

Summary
Background
EGFR overexpression occurs in 27–55% of oesophagogastric adenocarcinomas, and correlates with poor prognosis. We aimed to assess addition of the anti-EGFR antibody panitumumab to epirubicin, oxaliplatin, and capecitabine (EOC) in patients with advanced oesophagogastric adenocarcinoma.
Methods
In this randomised, open-label phase 3 trial (REAL3), we enrolled patients with untreated, metastatic, or locally advanced oesophagogastric adenocarcinoma at 63 centres (tertiary referral centres, teaching hospitals, and district general hospitals) in the UK. Eligible patients were randomly allocated (1:1) to receive up to eight 21-day cycles of open-label EOC (epirubicin 50 mg/m2 and oxaliplatin 130 mg/m2 on day 1 and capecitabine 1250 mg/m2 per day on days 1–21) or modified-dose EOC plus panitumumab (mEOC+P; epirubicin 50 mg/m2 and oxaliplatin 100 mg/m2 on day 1, capecitabine 1000 mg/m2 per day on days 1–21, and panitumumab 9 mg/kg on day 1). Randomisation was blocked and stratified for centre region, extent of disease, and performance status. The primary endpoint was overall survival in the intention-to-treat population. We assessed safety in all patients who received at least one dose of study drug. After a preplanned independent data monitoring committee review in October, 2011, trial recruitment was halted and panitumumab withdrawn. Data for patients on treatment were censored at this timepoint. This study is registered with ClinicalTrials.gov, number NCT00824785.
Findings
Between June 2, 2008, and Oct 17, 2011, we enrolled 553 eligible patients. Median overall survival in 275 patients allocated EOC was 11·3 months (95% CI 9·6–13·0) compared with 8·8 months (7·7–9·8) in 278 patients allocated mEOC+P (hazard ratio [HR] 1·37, 95% CI 1·07–1·76; p=0·013). mEOC+P was associated with increased incidence of grade 3–4 diarrhoea (48 [17%] of 276 patients allocated mEOC+P vs 29 [11%] of 266 patients allocated EOC), rash (29 [11%] vs two [1%]), mucositis (14 [5%] vs none), and hypomagnesaemia (13 [5%] vs none) but reduced incidence of haematological toxicity (grade ≥3 neutropenia 35 [13%] vs 74 [28%]).
Interpretation
Addition of panitumumab to EOC chemotherapy does not increase overall survival and cannot be recommended for use in an unselected population with advanced oesophagogastric adenocarcinoma.
Funding
Amgen, UK National Institute for Health Research Biomedical Research Centre.
Introduction
Gastric and oesophageal cancers are among the most common causes of cancer-related mortality, and were responsible for more than 1·1 million deaths worldwide in 2008.1 Combination chemotherapy is beneficial in perioperative and advanced disease settings, although overall survival is poor. In patients with metastatic disease, median overall survival with best supportive care is about 3 months, which can be extended to about 10 months with chemotherapy.2,3 No internationally accepted standard of care regimen exists for advanced oesophagogastric adenocarcinoma, although most centres use doublet or triplet chemotherapy combinations with a platinum-fluoropyrimidine backbone. The REAL2 non-inferiority study established epirubicin, oxaliplatin, and capecitabine (EOC) as a standard first-line regimen, and noted a median overall survival of 11·2 months.3 This result compared favourably with the alternative regimens assessed in REAL2, including a combination of epirubicin, cisplatin, and fluorouracil that had a median overall survival of 9·9 months.
In the past decade, the EGFR pathway has been recognised as one of the key proliferative pathways that is dysregulated during tumorigenesis. Preclinical data confirm that transfection of EGFR into human cancer cells is associated with an aggressive phenotype,4 and several molecular aberrations within this pathway can function as potent oncogenes. In oesophagogastric adenocarcinoma, EGFR overexpression is reported in 27–55% of cases in published literature,5,6 and has been associated with reduced overall survival in some series.5,7 Amplification of EGFR, measured by fluorescence in-situ hybridisation, is also reported in a smaller subset of oesophagogastric adenocarcinoma.5,8 This pathway therefore represents a potential therapeutic target in this disease setting, and single-drug phase 2 trials have reported a small number of responses with monoclonal antibodies9 or tyrosine-kinase inhibitors10 targeting EGFR in advanced oesophagogastric adenocarcinoma.
Panitumumab is a fully human immunoglobulin G2 monoclonal antibody directed against EGFR that has confirmed survival benefits in advanced colorectal cancer.11 We designed the REAL3 trial to assess the addition of panitumumab to EOC in molecularly unselected patients with advanced oesophagogastric adenocarcinoma.
Methods
Study design and participants
REAL3 was an open-label, multicentre, phase 3, randomised controlled trial, undertaken at 63 parti-cipating centres (tertiary referral centres, teaching hospitals, and district general hospitals) in the UK. Safety, response, and survival outcomes were compared between standard EOC and modified EOC plus panitumumab (mEOC+P). The mEOC+P schedule was established in an unplanned dose-finding study,12 which was done because excessive toxicity (primarily diarrhoea) was noted when the drugs were initially combined at full dose. As reported previously,12 eligible patients had histologically verified, untreated, metastatic or locally advanced inoperable adenocarcinoma or undifferentiated carcinoma of the oesophagus, gastro-oesophageal junction, or stomach. Patients were aged at least 18 years and had measurable disease on CT or MRI. Other eligibility criteria included WHO performance status of 0–2, and adequate cardiac, renal, liver, and bone marrow function. Cardiac function was not formally measured at baseline unless there were concerns based on previous medical history. Renal, liver, and bone marrow function were assessed via routine blood tests (full blood count and biochemistry). Exclusion criteria included previous chemotherapy (including adjuvant chemotherapy), previous anti-EGFR therapy, known brain metastases, and clinically significant cardiac disease or other significant comorbidity. Recruitment largely predated routine HER2 testing in oesophagogastric adenocarcinoma and therefore HER2 status did not affect eligibility.
The study was done in accordance with good clinical practice guidelines and the declaration of Helsinki, and was approved by the North West Research Ethics Committee. All patients provided written informed consent before screening investigations.
Randomisation and masking
We randomly allocated patients (1:1) to standard EOC chemotherapy (intravenous epirubicin 50 mg/m2 on day 1, intravenous oxaliplatin 130 mg/m2 on day 1, and oral capecitabine 1250 mg/m2 per day on days 1–21) or modified-dose EOC in combination with panitumumab (mEOC+P; intravenous epirubicin 50 mg/m2 on day 1, intravenous oxaliplatin 100 mg/m2 on day 1, oral capecitabine 1000 mg/m2 per day on day 1–21, and intravenous panitumumab 9 mg/kg on day 1) every 21 days. Randomisation was done independently at the Institute for Cancer Research Clinical Trials and Statistics Unit (ICR-CTSU) by random permuted blocks (block sizes of six and eight) and stratified by centre region (locations were divided into 11 regions), extent of disease (locally advanced vs metastatic disease), and performance status (0 vs 1 vs 2). Patients were enrolled by trials office staff at the Royal Marsden Hospital, who then faxed confirmation of the allocated treatment group to local site staff. The trial was open-label with no masking of patients or study staff to the treatment allocation. Patients received a maximum of eight cycles of treatment. Advice regarding dose modifications for toxic effects was provided in the protocol (appendix).
Procedures
The primary endpoint was overall survival, defined as the time from randomisation until death from any cause. Secondary endpoints were progression-free survival (PFS), defined as the time from randomisation until documented disease progression or death from any cause; response rate according to RECIST 1.0 criteria;13 toxicity graded according to National Cancer Institute Common Terminology Criteria for Adverse Events (NCI-CTCAE) version 3.0; patient-reported outcomes; and KRAS mutation status. CT scans were done every 12 weeks. No central imaging review was done in relation to response or PFS endpoints. Details of the translational research methods are available in the appendix. Results from patient-reported outcomes will be reported separately.
Data for patients recruited at Royal Marsden Hospital were subject to source data verification by trust-appointed monitoring staff. For other UK centres, the sponsor deemed it appropriate, in keeping with good clinical practice requirements, to undertake central monitoring and provide sites with training meetings and written guidance to ensure appropriate conduct of the trial. Participating centres were required to provide evidence confirming patient eligibility, including blood test results, histopathology reports, and imaging reports to ensure appropriate randomisation and stratification at trial entry. During trial treatment, any discrepancies in data received compared with expected were queried with sites in real time. Recorded toxicities were cross-referenced against reported serious adverse events to ensure that all toxicities were captured and that all events meeting the criteria for a serious adverse event were reported as such.
Statistical analysis
The trial was powered to detect a 10% improvement in 1 year survival, from 45% for EOC to 55% with mEOC+P, equating to a hazard ratio (HR) of 0·749. To achieve 90% power and a two-sided α of 0·05, we needed to include 509 events (deaths from any cause) and planned a total accrual of 730 patients. We planned to do an interim survival analysis after 255 events (50%). We also did a preplanned non-comparative interim analysis of response rate with mEOC+P after the first 200 patients were assessable for response (phase 2 population). These data were reviewed by the independent data monitoring committee (IDMC), and confirmed an acceptable response rate of 52% in the mEOC+P group, which exceeded the predefined futility threshold of 45%.
Throughout the trial, unmasked data were reviewed by the IDMC to examine the safety, scientific validity, and conduct of the trial. At annual review of the data in October, 2011, the IDMC noted a statistically inferior overall survival outcome in the mEOC+P group based on the occurrence of 169 events (HR 1·53, p=0·0062). In discussion with the trial management group, we decided to close the trial to further recruitment with immediate effect, withdraw panitumumab, and cross all patients over to full-dose EOC. We did our efficacy analyses in the intention-to-treat population, defined as all eligible randomised participants. Data for patients still on treatment were censored at the time of crossover to allow accurate comparison between the two trial groups. Toxicity analyses included all participants who received at least one dose of study drug.
We estimated overall survival and PFS with the Kaplan-Meier method. We compared groups with the log-rank test and used Cox regression analysis to generate HRs and 95% CIs. We compared response rates between groups with logistic regression, and generated odds ratios and 95% CIs. We used the χ2 test to compare incidence of toxic effects between groups. In multivariate analysis, we used forward stepwise Cox regression to calculate corrected HRs and 95% CIs. Statistical analyses were done with SPSS version 19.
This study is registered with ClinicalTrials.gov, number NCT00824785.
Role of the funding source
Amgen were involved in planning of the study design. The funding sources were not involved in collection, analysis, and interpretation of data, writing of the report, or the decision to submit the paper for publication. The corresponding author had full access to all the data in the study and had final responsibility for the decision to submit for publication.
Results
Between June 2, 2008, and Oct 17, 2011, we enrolled 575 patients, three of whom were withdrawn because they did not fulfil eligibility criteria. Additionally, 19 patients randomly allocated during the phase 1 dose-finding study were excluded from the intention-to-treat analysis.12 We included 553 eligible patients in the phase 3 intention-to-treat population, representing 76% of the planned accrual at the time of trial closure (figure 1; table 1). 494 (89%) patients had metastatic disease. Median follow-up in patients who were alive at the time of analysis was 4·6 months (IQR 1·8–10·1) in the EOC group and 5·3 months (2·6–9·5) in the mEOC+P group.
Figure 1.
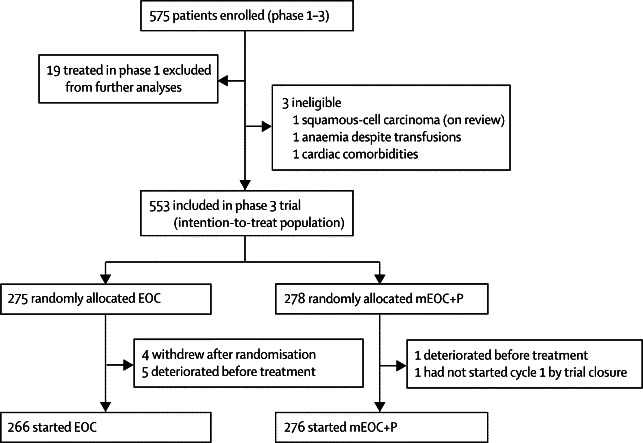
Trial profile
EOC=epirubicin, oxaliplatin, and capecitabine. mEOC+P=modified EOC plus panitumumab.
Table 1.
Demographics and baseline characteristics of the intention-to-treat population
| EOC group (n=275) | mEOC+P group (n=278) | ||
|---|---|---|---|
| Age, years | |||
| Median (range; IQR) | 62 (26–83; 54–68) | 63 (26–83; 56–68) | |
| <60 | 110 (40%) | 105 (38%) | |
| ≥60 | 165 (60%) | 173 (62%) | |
| Sex, male | 226 (82%) | 232 (83%) | |
| Performance status | |||
| 0 | 117 (43%) | 118 (42%) | |
| 1 | 143 (52%) | 144 (52%) | |
| 2 | 15 (5%) | 16 (6%) | |
| Site | |||
| Oesophagus | 111 (40%) | 106 (38%) | |
| Oesophagogastric junction | 75 (27%) | 94 (34%) | |
| Stomach | 89 (32%) | 78 (28%) | |
| Extent | |||
| Locally advanced | 25 (9%) | 34 (12%) | |
| Metastatic | 250 (91%) | 244 (88%) | |
| Histology | |||
| Adenocarcinoma | 272 (99%) | 273 (98%) | |
| Undifferentiated | 3 (1%) | 5 (2%) | |
Data are n (%), unless otherwise stated. EOC=epirubicin, oxaliplatin, and capecitabine. mEOC+P=modified EOC plus panitumumab.
Based on 251 events (EOC 110, mEOC+P 141) at the time of reporting, median overall survival was lower in the mEOC+P group than in the EOC group (HR 1·37, 95% CI 1·07–1·76, p=0·013; figure 2). Median overall survival was 8·8 months (95% CI 7·7–9·8) in the mEOC+P group compared with 11·3 months (9·6–13·0) in the EOC group (figure 2). 35 (13%) of 278 patients in the mEOC+P group and 54 (20%) of 275 patients in the EOC group were on treatment at the time of trial closure and were censored at this timepoint. 1-year overall survival was 33% (95% CI 26–41) in the mEOC+P group and 46% (38–54) in the EOC group. Subgroup analyses according to extent of disease, site of primary tumour, and KRAS mutation status were prespecified. Other subgroup analyses (age, sex, performance status, and number of metastatic sites) were exploratory. Figure 3 shows the forest plot analysis for subgroups tested, with a similar effect favouring EOC noted between all subgroups.
Figure 2.
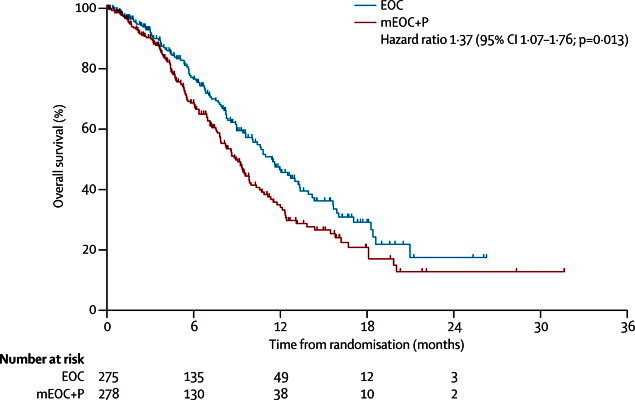
Overall survival in 553 patients in the intention-to-treat population, by treatment group
Patients still on treatment at the time of trial closure and treatment crossover were censored. EOC=epirubicin, oxaliplatin, and capecitabine. mEOC+P=modified EOC plus panitumumab.
Figure 3.
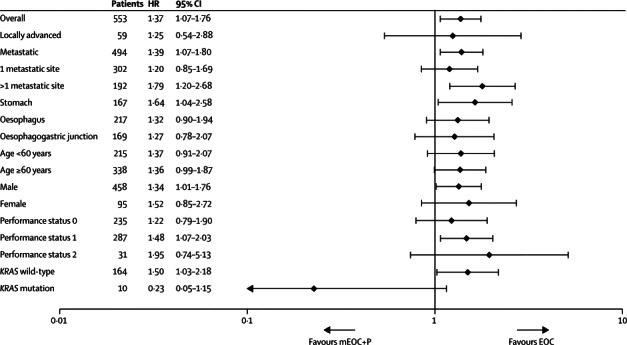
Forest plot of hazard ratios (HR) for overall survival according to baseline characteristics
EOC=epirubicin, oxaliplatin, and capecitabine. mEOC+P=modified EOC plus panitumumab.
Based on 333 events (EOC 153, mEOC+P 180), PFS did not differ between treatment groups (HR 1·22, 95% CI 0·98–1·52, p=0·068; figure 4). Median PFS was 6·0 months (95% CI 5·5–6·5) in the mEOC+P group and 7·4 months (6·3–8·5) in the EOC group. 1-year PFS was 20% (95% CI 14–26) in the mEOC+P group compared with 21% (14–27) in the EOC group.
Figure 4.
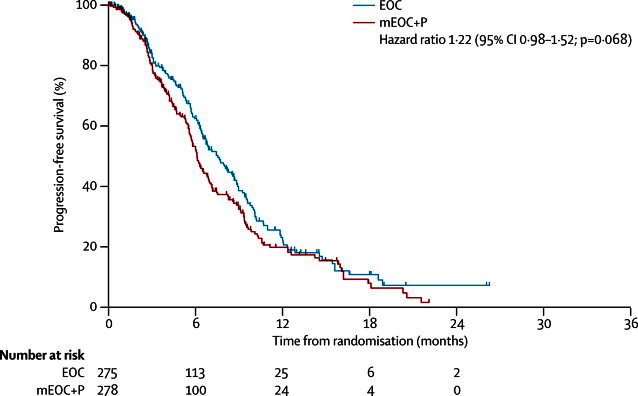
Progression-free survival in 553 patients in the intention-to-treat population, by treatment group
Patients still on treatment at the time of trial closure and treatment crossover were censored. EOC=epirubicin, oxaliplatin, and capecitabine. mEOC+P=modified EOC plus panitumumab.
61 patients who were still on treatment (37 in the EOC group and 24 in the mEOC+P group) and had not reached their first response assessment at the time of data censoring were excluded from the response analysis. Patients who died, progressed, or withdrew before response assessment were classed as non-responders. 116 (46%) of 254 patients in the mEOC+P group had a complete or partial response compared with 100 (42%) of 238 patients in the EOC group (odds ratio 1·16, 95% CI 0·81–1·66, p=0·42; table 2).
Table 2.
Responses by treatment group in 492 patients*
| EOC group (n=238) | mEOC+P group (n=254) | |
|---|---|---|
| Complete response | 5 (2%) | 8 (3%) |
| Partial response | 95 (40%) | 108 (43%) |
| Stable disease | 51 (21%) | 46 (18%) |
| Progressive disease | 19 (8%) | 30 (12%) |
| Not assessable | 68 (29%) | 62 (24%) |
EOC=epirubicin, oxaliplatin, and capecitabine. mEOC+P=modified EOC plus panitumumab.
61 patients were excluded because they were still on treatment and had not reached first response assessment at time of data censoring.
542 patients received at least one cycle of chemotherapy and were assessed for toxicity (table 3). Addition of panitumumab was associated with increased rates of grade 3–4 diarrhoea, mucositis, rash, and hypomagnesaemia. Patients in the EOC group had increased rates of grade 3–4 peripheral neuropathy, neutropenia, febrile neutropenia, and thrombocytopenia compared with patients in the mEOC+P group. Overall, we noted no significant difference between the two groups in terms of grade 3–5 toxicity when all toxicities were combined (p=0·21). However, when we excluded haematological and biochemical toxicities, more patients had grade 3–5 toxicities in mEOC+P group (p=0·035, table 3).
Table 3.
Reported toxicities according to treatment group in 542 assessable patients
|
EOC group (n=266) |
mEOC+P group (n=276) |
p value* | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Grade 1–2 | Grade 3 | Grade 4 | Grade 3–5 | Grade 1–2 | Grade 3 | Grade 4 | Grade 3–5 | ||
| Vomiting | 111 (42%) | 23 (9%) | 0 | 23 (9%) | 114 (41%) | 22 (8%) | 1 (<1%) | 23 (8%) | 1·00 |
| Mucositis | 85 (32%) | 0 | 0 | 0 | 162 (59%) | 14 (5%) | 0 | 14 (5%) | <0·0001 |
| Diarrhoea | 121 (45%) | 27 (10%) | 2 (1%) | 30 (11%) | 132 (48%) | 48 (17%) | 0 | 48 (17%) | 0·050 |
| Lethargy | 190 (71%) | 34 (13%) | 1 (<1%) | 35 (13%) | 183 (66%) | 48 (17%) | 0 | 48 (17%) | 0·19 |
| Hand-foot syndrome | 78 (29%) | 13 (5%) | 0 | 13 (5%) | 127 (46%) | 16 (6%) | 0 | 16 (6%) | 0·71 |
| Peripheral neuropathy | 188 (71%) | 18 (7%) | 0 | 18 (7%) | 194 (70%) | 4 (1%) | 0 | 4 (1%) | 0·002 |
| Deep vein thrombosis | 2 (1%) | 9 (3%) | 0 | 9 (3%) | 0 | 9 (3%) | 2 (1%) | 11 (4%) | 0·82 |
| Pulmonary embolism | NA | NA | 11 (4%) | 11 (4%) | NA | NA | 19 (7%) | 20 (7%) | 0·14 |
| Rash | 23 (9%) | 2 (1%) | 0 | 2 (1%) | 190 (69%) | 28 (10%) | 1 (<1%) | 29 (11%) | <0·0001 |
| Infection | 47 (18%) | 26 (10%) | 6 (2%) | 33 (12%) | 68 (25%) | 24 (9%) | 2 (1%) | 28 (10%) | 0·42 |
| Febrile neutropenia | NA | 26 (10%) | 9 (3%) | 37 (14%) | NA | 17 (6%) | 3 (1%) | 20 (7%) | 0·011 |
| Neutropenia | 56 (21%) | 46 (17%) | 28 (11%) | 74 (28%) | 61 (22%) | 23 (8%) | 12 (4%) | 35 (13%) | <0·0001 |
| Anaemia | 133 (50%) | 11 (4%) | 4 (2%) | 15 (6%) | 127 (46%) | 8 (3%) | 3 (1%) | 11 (4%) | 0·42 |
| Thrombocytopenia | 49 (18%) | 7 (3%) | 4 (2%) | 11 (4%) | 16 (6%) | 3 (1%) | 0 | 3 (1%) | 0·030 |
| Hypokalaemia | 28 (11%) | 12 (5%) | 4 (2%) | 16 (6%) | 29 (11%) | 7 (3%) | 3 (1%) | 10 (4%) | 0·23 |
| Hypomagnesaemia | 41 (15%) | 0 | 0 | 0 | 76 (28%) | 12 (4%) | 1 (<1%) | 13 (5%) | <0·0002 |
| Any grade 3–5 toxicity | NA | NA | NA | 166 (62%) | NA | NA | NA | 187 (68%) | 0·21 |
| Any grade 3–5 toxicity (excluding haematological or biochemical events) | NA | NA | NA | 143 (54%) | NA | NA | NA | 173 (63%) | 0·035 |
11 patients did not start chemotherapy after randomisation. The highest grade toxic effect in all treatment cycles received is reported for each patient. EOC=epirubicin, oxaliplatin, and capecitabine. mEOC+P=modified EOC plus panitumumab. NA=not applicable.
p values are for comparison of grade 3–5 toxicity between the two groups.
Four patients died from toxicities regarded as related to mEOC+P: one each of septicaemia, neutropenic sepsis, pulmonary embolism, and upper gastrointestinal haemorrhage. Six patients died from toxicities regarded as related to EOC: one each of pneumonia, diarrhoea and dehydration, pneumonitis, and myocardial infarction, and two cases of neutropenic sepsis.
For 542 patients who started treatment, the median number of cycles delivered was five (IQR three to eight) in both trial groups. 80 (30%) of 266 patients who started EOC received all eight planned cycles of chemotherapy, as did 74 (27%) of 276 patients who started mEOC+P. These findings are restricted by data censoring for the 89 patients still on treatment at the time of trial closure. Excluding these patients, the median number of cycles delivered was six (IQR three to eight) with EOC (80 [38%] of 212 patients completed eight cycles) and five (IQR three to eight) with mEOC+P (74 [31%] of 241 patients completed eight cycles).
Overall, 1307 cycles of EOC were administered compared with 1375 cycles of mEOC+P. Table 4 shows the median relative dose intensity for each drug according to treatment group and the proportion of patients in each group achieving a relative intensity of at least 80%.
Table 4.
Relative dose intensity in 542 patients who started chemotherapy
| EOC group (n=266) | mEOC+P group (n=276) | p value | |
|---|---|---|---|
| Median relative dose intensity | |||
| Epirubicin | 91% (76–99) | 92% (77–100) | 0·36 |
| Oxaliplatin | 89% (75–99) | 92% (78–100) | 0·086 |
| Capecitabine | 88% (77–97) | 88% (69–100) | 0·74 |
| Panitumumab | .. | 91% (77–100) | .. |
| Patients with relative dose intensity ≥80% | |||
| Epirubicin | 184 (69%) | 198 (72%) | 0·51 |
| Oxaliplatin | 177 (67%) | 200 (72%) | 0·13 |
| Capecitabine | 188 (71%) | 175 (63%) | 0·072 |
| Panitumumab | .. | 196 (71%) | .. |
Data are median (IQR) or n (%). EOC=epirubicin, oxaliplatin, and capecitabine. mEOC+P=modified EOC plus panitumumab.
In exploratory biomarker analyses of 276 patients treated with mEOC+P, development of any grade of rash (219 patients) was associated with significantly longer overall survival compared with patients with no rash (57 patients; figure 5). Median overall survival was 10·3 months (95% CI 8·9–11·6) and median PFS was 6·8 months (5·9–7·8) in patients with grade 1–4 rash compared with median overall survival of 4·3 months (3·3–5·4) and a median PFS of 3·7 months (2·3–5·1) in patients with grade 0 rash (p<0·0001 for overall survival and PFS).
Figure 5.
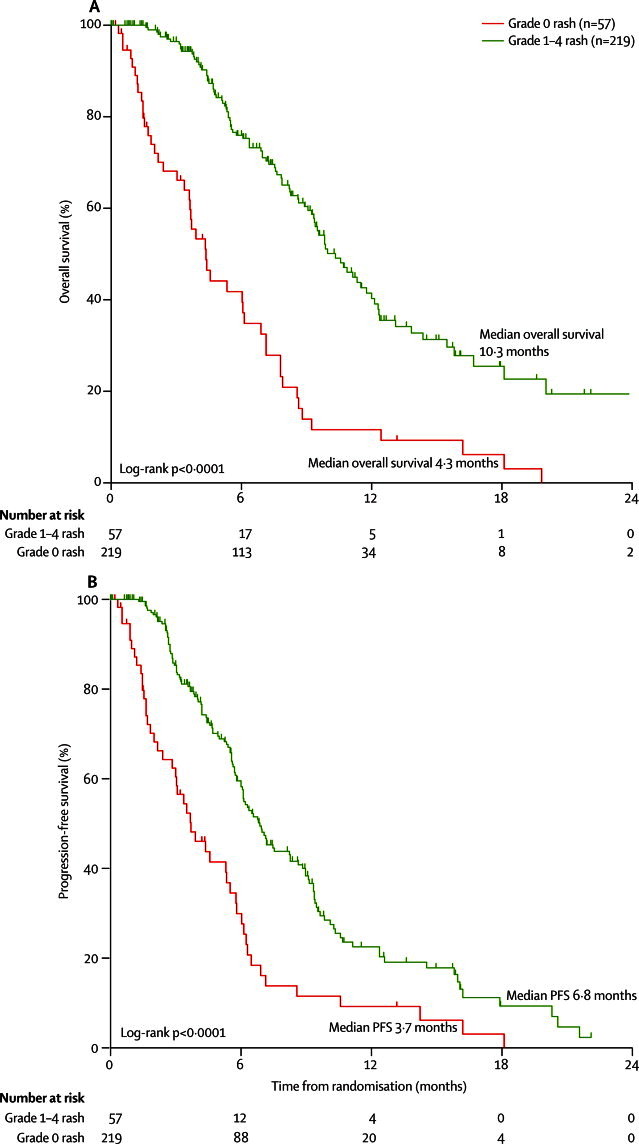
Overall and progression-free survival in 276 assessable patients in the mEOC+P group, by skin rash grade (grade 0 vs grade 1–4)
mEOC+P=modified epirubicin, oxaliplatin, and capecitabine plus panitumumab.
However, outcomes in the grade 0 rash population might be negatively skewed by patients with rapid disease progression who received insufficient panitumumab to develop rash. A further exploratory analysis was therefore done including only 194 patients (177 with rash, 17 with no rash) who received at least four cycles of chemotherapy. Although possibly restricted by the smaller number of patients, in this analysis overall survival did not differ between patients who developed rash (median 10·6 months [95% CI 9·0–12·2]) and those who did not (8·5 months [7·1–9·9]; HR 0·64 [95% CI 0·37–1·11], p=0·11).
Few tissue biomarker analyses have been undertaken to date. Frequencies of tested biomarkers in the first 200 patients are shown in table 5 and have been reported elsewhere.14 For ten patients with known KRAS mutant tumours, we noted a potential association with benefit from mEOC+P, although this association was not significant (figure 3). In multivariate overall survival analysis incorporating performance status and disease stage, both KRAS mutation (HR 2·21, 95% CI 1·15–4·26; p=0·017) and PIK3CA mutation (HR 3·63, 95% CI 1·13–11·67, p=0·030) were negatively prognostic. No prognostic effect was associated with HER2 or PTEN status, and no BRAF mutations were noted (data not shown). These biomarker analyses are exploratory and were limited by the small numbers of mutations identified to date. Further biomarker analyses are ongoing in the full trial cohort.
Table 5.
Biomarker frequencies
| EOC group (n=85) | mEOC+P group (n=90) | ||
|---|---|---|---|
| KRAS mutation | 3/85 (4%) | 7/89 (8%) | |
| Codon 12 | 2 | 5 | |
| Codon 13 | 1 | 2 | |
| Codon 61 | 0 | 0 | |
| BRAF mutation | 0/82 | 0/85 | |
| PIK3Ca mutation | 0/79 | 4/81 (5%) | |
| Exon 20 | 0 | 0 | |
| Exon 9 | 0 | 4 | |
| PTEN null | 9/80 (11%) | 16/87 (18%) | |
| HER2 positive | 17/86 (20%) | 11/88 (13%) | |
Data are n/number of patients with sufficient tissue available for analysis. We analysed samples from the first 200 patients enrolled (phase 2 population). EOC=epirubicin, oxaliplatin, and capecitabine. mEOC+P=modified EOC plus panitumumab.
Discussion
The REAL3 trial is one of two concurrent randomised phase 3 trials (the other being the EXPAND trial15) assessing the addition of anti-EGFR monoclonal antibodies to chemotherapy in first-line oesophagogastric cancer. Based on the findings of REAL3, use of panitumumab in combination with EOC cannot be recommended in an unselected population with advanced oesophagogastric adenocarcinoma, and was associated with inferior overall survival and PFS. Notably, this detrimental outcome in the experimental group was not predicted by the phase 2 endpoint of response rate (overall response rate 52% with mEOC+P). This trial does, however, confirm the efficacy of the EOC control group in this setting, with median overall survival and PFS results that are consistent with those previously reported in REAL2 (11·2 months for overall survival and 7·0 months for PFS).3
The poor outcome associated with mEOC+P in this trial did not seem to be attributable to increased treatment-related deaths, and therefore other potential explanations for our findings need to be considered. First, as reported previously,12 combination of panitumumab with full-dose EOC in the initial stages of the trial was associated with unacceptably high rates of grade 3 diarrhoea (four of the first five patients by cycle four). Therefore, we had to reduce the starting doses of oxaliplatin (by 23%) and capecitabine (by 20%) in the experimental group. Although these changes undoubtedly reduced the frequency of grade 3–4 diarrhoea with mEOC+P (17% in phase 3 population), they also served to reduce the dose intensity of chemotherapy, which is reflected in the reduced incidence of grade 3–4 neutropenia and peripheral neuropathy noted in the mEOC+P group. Additionally, the dose intensity data show a reduced proportion of patients achieving at least 80% of the planned capecitabine dose in the experimental group, suggesting that mEOC+P was still slightly more difficult to deliver than standard EOC.
Second, a negative interaction might have occurred between panitumumab and one or more of the EOC components. Evidence in the setting of colorectal cancer suggests that the chemotherapy partner for anti-EGFR therapy might be an important determinant of treatment efficacy, with oxaliplatin-containing regimens yielding inconsistent results. The OPUS16 and PRIME11 studies provide evidence of improved outcomes with the addition of cetuximab and panitumumab respectively, whereas no benefit was associated with the addition of cetuximab in the COIN17 and NORDIC VII18 studies in the same setting. Recent cell-line data also suggest that greater synergy might exist between anti-EGFR therapy and irinotecan than with oxaliplatin.19 Additionally, the COIN trial17 results suggest that there might be a differential benefit from cetuximab dependent on the fluoropyrimidine partner, with patients receiving oxaliplatin plus fluorouracil seemingly deriving increased benefit compared with those treated with oxaliplatin plus capecitabine. At present, the significance of these potential interactions is unknown, and has not been assessed in the setting of oesophagogastric cancer.
Third, our findings might have been affected because we assessed panitumumab therapy in a molecularly unselected population. During the years since the inception of the REAL3 trial, several studies have advanced our understanding of the EGFR signalling pathway and its role in oesophagogastric adenocarcinoma. Hot-spot mutations in key oncogenic drivers such as KRAS (common in colorectal cancer) and BRAF (common in malignant melanoma) are now known to be infrequent molecular events in oesophagogastric adenocarcinoma. Indeed, the 5·7% frequency of KRAS mutation in our population is in keeping with the 3–10% reported in other studies,20–22 and we did not note any BRAF mutations in 167 tumour samples tested. By contrast, gene copy number alterations (amplifications and deletions) seem to be a relatively frequent finding in oesophagogastric adenocarcinoma and are more likely to represent the key molecular alterations driving carcinogenesis. Two recent series23,24 of detailed genomic analyses in oesophagogastric adenocarcinoma reported that about 37% of tumours harbour copy number aberrations in genes that are deemed to be targetable, including KRAS, EGFR, HER2, and MET. Randomised clinical trials are therefore needed to establish whether targeting of these oncogenic signal transduction pathways can meaningfully improve outcomes for patients.
In preclinical studies, cetuximab can decrease EGFR pathway signalling via reduced phosphorylation of EGFR and AKT in oesophagogastric cancer cell lines.25 In combination with chemotherapy, cetuximab results in synergistic inhibition of cell proliferation and enhanced apoptosis.25–27 In hypoxic gastric cancer cell lines the addition of anti-EGFR therapy reversed oxaliplatin resistance.26 Additionally, a synergistic antitumour effect of combined cetuximab and S-1 was apparent in gastric cancer cell lines overexpressing EGFR.25,27 In colorectal cancer, somatic mutations in codon 12, 13, or 61 of the KRAS oncogene confer resistance to panitumumab therapy.11,28 MET amplification with or without KRAS mutations might be associated with resistance to cetuximab therapy in gastric cancer cell lines;29 however, no validated predictive biomarkers for this setting exist.
Unfortunately, despite preclinical data suggesting a role for anti-EGFR therapy in the treatment of oesophagogastric adenocarcinoma, the REAL3 trial findings are supported by two other phase 3 trials assessing anti-EGFR therapy in this disease setting. The EXPAND trial15 assessed the addition of cetuximab to a cisplatin-capecitabine doublet in 904 patients with previously untreated adenocarcinoma of the stomach and gastro-oesophageal junction, and did not meet its primary endpoint of improved PFS (HR 1·09, 95% CI 0·92–1·29, p=0·32).15 EXPAND also noted no improvement with the addition of cetuximab in either overall survival (HR 1·00, 95% CI 0·87–1·17, p=0·95) or overall response rate (30% in the experimental group vs 29% for controls). The COG trial30 assessed the anti-EGFR tyrosine-kinase inhibitor gefitinib compared with placebo in the second-line treatment of 450 patients with oesophageal and type I–II gastro-oesophageal junction cancers. This trial also did not meet its primary endpoint, with no improvement in overall survival (HR 0·90, p=0·285). However, improvements in PFS (HR 0·795, p=0·017) and disease control at 8 weeks (25·5% in the experimental group vs 16·0% in controls, p=0·014) were noted, suggesting some activity of gefitinib in a small undefined subset of patients.
Taken together, these relatively consistent findings suggest that the EGFR pathway is unlikely to represent an important therapeutic target in most patients with oesophagogastric cancer (panel). The presented biomarker analyses accompanying the REAL3 trial are restricted by small patient numbers and low rates of tested mutations. However, this work is ongoing in the full trial dataset and these translational analyses represent a unique opportunity to further assess the molecular biology of advanced oesophagogastric adenocarcinoma within a randomised trial setting. Techniques such as gene-expression profiling and next-generation sequencing might help to provide further information regarding the driver genetic events in this complex disease. Furthermore, the evaluation of genetic aberrations in pathways linked to EGFR signalling could still offer the prospect of identification of a low-frequency biomarker that identifies a subpopulation of patients benefiting from anti-EGFR targeted therapy in this setting.
Panel. Research in context.
Systematic review
We designed the REAL3 trial in an attempt to improve outcomes with systemic therapy in advanced oesophagogastric adenocarcinoma. The epirubicin, oxaliplatin, and capecitabine backbone was chosen after the REAL2 trial results. Emerging evidence had suggested EGFR overexpression was a relatively frequent occurrence in oesophagogastric adenocarcinoma, and possibly associated with inferior outcomes. Monoclonal antibodies directed against EGFR had already been shown to improve outcomes in combination with chemotherapy in advanced colorectal cancer, therefore there was interest in pursuing this combination as a possible therapeutic strategy in upper gastrointestinal malignancy. We searched PubMed for articles published in English with the MeSH terms “gastric cancer”, “oesophageal cancer”, “EGFR”, “panitumumab”, and “cetuximab”. Searches were not restricted by date.
Interpretation
Our results do not suggest a role for panitumumab in molecularly unselected patients with oesophagogastric adenocarcinoma. These findings are supported by negative results in two other phase 3 trials assessing anti-EGFR therapies in this disease setting (EXPAND15 and COG30 trials). Collectively, these findings suggest that the EGFR pathway does not represent a good therapeutic target in unselected patients with oesophagogastric adenocarcinoma. However, ongoing tissue analyses might identify a biomarker-defined subgroup of patients who could derive benefit from this treatment strategy.
This online publication has been corrected. The corrected version first appeared at thelancet.com/oncology on May 28, 2013
Acknowledgments
Acknowledgments
We thank all participating patients and staff at REAL3 trial sites, all UK principal investigators, and the Royal Marsden Hospital Research Data Management and Statistics Unit for additional statistical support. We also wish to acknowledge National Health Service (NHS) support provided through National Institute for Health Research Biomedical Research Centre funding at the Royal Marsden NHS Foundation Trust and Institute of Cancer Research.
Contributors
DC and IC were responsible for the initial concept and design of the trial. DC was the chief investigator for the trial and had oversight of the trial throughout. TW and AFCO were the trial physicians and were delegated responsibility for the day-to-day running of the study and real-time review of data on behalf of DC. CS was the senior trial co-ordinator. DC, IC, TW, AFCO, GM, JoW, DF, WM, TC, FC, DS, JuW, TI, SF, and SS were all responsible for the recruitment of patients and reporting of corresponding data. DG was responsible for doing the presented mutational analyses on tumour tissue. AW was responsible for histopathological analyses. CP and YB were the trial statisticians and did the final statistical analyses of the results. DC and TW led the interpretation of the data and writing of the manuscript. All authors had input into the data interpretation and preparation of the final report for publication.
Conflicts of interest
DC has received research funding from Amgen, Roche, Sanofi-Aventis, Merck-Serono, Novartis, and Celgene, and has had advisory roles (uncompensated) with Roche and Amgen. DF has received research funding from Amgen. IC has received research funding from Merck Serono and Roche. He has advisory roles with Merck Serono (uncompensated), Roche (compensated), and Sanofi-Aventis (compensated), and has received honoraria from Roche and Sanofi-Aventis. GM has received honoraria and travel grants from Roche, and honoraria from Sanofi. TI has received travel grants from Roche and has an advisory role (uncompensated) with Roche. TW, AFCO, JuW, and YB have received travel grants from Amgen. All other authors declare that they have no conflicts of interest.
Supplementary Material
References
- 1.Ferlay J, Shin HR, Bray F, Forman D, Mathers C, Parkin DM. Estimates of worldwide burden of cancer in 2008: GLOBOCAN 2008. Int J Cancer. 2010;127:2893–2917. doi: 10.1002/ijc.25516. [DOI] [PubMed] [Google Scholar]
- 2.Murad AM, Santiago FF, Petroianu A, Rocha PR, Rodrigues MA, Rausch M. Modified therapy with 5-fluorouracil, doxorubicin, and methotrexate in advanced gastric cancer. Cancer. 1993;72:37–41. doi: 10.1002/1097-0142(19930701)72:1<37::aid-cncr2820720109>3.0.co;2-p. [DOI] [PubMed] [Google Scholar]
- 3.Cunningham D, Starling N, Rao S, the Upper Gastrointestinal Clinical Studies Group of the National Cancer Research Institute of the United Kingdom Capecitabine and oxaliplatin for advanced esophagogastric cancer. N Engl J Med. 2008;358:36–46. doi: 10.1056/NEJMoa073149. [DOI] [PubMed] [Google Scholar]
- 4.Di Fiore PP, Pierce JH, Fleming TP. Overexpression of the human EGF receptor confers an EGF-dependent transformed phenotype to NIH 3T3 cells. Cell. 1987;51:1063–1070. doi: 10.1016/0092-8674(87)90592-7. [DOI] [PubMed] [Google Scholar]
- 5.Kim MA, Lee HS, Lee HE, Jeon YK, Yang HK, Kim WH. EGFR in gastric carcinomas: prognostic significance of protein overexpression and high gene copy number. Histopathology. 2008;52:738–746. doi: 10.1111/j.1365-2559.2008.03021.x. [DOI] [PubMed] [Google Scholar]
- 6.Langer R, Von Rahden BH, Nahrig J. Prognostic significance of expression patterns of c-erbB-2, p53, p16INK4A, p27KIP1, cyclin D1 and epidermal growth factor receptor in oesophageal adenocarcinoma: a tissue microarray study. J Clin Pathol. 2006;59:631–634. doi: 10.1136/jcp.2005.034298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lieto E, Ferraraccio F, Orditura M. Expression of vascular endothelial growth factor (VEGF) and epidermal growth factor receptor (EGFR) is an independent prognostic indicator of worse outcome in gastric cancer patients. Ann Surg Oncol. 2008;15:69–79. doi: 10.1245/s10434-007-9596-0. [DOI] [PubMed] [Google Scholar]
- 8.Miller CT, Moy JR, Lin L. Gene amplification in esophageal adenocarcinomas and Barrett's with high-grade dysplasia. Clin Cancer Res. 2003;9:4819–4825. [PubMed] [Google Scholar]
- 9.Chan JA, Blaszkowsky LS, Enzinger PC. A multicenter phase II trial of single-agent cetuximab in advanced esophageal and gastric adenocarcinoma. Ann Oncol. 2011;22:1367–1373. doi: 10.1093/annonc/mdq604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ferry DR, Anderson M, Beddard K. A phase II study of gefitinib monotherapy in advanced esophageal adenocarcinoma: evidence of gene expression, cellular, and clinical response. Clin Cancer Res. 2007;13:5869–5875. doi: 10.1158/1078-0432.CCR-06-1970. [DOI] [PubMed] [Google Scholar]
- 11.Douillard JY, Siena S, Cassidy J. Randomized, phase III trial of panitumumab with infusional fluorouracil, leucovorin, and oxaliplatin (FOLFOX4) versus FOLFOX4 alone as first-line treatment in patients with previously untreated metastatic colorectal cancer: the PRIME study. J Clin Oncol. 2010;28:4697–4705. doi: 10.1200/JCO.2009.27.4860. [DOI] [PubMed] [Google Scholar]
- 12.Okines AF, Ashley SE, Cunningham D. Epirubicin, oxaliplatin, and capecitabine with or without panitumumab for advanced esophagogastric cancer: dose-finding study for the prospective multicenter, randomized, phase II/III REAL-3 trial. J Clin Oncol. 2010;28:3945–3950. doi: 10.1200/JCO.2010.29.2847. [DOI] [PubMed] [Google Scholar]
- 13.Therasse P, Arbuck SG, Eisenhauer EA. New guidelines to evaluate the response to treatment in solid tumors. European Organization for Research and Treatment of Cancer, National Cancer Institute of the United States, National Cancer Institute of Canada. J Natl Cancer Inst. 2000;92:205–216. doi: 10.1093/jnci/92.3.205. [DOI] [PubMed] [Google Scholar]
- 14.Chau I, Okines AFC, Gonzalez de Castro D. REAL3: a multicenter randomized phase II/III trial of epirubicin, oxaliplatin, and capecitabine (EOC) versus modified (m) EOC plus panitumumab (P) in advanced oesophagogastric (OG) cancer–response rate (RR), toxicity, and molecular analysis from phase II. Proc Am Soc Clin Oncol. 2011;29(suppl) abstr 4131. [Google Scholar]
- 15.Lordick F, Kang Y-K, Chung H-C, on behalf of the Arbeitsgemeinschaft Internistische Onkologie (AIO) and EXPAND Investigators Cetuximab and cisplatin with or without cetuximab for patients with previously untreated advanced gastric cancer (EXPAND): a randomised, open-label phase 3 trial. Lancet Oncol. 2013 doi: 10.1016/S1470-2045(13)70102-5. publihsed online April 15. http://dx.doi.org/10.1016/S1470-2045(13)70102-5. [DOI] [PubMed] [Google Scholar]
- 16.Bokemeyer C, Bondarenko I, Makhson A. Fluorouracil, leucovorin, and oxaliplatin with and without cetuximab in the first-line treatment of metastatic colorectal cancer. J Clin Oncol. 2009;27:663–671. doi: 10.1200/JCO.2008.20.8397. [DOI] [PubMed] [Google Scholar]
- 17.Maughan TS, Adams RA, Smith CG, the MRC COIN Trial Investigators Addition of cetuximab to oxaliplatin-based first-line combination chemotherapy for treatment of advanced colorectal cancer: results of the randomised phase 3 MRC COIN trial. Lancet. 2011;377:2103–2114. doi: 10.1016/S0140-6736(11)60613-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tveit KM, Guren T, Glimelius B. Phase III trial of cetuximab with continuous or intermittent fluorouracil, leucovorin, and oxaliplatin (Nordic FLOX) versus FLOX alone in first-line treatment of metastatic colorectal cancer: the NORDIC-VII study. J Clin Oncol. 2012;30:1755–1762. doi: 10.1200/JCO.2011.38.0915. [DOI] [PubMed] [Google Scholar]
- 19.Vora N, Evans S, Bernard PJ, Francisco F, Nagourney RA. Optimal combination therapy for advanced colorectal cancer: An examination of synergy and antagonism with EGFR inhibition. Proc Am Soc Clin Oncol. 2012;30(suppl) abstr e14075. [Google Scholar]
- 20.Moehler M, Mueller A, Trarbach T, the German Arbeitsgemeinschaft Internistische Onkologie Cetuximab with irinotecan, folinic acid and 5-fluorouracil as first-line treatment in advanced gastroesophageal cancer: a prospective multi-center biomarker-oriented phase II study. Ann Oncol. 2011;22:1358–1366. doi: 10.1093/annonc/mdq591. [DOI] [PubMed] [Google Scholar]
- 21.Pinto C, Di Fabio F, Barone C. Phase II study of cetuximab in combination with cisplatin and docetaxel in patients with untreated advanced gastric or gastro-oesophageal junction adenocarcinoma (DOCETUX study) Br J Cancer. 2009;101:1261–1268. doi: 10.1038/sj.bjc.6605319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lordick F, Luber B, Lorenzen S. Cetuximab plus oxaliplatin/leucovorin/5-fluorouracil in first-line metastatic gastric cancer: a phase II study of the Arbeitsgemeinschaft Internistische Onkologie (AIO) Br J Cancer. 2010;102:500–505. doi: 10.1038/sj.bjc.6605521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Deng N, Goh LK, Wang H. A comprehensive survey of genomic alterations in gastric cancer reveals systematic patterns of molecular exclusivity and co-occurrence among distinct therapeutic targets. Gut. 2012;61:673–684. doi: 10.1136/gutjnl-2011-301839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dulak AM, Schumacher SE, van Lieshout J. Gastrointestinal adenocarcinomas of the esophagus, stomach, and colon exhibit distinct patterns of genome instability and oncogenesis. Cancer Res. 2012;72:4383–4393. doi: 10.1158/0008-5472.CAN-11-3893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fukuda K, Saikawa Y, Takahashi M. Antitumor effect of cetuximab in combination with S-1 in EGFR-amplified gastric cancer cells. Int J Oncol. 2012;40:975–982. doi: 10.3892/ijo.2011.1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Luo HY, Wei W, Shi YX. Cetuximab enhances the effect of oxaliplatin on hypoxic gastric cancer cell lines. Oncol Rep. 2010;23:1735–1745. doi: 10.3892/or_00000819. [DOI] [PubMed] [Google Scholar]
- 27.Kobunai T, Watanabe T, Fukusato T. Antitumour activity of S-1 in combination with cetuximab on human gastric cancer cell lines in vivo. Anticancer Res. 2011;31:3691–3696. [PubMed] [Google Scholar]
- 28.Peeters M, Price TJ, Cervantes A. Randomized phase III study of panitumumab with fluorouracil, leucovorin, and irinotecan (FOLFIRI) compared with FOLFIRI alone as second-line treatment in patients with metastatic colorectal cancer. J Clin Oncol. 2010;28:4706–4713. doi: 10.1200/JCO.2009.27.6055. [DOI] [PubMed] [Google Scholar]
- 29.Heindl S, Eggenstein E, Keller S. Relevance of MET activation and genetic alterations of KRAS and E-cadherin for cetuximab sensitivity of gastric cancer cell lines. J Cancer Res Clin Oncol. 2012;138:843–858. doi: 10.1007/s00432-011-1128-4. [DOI] [PubMed] [Google Scholar]
- 30.Ferry DR, Dutton SJ, Mansoor W. Phase III multi-centre, randomised, double-blind, placebo-controlled trials of gefitinib versus placebo in esophageal cancer progressing after chemotherapy, COG (cancer oesophagus gefitinib) Ann Oncol. 2012;23(suppl 9) ixe12. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.