Statistical analysis
Survival probabilities were estimated by the Kaplan-Meier method and analysis comparison was performed using a log rank (Mantel-Cox’s) test. Association of TP53 and cytogenetic abnormalities was determined using the χ2 test.
Results and Discussion
The study cohort included 38 t-MDS/AML patients with cryopreserved mononuclear cells (n=23 bone marrow aspirate, n=15 peripheral blood) and a history of primary solid tumor or hematologic malignancies. All patients had received prior chemotherapy or radiation, or a combination of therapies including chemotherapy and radiation with or without autologous stem cell transplant. The median age was 65 years (range 34–83). The median latency time from the start of therapy for the primary malignancy to the diagnosis of t-MDS/AML was 5.7 years (range 0.7–30.8). Patients’ characteristics are shown in Table 1.
Table 1.
Patients’ characteristics at time of sample collection.

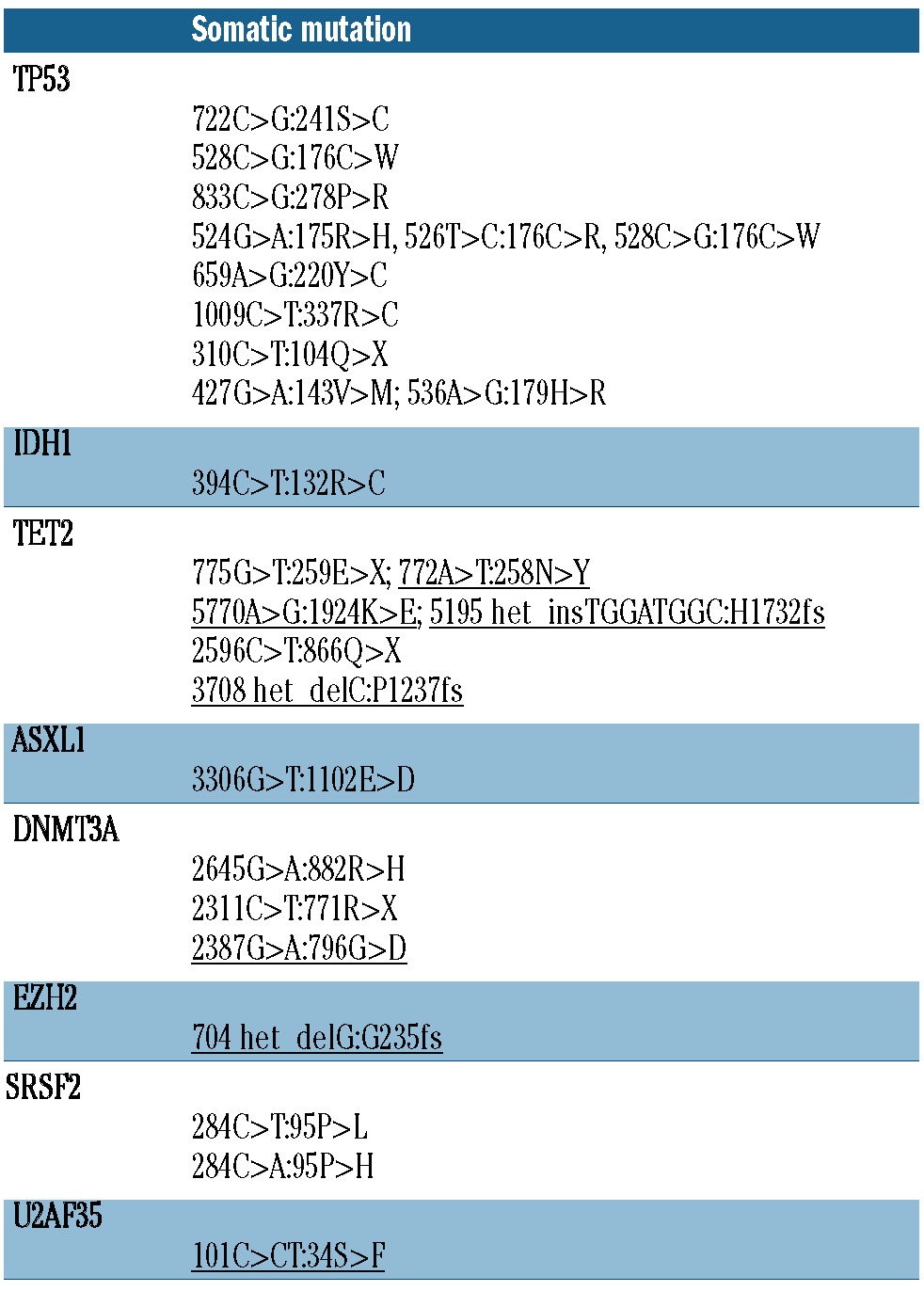
Mutations were identified in 8 of the 13 genes analyzed (Table 2). Four cases had more than one somatic point mutation in the same sample (Figure 1 and Table 3). Mutations in TP53 and TET2 were most common, occurring in 21% (8 of 38) and 10% (4 of 38) of cases, respectively. Mutations in the other target genes occurred at a frequency of less than 10% (Table 2). TP53 mutations were more common in our t-MDS/AML cohort compared to previous reports of de novo AML.13,17,18 No SF3B1 mutations were observed; however, no patients in this cohort had ringed sideroblast-associated morphology, which is closely associated with SF3B1 mutations.18 Samples with a higher (>5%) blast percentage were more likely to have somatic point mutations or loss of TP53 (10 of 12) compared to those with lower (≤5%) blast percentage (10 of 26). This finding may be explained by differences in disease biology, or by a limitation of Sanger sequencing in detecting rare clones. Most mutations were found in a heterozygous state, except in 2 patients for TP53 and 2 patients for TET2 in which more than one mutation was identified suggesting biallelic compound heterozygous mutations.
Table 2.
Mutation frequency in t-MDS/AML (n of 38) compared to reported frequencies in de novo MDS13, 17–19 and de novo AML.14–16, 18, 20–22
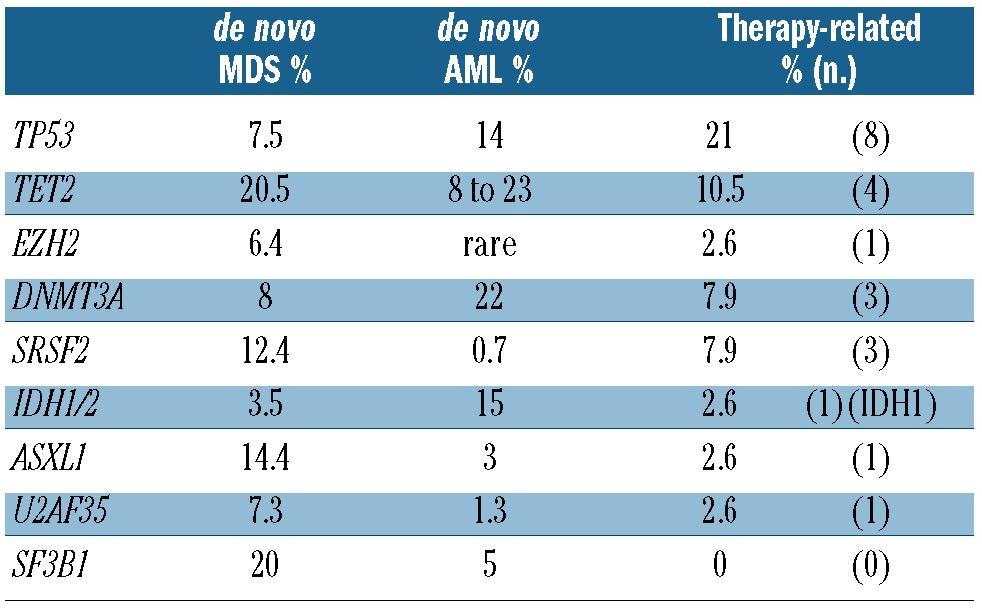
Figure 1.

Somatic point mutations and cytogenetic abnormalities in tMDS/AML patients. Each column represents 1 patient. Gray, present or received (treatment agent or modality). X, unavailable data. 17p loss detected by karyotype or FISH. #11q23 rearrangement or amplification. *AML sample. For “Sample type”: gray, BM sample; white, peripheral blood. For “Blast percentage”: black, greater than or equal to 20%; gray, between 5% and 20%; white, less than 5% (in sample at time of collection).
Table 3.
Identified somatic mutations. Mutations not in COSMIC underlined.
Somatic mutations were identified in 16 of 38 (42%) of cases studied and among these, 8 received prior chemotherapy alone, 3 received radiation alone, and 5 received combined modality therapy. We noted that only one of 9 patients exposed to alkylating agents without topoisomerase inhibitors developed a somatic mutation in the specific genes examined, compared to 11 of 22 patients who received both alkylating agents and topoisomerase inhibitors (Figure 1). A larger cohort is needed to verify whether the combination of alkylator agents and topoisomerase inhibitors is associated with an increased frequency of somatic mutations in t-MDS/AML.
Given the increased frequency of TP53 mutations (8 of 38) in our series, we also assessed for evidence of TP53 loss as a result of 17p deletion by performing fluorescence in situ hybridization (FISH) and through analysis of karyotype results. Deletion of chromosome 17p was observed in 15% (6 of 38) of cases. Two of these cases had concurrent somatic TP53 mutations, and thus 25% (2 of 8) of patients with somatic TP53 mutations had evidence of loss of heterozygosity and biallelic TP53 inactivation. In addition, 2 of the 8 samples with mutated TP53 harbored more than one mutation in TP53, consistent with functional loss of the wild-type allele. Thus 10.5% (4 of 38) of cases had possible complete loss of TP53 function.
Of the 12 cases with either a somatic TP53 mutation or cytogenetic abnormality resulting in loss of TP53, 91% (11 of 12) had poor risk (≥ 3 abnormalities) cytogenetics (P<0.01) and 83% (10 of 12) had monosomy 5 or del5q (P<0.01). Fifty percent (6 of 12) had concurrent loss of chromosome 7 or 7q (P=0.16). There were not enough cases with other individual mutations to assess an association with cytogenetics. Of the 8 remaining cases with mutations other than TP53, one had a chromosome 7 abnormality, one had complex cytogenetics, and none had chromosome 5 abnormalities. Of the 4 cases with 2 or more somatic mutations that did not include mutated TP53, 2 had normal cytogenetics. This suggests that t-MDS/AML with mutations in the genes we studied, other than TP53, may have a distinct biology compared to TP53 mutant disease.
We next examined overall survival in patients with and without TP53 loss (del17p by karyotype or FISH) or mutation (Figure 2). Patients with TP53 loss or mutation had a median survival of 8.8 months compared to 37.4 months in those with wild-type TP53 (P=0.0035). These data confirm the poor outcomes reported by others in patients with solid tumors, de novo MDS, and t-MDS/AML harboring TP53 mutations.13,23 There were insufficient cases to determine whether TP53 alterations were associated with a worse survival independent of IPSS category, as has been shown for de novo MDS,13 although IPSS was associated with prognostic relevance in this cohort (data not shown). There were not enough cases to draw conclusions regarding prognosis associated with other somatic mutations.
Figure 2.
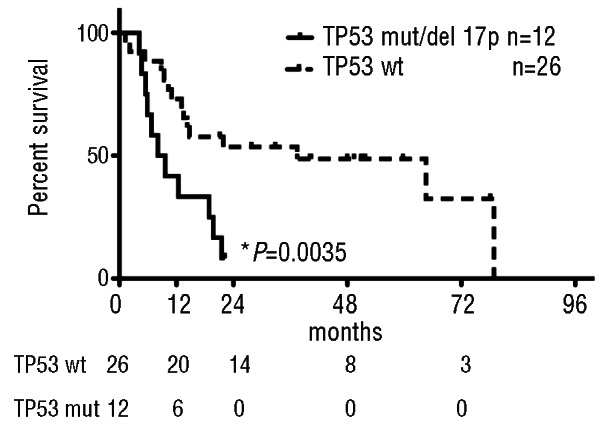
Survival of t-MDS/AML patients worsens with TP53 alterations. Median Survival of TP53 mutant/17p loss patients 8.8 months vs. TP53 wild type 37.4 months. (P=0.0035) by log-rank (Mantel-Cox) test. Table below graph, number of patients at risk at designated time points.
As expected, we noted that of the 30 patients exposed to alkylating agents, 67% (20 of 30) had abnormalities in chromosome 5 and/or 7. Of the 23 patients exposed to topoisomerase II inhibitors (n=23 anthracyclines, n=6 etoposide), only 2 had abnormalities involving the MLL gene at chromosome 11q23 by FISH analysis, and 20 were FISH negative. An additional 2 patients in our cohort had 11q23 abnormalities by FISH, one who received radiation alone and the other, radiation with an alkylating agent. It does not appear that exposure to topoisomerase II inhibitors in this cohort was a significant predisposing factor in developing MLL gene rearrangements. When patients with molecular and cytogenetic abnormalities are combined, 89% (34 of 38) had at least one genetic alteration.
This study suggests that t-MDS/AML has a distinct molecular profile compared to de novo MDS/AML. Although the genetic lesions and cytogenetic abnormalities may be similar, the mutation frequencies are different in classically mutated genes, such as TP53, and in some of the epigenetic modifiers and splicing genes included in our study. In other cancer types, molecular profiling has been used to improve the accuracy of prognostic models and predict the likelihood of response to therapy. In t-MDS/AML, it is clear that mutations in TP53 are common and are associated with inferior clinical outcomes, which may be related to the accumulation of genetic abnormalities associated with its loss of function.23 Recent reports on the predictive value of TET2 mutations in de novo MDS patients responding to 5-azacytidine and DNMT3A and NPM1 mutations in AML patients receiving high-dose daunorubicin suggest that mutational profiling can predict the pre-treatment likelihood of response to chemotherapy.16,24 An extended survey of gene mutations in a larger number of t-MDS/AML patients receiving chemotherapy or hypomethylating agents may likewise determine if response can be predicted on the basis of mutational profiling.
As cancer survivorship improves, the incidence of t-MDS/AML will likely increase. Through a better understanding of the molecular underpinnings of this poor-risk MDS/AML subtype, we hope to improve prognostic modeling and treatment options for these patients.
Acknowledgments
The authors would like to thank Tony Deblasio for processing patient samples. The authors also wish to acknowledge the support of the Geoffrey Beene Translational Oncology Core at MSKCC.
Footnotes
Funding
This work was supported by the Leukemia and Lymphoma Society (VMK, RLL, SDN), the Charles A. Dana Foundation (AHS, SSC), the American Society of Hematology (OA-W), and by grants 1U54CA143798-01 NIH/NCI and 1R01CA138234-01 NIH/NCI (RLL).
Authorship and Disclosures
Information on authorship, contributions, and financial & other disclosures was provided by the authors and is available with the online version of this article at www.haematologica.org.
References
- 1.Godley LA, Larson RA. Therapy-related myeloid leukemia. Semin Oncol. 2008;35 (4):418–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Smith RE, Bryant J, DeCillis A, Anderson S. Acute myeloid leukemia and myelodysplastic syndrome after doxorubicin-cyclophosphamide adjuvant therapy for operable breast cancer: the National Surgical Adjuvant Breast and Bowel Project Experience. J Clin Oncol. 2003;21(7):1195–204 [DOI] [PubMed] [Google Scholar]
- 3.Armitage JO, Carbone PP, Connors JM, Levine A, Bennett JM, Kroll S. Treatment-related myelodysplasia and acute leukemia in non-Hodgkin’s lymphoma patients. J Clin Oncol. 2003;21(5):897–906 [DOI] [PubMed] [Google Scholar]
- 4.Swerdlow SH, Campo E, Harris NL, Jaffe ES, Pileri SA, Stein H, et al. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues, Fourth Edition. 4 ed: WHO Press, 2008 [Google Scholar]
- 5.Rowley JD, Golomb HM, Vardiman JW. Nonrandom chromosome abnormalities in acute leukemia and dysmyelopoietic syndromes in patients with previously treated malignant disease. Blood. 1981;58(4):759–67 [PubMed] [Google Scholar]
- 6.Pedersen-Bjergaard J, Andersen MK, Andersen MT, Christiansen DH. Genetics of therapy-related myelodysplasia and acute myeloid leukemia. Leukemia. 2008;22(2):240–8 [DOI] [PubMed] [Google Scholar]
- 7.Klimek VM, Dolezal EK, Tees MT, Devlin SM, Stein K, Romero A, et al. Efficacy of hypomethylating agents in therapy-related myelodysplastic syndromes. Leukemia Res. 2012;36(9):1093–7 [DOI] [PubMed] [Google Scholar]
- 8.De Roos AJ, Deeg HJ, Davis S. A population-based study of survival in patients with secondary myelodysplastic syndromes (MDS): impact of type and treatment of primary cancers. Cancer Causes Control. 2007;18(10):1199–208 [DOI] [PubMed] [Google Scholar]
- 9.Smith SM, Le Beau MM, Huo D, Karrison T, Sobecks RM, Anastasi J, et al. Clinical-cytogenetic associations in 306 patients with therapy-related myelodysplasia and myeloid leukemia: the University of Chicago series. Blood. 2003;102(1):43–52 [DOI] [PubMed] [Google Scholar]
- 10.Kroger N, Brand R, van Biezen A, Zander A, Dierlamm J, Niederwieser D, et al. Risk factors for therapy-related myelodysplastic syndrome and acute myeloid leukemia treated with allogeneic stem cell transplantation. Haematologica. 2009;94(4):542–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yokoyama H, Mori S, Kobayashi Y, Kurosawa S, Saito B, Fuji S, et al. Hematopoietic stem cell transplantation for therapy-related myelodysplastic syndrome and acute leukemia: a single-center analysis of 47 patients. Int J Hematol. 2010;92 (2):334–41 [DOI] [PubMed] [Google Scholar]
- 12.Armand P, Deeg HJ, Kim HT, Lee H, Armistead P, de Lima M, et al. Multicenter validation study of a transplantation-specific cytogenetics grouping scheme for patients with myelodysplastic syndromes. Bone Marrow Transplant. 2010;45(5):877–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bejar R, Stevenson K, Abdel-Wahab O, Galili N, Nilsson B, Garcia-Manero G, et al. Clinical effect of point mutations in myelodysplastic syndromes. N Engl J Med. 2011;364(26):2496–506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yoshida K, Sanada M, Shiraishi Y, Nowak D, Nagata Y, Yamamoto R, et al. Frequent pathway mutations of splicing machinery in myelodysplasia. Nature. 2011;478 (7367):64–9 [DOI] [PubMed] [Google Scholar]
- 15.Ley TJ, Ding L, Walter MJ, McLellan MD, Lamprecht T, Larson DE, et al. DNMT3A mutations in acute myeloid leukemia. N Engl J Med. 2010;363(25):2424–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Patel JP, Gonen M, Figueroa ME, Fernandez H, Sun Z, Racevskis J, et al. Prognostic relevance of integrated genetic profiling in acute myeloid leukemia. N Engl J Med. 2012;366(12):1079–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Walter MJ, Ding L, Shen D, Shao J, Grillot M, McLellan M, et al. Recurrent DNMT3A mutations in patients with myelodysplastic syndromes. Leukemia. 2011;25(7):1153–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Papaemmanuil E, Cazzola M, Boultwood J, Malcovati L, Vyas P, Bowen D, et al. Somatic SF3B1 mutation in myelodysplasia with ring sideroblasts. N Engl J Med. 2011;365(15):1384–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Thol F, Kade S, Schlarmann C, Loffeld P, Morgan M, Krauter J, et al. Frequency and prognostic impact of mutations in SRSF2, U2AF1, and ZRSR2 in patients with myelodysplastic syndromes. Blood. 2012; 119(15):3578–84 [DOI] [PubMed] [Google Scholar]
- 20.Metzeler KH, Maharry K, Radmacher MD, Mrozek K, Margeson D, Becker H, et al. TET2 mutations improve the new European LeukemiaNet risk classification of acute myeloid leukemia: a Cancer and Leukemia Group B study. J Clin Oncol. 2011;29(10):1373–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gaidzik VI, Paschka P, Spath D, Habdank M, Kohne CH, Germing U, et al. TET2 mutations in acute myeloid leukemia (AML): results from a comprehensive genetic and clinical analysis of the AML study group. J Clin Oncol. 2012;30(12): 1350–7 [DOI] [PubMed] [Google Scholar]
- 22.Haferlach C, Dicker F, Herholz H, Schnittger S, Kern W, Haferlach T. Mutations of the TP53 gene in acute myeloid leukemia are strongly associated with a complex aberrant karyotype. Leukemia. 2008;22(8):1539–41 [DOI] [PubMed] [Google Scholar]
- 23.Christiansen DH, Andersen MK, Pedersen-Bjergaard J. Mutations with loss of heterozygosity of p53 are common in therapy-related myelodysplasia and acute myeloid leukemia after exposure to alkylating agents and significantly associated with deletion or loss of 5q, a complex karyotype, and a poor prognosis. J Clin Oncol. 2001;19(5):1405–13 [DOI] [PubMed] [Google Scholar]
- 24.Itzykson R, Kosmider O, Cluzeau T, Mansat-De Mas V, Dreyfus F, Beyne-Rauzy O, et al. Impact of TET2 mutations on response rate to azacitidine in myelodysplastic syndromes and low blast count acute myeloid leukemias. Leukemia. 2011; 25(7):1147–52 [DOI] [PubMed] [Google Scholar]