

Abstract
Targeting gene- or drug-loaded nanoparticles (NPs) to tumors and ensuring their intratumoral retention after systemic administration remain key challenges to improving the efficacy of NP-based therapeutics. Here, we investigate a novel targeting approach that exploits changes in lipid metabolism and cell membrane biophysics that occur during malignancy. We hypothesized that modifications to the surface of NPs that preferentially increase their biophysical interaction with the membrane lipids of cancer cells will improve intratumoral retention and in vivo efficacy upon delivery of NPs loaded with a therapeutic gene. We have demonstrated that different surfactants, incorporated onto the NPs’ surface, affect the biophysical interactions of NPs with the lipids of cancer cells and normal endothelial cells. NPs surface modified with didodecyldimethylammoniumbromide (DMAB) demonstrated greater interaction with cancer cell lipids, which was 6.7-fold greater than with unmodified NPs and 5.5-fold greater than with endothelial cell lipids. This correlated with increased uptake of DMAB-modified NPs with incubation time by cancer cells compared to other formulations of NPs and to uptake by endothelial cells. Upon systemic injection, DMAB-NPs demonstrated a 4.6-fold increase in tumor accumulation compared to unmodified NPs which also correlated to improved efficacy of p53 gene therapy. Characterization of the biophysical interactions between NPs and lipid membranes of tumors or other diseased tissues/organs may hold promise for engineering targeted delivery of therapeutics.
Keywords: gene therapy, nanoparticles, tumor targeting, membrane model
1. Introduction
The success of NP-based delivery systems for cancer therapy relies on effective localization and uptake of NPs within tumors. Conventional strategies for tumor targeting of NPs typically exploit the proteins/receptors overexpressed on tumor cells that distinguish them from normal cells. Ligands, such as antibodies, peptides, or aptamers, may be conjugated to the NP surface to actively bind to specific molecules on the surface of cancer cells or the tumor vasculature [1]. This approach has met with some success compared with similar nontargeted/passively-targeted drug delivery systems [2]; however, several limitations exist: a) the number of targeting sites available or accessible may be limited and insufficient for achieving a therapeutic dose of NPs in tumor tissue; b) ligands with low affinity binding for their receptor can limit targeting efficiency; and c) slow recycling of receptors may limit the dose of NPs that can be delivered to the tumor site at one time.
To address these limitations, we investigated a targeting strategy that takes advantage of the overall differences in the biophysical properties of cell membranes of malignant vs. normal cells, rather than aiming at specific molecules. Cell membrane properties are determined, to a large extent, by their lipid composition. Lipid metabolism is altered during the malignant transformation of cells; in this context, tumor cells undergo increased metabolic demands, adapt to hypoxia, and respond to specific oncogenic pathways [3]. Consequently, the lipid composition and biophysical characteristics of the cell membranes of cancerous vs. normal cells differ substantially. Recently, we also found differences in lipid membrane composition and biophysical characteristics of drug-sensitive vs. drug-resistant breast cancer cells that affect their uptake of Doxil, a liposomal NP formulation of doxorubicin. Drug-resistant cells had rigid cell membranes that impaired their endocytic function, resulting in lower Doxil uptake compared with drug-sensitive cells. When drug-resistant cells were treated with an epigenetic drug that altered lipid metabolism, resulting in more fluid cell membranes, the uptake of Doxil® improved [4]. This recent study illustrated the role of lipids in cell membrane biophysics and NP uptake. A number of drugs that target lipid metabolic pathways have been investigated as anticancer therapies [3, 5]. However, in the field of NP-based therapeutic strategies, scant consideration has been given to lipid metabolism and how it can be exploited to improve NP tumor localization.
We have developed and extensively studied polymeric NPs based on poly(lactic co-glycolic acid) (PLGA) for anti-cancer drug and gene delivery. We previously demonstrated that NPs provide sustained intracellular release of a tumor suppressor gene, p53, in vitro and in vivo thereby reducing tumor cell proliferation and tumor growth [6–8]. However, systemic administration was not as effective for tumor inhibition and animal survival as intratumoral injection of p53-loaded NPs (p53NPs); this is because a significant fraction of the administered NP dose was lost to other body compartments [7]. We hypothesized that modifications to the surface of NPs that preferentially increase their biophysical interaction with the lipids of cancer cells would improve tumor localization and efficacy of p53 gene-loaded NPs in vivo. To study lipid-NP interactions, we have developed methods using a Langmuir balance. In our earlier studies, we demonstrated, with a model lipid membrane of an endothelial cell, that the molecular structure of surfactants on the surface of NPs affects the biophysical interaction of NPs [9]. Here, we investigated the interactions of surfactant-modified NPs with cancer cell lipids vs. normal endothelial cell lipids and correlated the biophysical interactions with the in vivo performance of the NPs.
DNA-loaded NPs (DNA-NPs) were formulated with two cationic surfactants differing in their molecular structure. Our objectives were to 1) evaluate if these surface modifications enable DNA-NPs to preferentially interact with the membrane lipids of malignant vs. normal cells and 2) determine if these biophysical characteristics improved tumor localization and efficacy of p53NPs in vivo to inhibit tumor growth and disease progression. Ultimately, our goal is to assess the feasibility of screening and selecting NP formulations for therapeutic potential based on their biophysical interaction patterns and, in this case, apply our findings to developing an effective nonviral gene therapy system for cancer treatment.
2. Materials and Methods
Formulation of NPs containing plasmid DNA
The recombinant pCEP4 vector containing a cytomegalovirus-driven wild-type wt-human p53 cDNA was used [10]. DNA-loaded PLGA (copolymer ratio 50:50, inherent viscosity 1.24 g/dL, Durect Corporation) were formulated by our previously reported double emulsion-solvent evaporation technique [7]. The secondary emulsion was done in a solution containing 2% w/v of PVA (molecular weight 30,000–70,000, 87–90% hydrolyzed, Sigma) in water for unmodified-NPs, and 1% w/v PVA and 4 mM of eitherdidodecyldimethylammoniumbromide (DMAB) (Sigma) or cetyltrimethylammonium bromide (CTAB) (Sigma) to yield DMAB-modified p53NPs (DMAB-p53NPs) and CTAB-modified p53NPs (CTAB-p53NPs). NPs were recovered after chloroform evaporation by ultracentrifugation at 35,000 rpm (Beckman L80, Beckman Instruments, Inc), washed three times with water, resuspended in 3% sucrose solution, and lyophilized. The washings following the recovery of NPs were saved to determine DNA encapsulation using an indirect method [11]. NP diameter was measured by a dynamic light-scattering technique, and zeta potentials were determined with a phase analysis light-scattering technique using a commercial particle-sizing system (PSS/NICOMP 380/ZLS Particle Sizing Systems, Santa Barbara, CA).
Cell Culture
PC-3 human prostate carcinoma cells (American Type Culture Collection) were cultured in RPMI 1640 supplemented with 10% FBS. Human umbilical vein endothelial cells (HUVECs) were cultured in endothelial basal medium with growth factors supplied by Lonza. Prior to use, cells were detached using trypsin/EDTA at 37 °C.
Biophysical Interaction Studies
A Langmuir film balance (Minimicro 2, KSV Instruments, Helsinki, Finland) was used to study the interactions of NPs with HUVEC and PC-3 cell membrane lipids. To extract lipids, both HUVEC and PC-3 cells were cultured in six plates (150 × 25 mm) using the media described above. At 80–90% confluence, the cells were harvested by scraping into 10 mL of sterile water. Cell suspensions were combined and centrifuged at 1300 rpm and 4 °C for 7 min. The resulting cell pellet was suspended in 3 mL of sterile water and lyophilized for 48 hrs, and stored at −80 °C until use. Lipids from the lyophilized cell mass were extracted by using a modified Bligh and Dyer method as described in our previous study [12]. Contaminating hydrophobic proteins were removed via column chromatography as previously described.
A lipid monolayer was created by adding either HUVEC (14 μL) of PC-3 (10 μL) lipid extract dissolved at 5mg/mL in chloroform:methanol 4:1 v/v solvent mixture on the buffer surface (PBS at pH 7.4). Solvent was allowed to evaporate for 10 min. Then, lipids on the buffer surface were compressed until the SP reached 30 mN/m, as described in our previous studies [9]. The SP is equivalent to the lateral pressure of a red blood cell membrane; therefore at this SP the arrangement of lipids in the monolayer mimics the arrangement of lipids in the cell membrane bilayer [13]. The intrinsic ΔSP was monitored for 5 min. Thirty microliters of NP suspension (3 mg/100 mL saline) was then injected below the model membrane and SP was monitored for 20 min. Uniform distribution of NPs was ensured by stirring the subphase with the magnetic stir plate under the Langmuir trough. Temperature was maintained at 37 °C. As a control, ΔSP was also monitored for saline injections. Experiments were repeated in triplicate.
Cell Uptake Studies
PC-3 and HUVEC cells were seeded separately in 24-well plates at a density of 100,000 cells/well. After 48 hrs cells were washed with PBS and incubated witheither unmodified-NPs, DMAB-NPs, or CTAB-NPs (25 μg/mL/well) for 15 and 30 min. The NPs were formulated using the protocol described above but without DNA and with the addition of 6-coumarin (50 μg/90 mg PLGA) in the polymer solution, which acts as a marker for NPs [14]. The DNA was excluded due to potential interference with the fluorescent dye [15, 16]. Size, surface charge, and biophysical interactions were the same in the 6-coumarin-loaded NPs and DNA-loaded NPs in all cases. After incubation, NPs were removed, the cells were washed with PBS and lysed with cold radioimmuno precipitation assay (RIPA) buffer (Sigma) containing 1X protease inhibitor (Calbiochem, Gibbstown, NJ). Cell lysate (20 μL) was used to determine the total protein content using the Pierce BCA protein assay kit (Pierce Biotechnology, Rockford, IL). The remaining cell lysate (180 μL) was lyophilized for 48 hrs. To quantify the 6-coumarin (and hence NPs) the dye was extracted with methanol (1 mL/lysate) overnight. Samples were centrifuged in a microcentrifuge at 14,000 rpm for 10 min at 4 °C. The supernatant was collected and analyzed for 6-coumarin using high-performance liquid chromatography (Shimadzu Scientific Instruments, Columbia, MD) [9]. Loading efficiency of 6-coumarin was 88% for unmodified NPs, 83% for CTAB-NPs, and 80% for DMAB-NPs.
Confocal Microscopy Imaging of NP Uptake
Live cell imaging was performed using UltraVox spinning disk confocal microscope (Perkin Elmer, Waltham, MA). The system was equipped with six different lasers for excitation (at 405, 445, 488, 514, 561, and 640 nm wavelengths), two Hamamtsu EMCCD cameras (512 * 512 C9100-13 and 1024 * 1024 c9100-50), XYZ piezo stage for rapid z-stack acquisition, and an environmental chamber for maintaining the CO2, temperature and humidity for cells. PC-3 cells and HUVECs were seeded on separate 35mm glass bottom culture dishes (MatTek Corporation, Ashland, MA) at a density of 100 000 cell/cm2. After 48 hours, cells were washed with PBS and incubated in a 2mL suspension of 6-coumarin-labelled NPs (100 μg/2mL media) for 30 min. To stain the cell membrane, 5 μg/mL of dye solution (Deep red plasma, Invitrogen) was added to the dish 5 minutes prior to the end of incubation time. Prior to imaging, the cells were washed twice with PBS to remove the excess NPs and dye, and replaced with their respective media. Samples were illuminated with respective lasers in an alternating fashion.
PC-3 Prostate Carcinoma Mouse Model
Male, athymic nude mice (nu/nu, 5–6 wks old) were purchased from Charles River Laboratories (Wilmington, MA). Animal protocols were approved by the Institutional Animal Care and Use Committee. One million PC-3 cells were suspended in 200 μL of 50:50 phosphate buffered saline (PBS, Gibco) and Matrigel (Growth Factor Reduced, BD Biosciences, San Jose, CA), and injected subcutaneously into the left flank of each mouse. Tumor size was measured using digital calipers. Tumor volume was calculated as 0.5 × length × width2. For biodistribution and efficacy studies, animals were treated by intravenous injection of NPs when tumor volume reached ~300 mm3.
Imaging and Evaluation of Biodistribution of Nanoparticles
The biodistribution of NPs was determined using the Cambridge Research and Instrumentation Maestro EX fluorescence imaging system (Woburn, MA). NPs were made as described above but without DNA, and with the addition of the NIR dye SDB 5491 (HW Sands, Jupiter, FL) to the PLGA solution. This dye remains incorporated within the NP for the timeframe of the experiment and therefore NIR signal can be used as a marker for the NPs. Exclusion of DNA from these formulations was done because the DNA was found to inhibit the dye signal in vivo. Optimization of dye loading for each formulation was performed to achieve a suitable signal for in vivo imaging. For the unmodified NP formulation, 100 μg dye/3 mL PLGA solution was used; for CTAB-modified NPs, 400 μg dye/3 mL PLGA solution was used; for DMAB-modified NPs, 600 μg dye/3 mL PLGA solution was used. Different dye concentrations were used to account for differences in dye loading during formulation (approximately 86% efficiency for unmodified-NPs, 22% efficiency for CTAB-NPs, and 10–14% efficiency for DMAB-NPs). The surface-modified NPs demonstrated lower dye loading than unmodified NPs due to the interaction of excess surfactant (i.e., surfactant not bound to the NP) with the dye, causing some of the dye to partition into surfactant micelles. This effect was more pronounced during DMAB-NP formulation, since DMAB is more hydrophobic and interacted more strongly with the dye. The resulting dye-loaded NPs were similar in size and surface charge as the respective DNA-loaded NPs. To evaluate the biodistribution of NPs, dye-loaded NPs were injected via tail vein in tumor-bearing mice at the same dose as that used in the efficacy studies (3 mg NPs; 110 mg/kg body weight). Mice were imaged prior to and post injection over a 96-hr period. The cubes (images) obtained were unmixed to obtain an image showing fluorescing regions. Regions of interest (ROIs) were created on anatomical locations corresponding to the tumor and skin (as background) to obtain average signal counts. Some animals were sacrificed after 48 hrs and the tumors removed for ex vivo evaluation. Tumors were homogenized, lyophilized, and mixed with methanol at 4 °C for 2 days to extract the NIR dye. Standards were prepared using the same process but by adding known amounts of NPs to tissue homogenate (from untreated mouse). Samples were centrifuged and 100 μL of supernatant was placed in white 96-well plates (Nunc Brand, Thermo Fisher Scientific). Plates were imaged with the Maestro using the NIR filter set at auto-exposure. ROIs were created around each well and the average signal recorded. The standards were used to obtain a standard curve for each NP formulation, which was used to calculate the amount of NPs in the tumors for each group.
p53NP Efficacy Studies
Treatment groups were as follows: (1) unmodifiedp53NP, (2) DMAB-p53NP, (3) CTAB-p53NP, (4) DMAB-NPs made with control plasmid DNA, which does not encode for p53 (DMAB-CNP), and (5) saline. A dose equivalent to 60 μg of DNA (approximately 3 mg of NPs; 110 mg/kg body weight) suspended in 100 μL of saline was administered at day 0 and day 11 via the tail vein. This dosing regimen was selected based on preliminary studies in which tumor growth inhibition, without nonspecific toxic effects, occurred after a single dose of DMAB-NPs but tumor growth increased approximately 11–14 days after this. A second dose was administered to maintain a longer duration of tumor inhibition. Animals were monitored for tumor size and body weight every other day post-treatment. Animals were euthanized if tumors reached >10% of body weight or if animals had lost >20% of their original body weight. Tumor growth curves were analyzed by calculating the area under the curve from day 0 through day 17, which was when animals started reaching their endpoint (based on tumor size or weight loss). The area under the curve provided estimates of percentage inhibition in tumor growth compared to control groups over the time course of the study, rather than at a single time point [17].
Statistical Analysis
All numerical data were expressed as mean ± SEM, unless otherwise noted. Analysis of the Kaplan-Meier survival curve was conducted by a log-rank statistical test. In all other experiments statistical significance was determined by Student’s t test (without Bonferroni correction). We considered P ≤ 0.05 as significant.
3. Results
Biophysical Characterization NP-Lipid Interactions
Unmodified DNA-loaded PLGA NPs were prepared using the previously established double-emulsion technique, whereby the non-ionic surfactant poly(vinyl) alcohol (PVA) is used as an emulsifier during the second emulsion [7]. The resulting unmodified NPs have plasmid DNA encapsulated within the polymer matrix and PVA at the surface. The surface of DNA-loaded NPs was modified by incorporation of either the dichain surfactant didodecyldimethylammoniumbromide (DMAB) or the single-chain surfactant cetyltrimethylammonium bromide (CTAB) into the secondary emulsion, in combination with PVA (Fig. 1a). The DMAB- and CTAB-modified NPs had cationic surface charges, whereas unmodified NPs were anionic (Table 1). Formulations were optimized to achieve size and DNA-loading similar to that of unmodified NPs so that we could study the effects of the surfactant properties only.
Figure 1. NP-Lipid biophysical interaction studies.

The different surfactants used in PLGA NP formulation (a). Unmodified NPs were formulated with PVA alone; surface-modified NPs were formulated with PVA and either DMAB or CTAB. To study NP-lipid interactions, representative lipid monolayers were created over a buffer subphase into which NPs were injected (b). The change in surface pressure (ΔSP) was monitored over time for PC-3 lipids (c) and HUVEC lipids (d). Curves represent the median result. The positive ΔSP at 20 min after NP injection demonstrates quantitatively that DMAB-NPs had greater interaction with PC-3 lipids than did CTAB- and unmodified NPs and that DMAB-NPs also had greater interaction with PC-3 cells than with HUVECs (e). Data are shown as mean ± standard deviation; n = 3;*p< 0.05.
Table I.
Physical Characteristics of Nanoparticles
| NP Formulation | Size(nm) | ζ Potential (mV) | DNA Loading Efficiency (%) |
|---|---|---|---|
|
| |||
| unmodified-NPs | 310.6 ± 25.4 | −12.2 ± 8.6 | 76.0 ± 9.10 |
| CTAB-NPs | 285.0 ± 47.0 | +11.0 ± 3.2 | 78.0 ± 11.0 |
| DMAB-NPs | 289.3 ± 39.4 | +19.5 ± 4.8 | 80.7 ± 13.0 |
Biophysical interactions between NPs and either lipids extracted from prostate cancer cells (PC-3) or normal human vascular endothelial cells (HUVECs) were studied using a Langmuir balance. PC-3 cells were selected because they are p53 deficient and therefore relevant targets for p53 gene therapy. HUVECs were selected to represent normal endothelial cells, which are among the first cells that NPs encounter in vivo upon intravenous injection. In the Langmuir balance, a lipid monolayer was created over a buffer subphase and compressed to a physiologically relevant surface pressure (SP) of 30 mN/m (Fig. 1b). The biophysical interaction between NPs and membrane lipids was determined as the change in SP (ΔSP) in the lipid monolayer after injection of NPs into the buffer phase. This connection between the ΔSP and NP-lipid membrane interaction was validated in a previous study in which we used atomic force microscopic imaging to confirm the presence (positive pressure change) or absence (negative pressure change) of NPs in lipid membrane [9]. A positive ΔSP correlated to the presence of NPs in lipid membranes, hence their increased interaction with the membrane. DMAB-NPs showed a sharp increase in SP of PC-3 lipid membranes after initial injection of the NPs; this increase became more gradual after 15 min (Fig. 1c). CTAB-NPs demonstrated a lesser increase in SP of PC-3 lipids than DMAB-NPs, especially with increasing time. In contrast, unmodified NPs showed a slight, gradual increase in SP of PC-3 lipids. The overall ΔSP caused by DMAB-NPs of 7.73 mN/m was significantly greater than that of CTAB-NPs (5.7 mN/m, p< 0.005) and PVA-NPs (1.15 mN/m, p< 0.005). DMAB-NP and CTAB-NP demonstrated similar Δ SPs, and hence interactions, with HUVEC lipids, but these were much lower compared with PC-3 lipids (Fig. 1d,e). Unmodified NPs demonstrated a slight decrease in SP with HUVEC lipids, similar to that of saline controls (Fig. 1d).
To determine if there were fundamental differences in the biophysical characteristics of membrane lipids from PC-3 cells vs. HUVECs, the compressive modulus of the lipid monolayers alone was evaluated from the respective isotherms as per our previously described protocol [4]. The previous study demonstrated that higher membrane fluidity correlates to increased endocytic function. At a biologically relevant SP range (30–35 mN/m), PC-3 cell lipid monolayers demonstrated a significantly lower compression modulus (31.5–40 mN/m) indicating higher membrane fluidity than HUVEC cell lipid monolayers (47.5–75 mN/m).
In vitro Cell Uptake
Results from the cell uptake and the NP-lipid biophysical interaction studies were consistent (Fig. 2). Cell uptake was evaluated after 15 and 30 min of incubation, which corresponds to a timeframe in which uptake is most affected by biophysical interactions. After 15 min (Fig. 2a), DMAB-NPs demonstrated the highest uptake by PC-3 cells, followed by CTAB-NPs and unmodified NPs. Uptake by HUVECs was similar for DMAB- and CTAB-NPs but significantly lower for unmodified NPs. DMAB-NP uptake was 3-fold higher in PC-3 cells than in HUVEC cells, whereas CTAB-NPs and unmodified NPs demonstrated a 1.7-fold increase in uptake by malignant vs. normal cells. Cell uptake of unmodified NPs by both HUVEC and PC-3 cells increased from 15 to 30 min such that they approached levels similar to those of CTAB-NPs (Fig. 2b). For CTAB-NPs, there was an increase in uptake by HUVECs from 15 to 30 min, but uptake by PC-3 cells did not change. The DMAB-NP group showed a significant increase in uptake by both cancerous and normal cells after 30 min, but still demonstrated a 2-fold increase in cancer cell uptake relative to normal endothelial cell uptake. In contrast, uptake of the CTAB-NPs and PVA-NPs were similar by both cell types. Confocal images also show greater uptake of DMAB-NPs by PC-3 cells compared to HUVECs (Fig 2c). Altogether, these results suggest that the DMAB-NPs interact preferentially with the lipids of PC-3 cells, thereby enhancing their uptake. Differences in the levels and kinetics of cell uptake among NPs may reflect different mechanisms of NP-membrane interaction and cell uptake.
Figure 2. Cell uptake of NPs.

Uptake of NPs by HUVECs and PC-3 cells after 15 min (a) and 30 min (b) of incubation. Data are shown as mean ± standard deviation; n = 3. Brackets indicate p< 0.05 between groups. Confocal microscopy (c) shows NP (green) uptake in PC-3 and HUVEC cells after 30 min of incubation (cell membranes labeled red).
NP Localization to Tumors
Biodistribution studies were conducted to determine if the biophysical interactions and cell uptake observed in vitro correlated to tumor localization in vivo. Human PC-3 xenografts were created subcutaneously in athymic nude mice. NPs loaded with near-infrared (NIR) dye were injected intravenously, and animals were imaged over 4 days using methods validated in previous studies [7, 18]. The tumor signal profile of the DMAB-NP group differed from that of the CTAB-NP and unmodified NP groups. With CTAB-NPs and unmodified NPs, the tumor signal peaked at 24 hours, after which a significant decline was observed (Fig. 3a,b; p = 0.04 in both groups after background subtraction). In the DMAB-NP group, the tumor signal remained relatively stable over the 4-day period post injection (Fig. 3c). In addition, the “skin” signal, which is the background signal in the animal and reflects NPs in the circulation, also differed among the groups. In the DMAB group, the skin signal dropped to near zero within the first hours after injection. In the CTAB-NP and unmodified NP groups, the skin signal also dropped after the first 3 hours; however, a notable signal remained for the duration of the experiment. The dye loading was not exactly the same for the different NP formulations, with DMAB-NPs having lower dye loading compared with CTAB-NPs and unmodified NPs. Therefore, to directly compare tissue accumulation of NPs between groups, tissues were removed after 48 hrs for ex vivo quantification of dye content using standard curves for each of the NP formulations. This analysis demonstrated a 3-fold increase in NPs in tumors from mice treated with DMAB-NPs compared with CTAB-NPs and a 4.6-fold increase compared with unmodified NPs. In all cases, more NPs accumulated in liver than in tumor, but DMAB-NPs demonstrated the highest tumor-to-liver ratio of NPs normalized to tissue weight (0.51 DMAB-NPs > 0.18 CTAB-NPs > 0.10 unmodified NPs).
Figure 3. Biodistribution of surface-modified NPs.
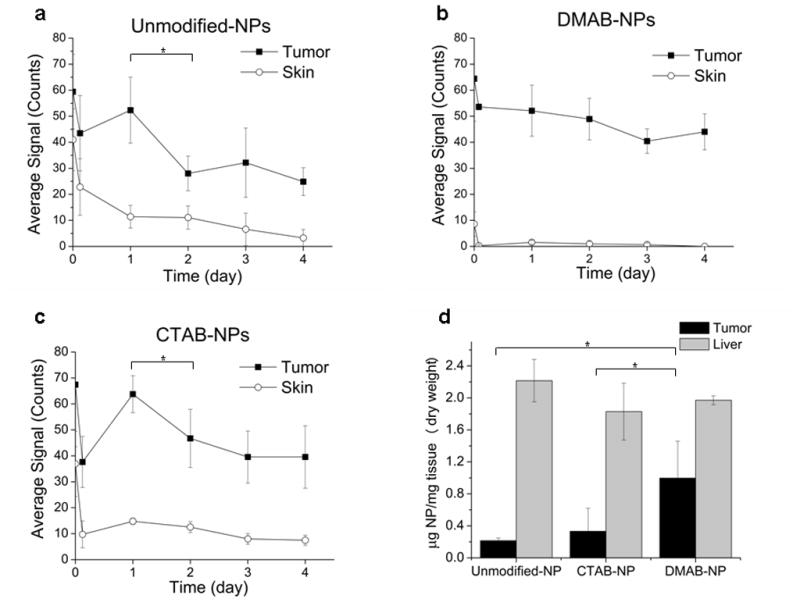
In vivo NIR signal from the tumor and from a skin region further away from the tumor (background tissue signal) was monitored over 4 days after injection of NIR-dye loaded unmodified NPs (a), DMAB-NPs (b), and CTAB-NPs (c); n = 3–4. Statistical analysis was performed on tumor signal with skin signal subtracted (*p<0.05). Quantification of NPs in tumor and liver for each group was also performed ex vivo 48 hrs post-injection (d, *p< 0.05).
Efficacy of p53 Gene Therapy
We tested if the different surface modifications affected the efficacy of NP-mediated p53 gene therapy upon systemic administration. Tumors in animals treated with DMAB-p53NPs did not grow for 11 days after the first dose, then grew slowly relative to other groups after the second dose (Fig. 4a). In contrast, steady tumor growth was observed in all other groups. Control DMAB-NPs (DMAB-CNPs), which were formulated with a control plasmid that did not encode for p53, did not demonstrate any tumor inhibition (tumor growth was similar to that of saline controls). CTAB-p53NP and unmodified p53NP groups showed similar tumor growth profiles, which were in between the DMAB-p53NP group and control groups. The DMAB-p53NP group demonstrated a 48% reduction in tumor growth compared to saline controls (p< 0.005); unmodified p53NPs and CTAB-p53NPs demonstrated a 13% and 17% reduction in tumor growth, respectively (Fig. 4b).
Figure 4. Efficacy of p53-gene therapy by surface-modified NPs in prostate tumors.
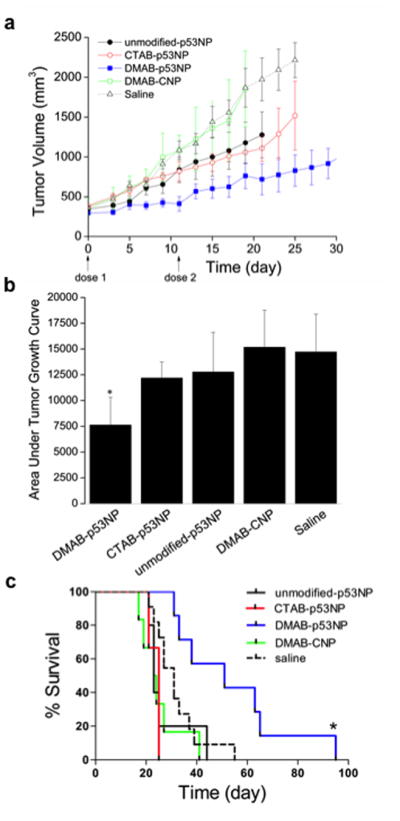
Tumor-bearing mice were treated with two doses of unmodified p53NPs, CTAB-p53NPs, and DMAB-p53NPs, with each dose equivalent to 60 μg of p53 plasmid DNA. Controls included saline and DMAB-CNPs made with control plasmid. Tumor volume was monitored over time (a). Tumor volume is shown up to the time point when most animals were still alive. The area under the curve (calculated 0 through day 17, the day the first animal died) demonstrated significantly greater reduction in overall tumor growth in the DMAB-p53NP group vs. all other treated and control groups (*p< 0.05) (b). This Kaplan-Meier plot shows improvement in animal survival after treatment with DMAB-p53NPs vs. other treatment and control groups. Log-rank test of DMAB-p53NP and each group yields *p ≤ 0.05 (c).
The reduction in tumor growth by DMAB-p53NPs corresponded with improved animal survival compared with other treatment groups and controls (P ≤ 0.01; Fig. 4c). DMAB-p53NP-treated animals had a median survival of 51 days, compared with 35 days for the unmodified p53NP group, 25 days for the CTAB-p53NP groups, 24 days for the DMAB-CNP group, and 32 days for saline controls.
4. Discussion
This study demonstrated that surface modifications that selectively enhanced biophysical interactions between NPs and cancer cell lipids also enhanced cancer cell uptake in vitro and improved tumor localization and efficacy in vivo for NP-mediated p53 gene therapy. Our group has extensively studied the role of biophysics of cell membranes in cancer and drug delivery [4, 9, 12, 19]; however, this is the first study to demonstrate the in vivo correlation and applicability of this biophysical model for optimizing an NP-based drug delivery system. Our findings suggest that exploiting the lipid membrane characteristics of cancer cells may be an effective strategy for developing tumor-targeted drug/gene delivery systems. To our knowledge, no one has previously evaluated the biophysical characteristics of malignant versus normal cell lipids for the purpose of NP-based targeted gene delivery.
Cell membranes are complex, dynamic arrangements of lipids, proteins, and polysaccharides, all of which are involved in endocytosis and cell membrane characteristics. We use the Langmuir balance method to create simplified models of specific cell membranes using lipid extracts from cells of interest. The biophysical interactions between the NPs and the model lipid membrane therefore cannot replicate all aspects of the cellular uptake process. However, this approach has been useful for understanding mechanisms of drug resistance in cancer cells [4]. Because of their dynamic nature, it is difficult to study such NP-lipid interactions in live cells. Similar lipid membrane models have also proven useful in explaining the therapeutic efficacy of a number of agents, including antibiotics and antifungal, antipsychotic, and anticancer drugs [20].
Most of these models, including monolayers, supported bilayers, and liposomes, use a well-defined mixture of lipids. By using lipid extracts from specific cell types, we aim to more closely mimic the lipid profile in those cell membranes, recapitulate the unique biophysical properties of lipid membranes from different cells, and assess cell-specific NP-lipid interactions. The findings from our Langmuir balance studies, cell uptake, biodistribution, and in vivo efficacy were generally consistent despite the increased biological complexity in each experiment. Our study was conducted on one prostate cancer model; future studies will continue to investigate correlations between the behavior of NPs in our in vitro models of membrane lipids and in vivo disease models for other types of cancers. If strong correlations continue to be found, lipid model membranes may have the potential to become a powerful, rapid, and predictive technique for the development of targeted drug delivery systems.
DMAB and CTAB are surfactants commonly used in NP formulations to improve the stability, size distribution, and cell interactions of NPs [21, 22]. DMAB-coated NPs have been shown to improve NP uptake by various cells including arterial, intestinal mucosal, and cancer cells [23, 24]. This increased uptake is typically attributed to the electrostatic interactions between the positive surface charge of the NPs and negative charge of the cell membranes. Similarly, NP-based gene carriers commonly use cationic surfactants and polymers to form complexes with negatively charged DNA and to enhance cellular uptake [25]. In a previous study, we evaluated several cationic surfactants, with different carbon chain lengths and numbers of chains, adsorbed onto the surface of polystyrene NPs. We discovered that the molecular structure of the surfactant affected the manner in which the NPs interacted with an endothelial model membrane, leading to differences in cell uptake [9].
Our current study also demonstrated that surface charge alone does not explain improved cell uptake or the differences in NP uptake between the PC-3 and HUVECs. CTAB- and DMAB-NPs both have positive charges, yet demonstrated different levels of uptake by cancer cells. Cell uptake is affected by how efficiently the NPs can penetrate into cell membranes. The success of such penetration depends on cell membrane biophysical characteristics such as membrane tension, membrane fluidity/rigidity, and adhesion strength/binding energy between the NPs and the cell membrane [24]. The DMAB-NPs demonstrated greater interaction with PC-3 lipids than CTAB-NPs, which likely contributed to the increase in uptake. DMAB- and CTAB-NPs demonstrated similar biophysical interactions with endothelial cells and similar uptake. The PC-3 lipid monolayers demonstrated a lower compressive modulus than HUVEC lipids. This finding confirms that there are differences in the biophysical properties of cancer cells compared with normal cells, and thus, studies are ongoing to characterize the lipid composition in the two cell types. In addition, a lower compressive modulus indicates greater membrane fluidity, which facilitates endocytosis. Both the increased interaction of DMAB-NPs with PC-3 cell lipids and PC-3 cell membrane fluidity likely played a role in increasing PC-3 cell uptake in vitro and in vivo. This study demonstrates the relevance of lipid-NP interactions for NP targeting. Further studies are needed to understand the nature of these interactions, specifically how DMAB-NPs selectively interact with PC-3 cancer cells versus normal endothelial cells in contrast to CTAB- and unmodified-NPs. For example, the molecular structure of the DMAB on the surface of the NP may facilitate interactions with specific lipids or lipid domains on the surface of cancer cells.
CTAB-NPs demonstrated greater interaction with PC-3 lipids compared with unmodified NPs and greater cell uptake after 15 min incubation; however, cell uptake after 30 min as well as in vivo biodistribution and efficacy were similar in these two groups. This achievement of eventual parity may be related to the pattern of NP-lipid interaction we previously observed using polystyrene NPs coated with CTAB. In that study, we showed that CTAB-coated NPs adhered to specific lipid domains of a model membrane, whereas DMAB-coated NPs readily penetrated the lipid membrane [9]. The source of lipids used differed from those used in the current study; however, this suggests further characterization of the pattern of interaction may be useful in understanding what additional biophysical parameters may be required to predict in vivo efficacy or if there is a threshold value or range of ΔSP that correlates with increased cell uptake in vitro and in vivo. Protein interactions from the serum present during in vitro cell uptake studies and the in vivo environment may also play a role.
In vivo biodistribution studies showed a positive correlation with the in vitro biophysical interaction and cell uptake studies. We observed greater tumor accumulation and retention with DMAB-NPs than CTAB-NPs and unmodified NPs. NPs accumulate into tumors primarily due to the “enhanced permeability and retention” effect. Once inside the tumor, interaction with tumor cells is required to facilitate uptake. This interaction is of particular importance for gene therapy since DNA must be delivered intracellularly for efficacy. Based on our in vitro findings, we believe that the improved tumor targeting is due to enhanced biophysical interactions between the DMAB-NPs and cancer cells, resulting in improved tumor cell uptake compared with the other NP formulations. This offers the potential to delivery a higher concentration of a given therapeutic payload, in this case the p53 tumor suppressor gene, to tumors.
A significant amount of NPs were also lost to the organs of the reticuloendothelial system (RES), such as the liver. Further optimization of NP formulation parameters, such as size, surfactant density, and/or additional modifications, may improve the bioavailability of DMAB-NPs to the tumor. Future biophysical interactions studies should include evaluation of interactions with lipids from cells of the RES, such as Kupffer cells, to further refine the development and selection of NP surface modifications. In addition, characterization of intra-tumoral distribution of the NPs will further our understanding cellular uptake in vivo. Current tumor targeting strategies employ “stealth” technologies that involve coating of NPs with hydrophilic polymers such polyethylene glycol or block copolymers to reduce protein absorption and evade recognition by the RES. However, the hydrophilic polymer coating also reduces interactions between NPs and tumor cells and interferes with trafficking involved in intracellular delivery, resulting in decreased biological activity and efficacy of NP-based gene carriers [26, 27].
An important goal of this study was to identify surface modifications that improve the efficacy of NP-mediated p53 gene delivery to tumors upon systemic administration based on inhibition of tumor progression. Efficacy results in a PC-3 tumor model supported the findings from the biodistribution and in vitro studies. DMAB-p53NPs demonstrated greater tumor inhibition and improved survival compared with the CTAB-NPs and unmodified NPs. Surfactants such as DMAB and CTAB are known to be cytotoxic themselves in vitro [22, 28], and may cause immune activation and/or immunotoxicity in vivo [29]. This can contribute to non-specific effects on tumor growth inhibition as is observed with many cationic lipid-based nanoparticle formations [30, 31]. Future mechanistic studies will include evaluation of immune response to surface-modified NPs. However, at the doses used here, we did not observe any signs of toxicity to the animals. In addition, no effect on tumor growth was observed in our study with control DMAB-NPs, therefore the improved efficacy in the DMAB-p53NP group was attributed to successful delivery of the p53 gene. Our previous studies extensively characterized NP-induced p53 gene expression in vitro and in vivo and demonstrated an inverse relationship between tumor growth and p53 gene and protein levels. Based on this relationship, the increased tumor inhibition in the DMAB-p53NP group is likely related to increased levels of intratumoral p53 compared with the other groups.
It should be noted that use of the unmodified p53NP group did not significantly improve survival in this study, as was reported in our previous study where three times higher dose of p53NPs was used [7]. The significant improvement in survival of animals with DMAB-modified p53NPs even at lower doses suggests improved tumor localization of modified NPs and gene delivery.
The gene p53 is one of the most commonly mutated genes in human cancer and its mutant form results in more aggressive disease and greater resistance to conventional treatment. As a result, there is great interest in therapeutic strategies aimed at restoring the function of p53 for the treatment of cancer [32]. Viral vectors for p53 gene therapy have been investigated; however, serious safety issues, such as toxicity, insertional mutagenesis, and immunogenicity (especially with multiple administration of adenoviral vector), often limit their use to local injections rather than systemic delivery [25, 33, 34]. As a result, clinical trials of adenoviral vector-mediated p53 gene therapy have been conducted for cancers in which intratumoral injections were feasible [35]. Our goal is to develop a safe nonviral p53 gene vector that can be delivered systemically and selectively target primary as well as metastatic tumors. This study moves us closer to that goal by identifying techniques with which to improve cancer cell targeting of NP-based gene carriers.
Conclusions
We have demonstrated that the surface of NPs can be modified with a surfactant, DMAB, to preferentially interact with the lipids of cancer cells compared to normal cells. Increased biophysical interactions with cancer cell lipids correlated with increased NP uptake by cancer cells in vitro, enhanced tumor accumulation in vivo, and improved efficacy of gene therapy. This study suggests a role of NP-lipid biophysical interactions in developing NP-based therapeutics. Characterization of the biophysical interactions between NPs and lipid membranes of tumors or other diseased tissues/organs may be a promising approach for screening and developing targeted delivery systems.
Acknowledgments
Funding for this study is gratefully acknowledged: Grants 1R01CA149359 and 1R01EB003975 (to V.L.) from the National Cancer Institute (NCI) and National Institute of Biomedical Imaging and Bioengineering of the National Institutes of Health. Isaac Adjei is funded by the “Med into Grad” initiative of the Howard Hughes Medical Institute and predoctoral fellowship 5F31CA150566 from the NCI. Confocal microscopy studies were performed at the Cleveland Clinic Imaging Core with guidance from Dr. Judith Drazba.
Footnotes
Conflict of Interest: None.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Ruoslahti E, Bhatia SN, Sailor MJ. Targeting of drugs and nanoparticles to tumors. J Cell Biol. 2010;188:759–768. doi: 10.1083/jcb.200910104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cheng Z, Al Zaki A, Hui JZ, Muzykantov VR, Tsourkas A. Multifunctional nanoparticles: cost versus benefit of adding targeting and imaging capabilities. Science. 2012;338:903–910. doi: 10.1126/science.1226338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Santos CR, Schulze A. Lipid metabolism in cancer. FEBS J. 2012;279:2610–2623. doi: 10.1111/j.1742-4658.2012.08644.x. [DOI] [PubMed] [Google Scholar]
- 4.Vijayaraghavalu S, Peetla C, Lu S, Labhasetwar V. Epigenetic modulation of the biophysical properties of drug-resistant cell lipids to restore drug transport and endocytic functions. Mol Pharmaceutics. 2012;9:2730–2742. doi: 10.1021/mp300281t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schug ZT, Frezza C, Galbraith LC, Gottlieb E. The music of lipids: how lipid composition orchestrates cellular behaviour. Acta Oncol. 2012;51:301–310. doi: 10.3109/0284186X.2011.643823. [DOI] [PubMed] [Google Scholar]
- 6.Prabha S, Labhasetwar V. Nanoparticle-mediated wild-type p53 gene delivery results in sustained antiproliferative activity in breast cancer cells. Mol Pharmaceutics. 2004;1:211–219. doi: 10.1021/mp049970+. [DOI] [PubMed] [Google Scholar]
- 7.Sharma B, Ma W, Adjei IM, Panyam J, Dimitrijevic S, Labhasetwar V. Nanoparticle-mediated p53 gene therapy for tumor inhibition. Drug Deliv Transl Res. 2011;1:43–52. doi: 10.1007/s13346-010-0008-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Prabha S, Sharma B, Labhasetwar V. Inhibition of tumor angiogenesis and growth by nanoparticle-mediated p53 gene therapy in mice. Cancer Gene Ther. 2012;19:530–537. doi: 10.1038/cgt.2012.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Peetla C, Labhasetwar V. Effect of molecular structure of cationic surfactants on biophysical interactions of surfactant-modified nanoparticles with a model membrane and cellular uptake. Langmuir. 2009;25:2369–2377. doi: 10.1021/la803361y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Seki M, Iwakawa J, Cheng H, Cheng PW. p53 and PTEN/MMAC1/TEP1 gene therapy of human prostate PC-3 carcinoma xenograft, using transferrin-facilitated lipofection gene delivery strategy. Hum Gene Ther. 2002;13:761–773. doi: 10.1089/104303402317322311. [DOI] [PubMed] [Google Scholar]
- 11.Prabha S, Labhasetwar V. Critical determinants in PLGA/PLA nanoparticle-mediated gene expression. Pharm Res. 2004;21:354–364. doi: 10.1023/b:pham.0000016250.56402.99. [DOI] [PubMed] [Google Scholar]
- 12.Peetla C, Bhave R, Vijayaraghavalu S, Stine A, Kooijman E, Labhasetwar V. Drug resistance in breast cancer cells: biophysical characterization of and doxorubicin interactions with membrane lipids. Mol Pharmaceutics. 2010;7:2334–2348. doi: 10.1021/mp100308n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zwaal RFA, Demel RA, Roelofsen B, van Deenen LLM. The lipid bilayers concept of the cell membranes. Trends Biochem Sci. 1976;1:112–114. [Google Scholar]
- 14.Panyam J, Sahoo SK, Prabha S, Bargar T, Labhasetwar V. Fluorescence and electron microscopy probes for cellular and tissue uptake of poly(D,L-lactide-co-glycolide) nanoparticles. Int J Pharm. 2003;262:1–11. doi: 10.1016/s0378-5173(03)00295-3. [DOI] [PubMed] [Google Scholar]
- 15.Nazarenko I, Pires R, Lowe B, Obaidy M, Rashtchian A. Effect of primary and secondary structure of oligodeoxyribonucleotides on the fluorescent properties of conjugated dyes. Nucleic Acids Res. 2002;30:2089–2195. doi: 10.1093/nar/30.9.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Seidel CAM, Schulz A, Sauer MHM. Nucleobase-specific quenching of fluorescent dyes .1. Nucleobase one-electron redox potentials and their correlation with static and dynamic quenching efficiencies. J Phys Chem-Us. 1996;100:5541–5553. [Google Scholar]
- 17.Dings RP, Van Laar ES, Loren M, Webber J, Zhang Y, Waters SJ, Macdonald JR, Mayo KH. Inhibiting tumor growth by targeting tumor vasculature with galectin-1 antagonist anginex conjugated to the cytotoxic acylfulvene, 6-hydroxylpropylacylfulvene. Bioconjug Chem. 2010;21:20–27. doi: 10.1021/bc900287y. [DOI] [PubMed] [Google Scholar]
- 18.Foy SP, Manthe RL, Foy ST, Dimitrijevic S, Krishnamurthy N, Labhasetwar V. Optical imaging and magnetic field targeting of magnetic nanoparticles in tumors. ACS Nano. 2010;4:5217–5224. doi: 10.1021/nn101427t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Peetla C, Rao KS, Labhasetwar V. Relevance of biophysical interactions of nanoparticles with a model membrane in predicting cellular uptake: study with TAT peptide-conjugated nanoparticles. Mol Pharmaceutics. 2009;6:1311–1320. doi: 10.1021/mp900011h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Peetla C, Stine A, Labhasetwar V. Biophysical interactions with model lipid membranes: applications in drug discovery and drug delivery. Mol Pharmaceutics. 2009;6:1264–1276. doi: 10.1021/mp9000662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li D, Li G, Li P, Zhang L, Liu Z, Wang J, Wang E. The enhancement of transfection efficiency of cationic liposomes by didodecyldimethylammonium bromide coated gold nanoparticles. Biomaterials. 2010;31:1850–1857. doi: 10.1016/j.biomaterials.2009.11.027. [DOI] [PubMed] [Google Scholar]
- 22.Fay F, Quinn DJ, Gilmore BF, McCarron PA, Scott CJ. Gene delivery using dimethyldidodecylammonium bromide-coated PLGA nanoparticles. Biomaterials. 2010;31:4214–4222. doi: 10.1016/j.biomaterials.2010.01.143. [DOI] [PubMed] [Google Scholar]
- 23.Labhasetwar V, Song C, Humphrey W, Shebuski R, Levy RJ. Arterial uptake of biodegradable nanoparticles: effect of surface modifications. J Pharm Sci. 1998;87:1229–1234. doi: 10.1021/js980021f. [DOI] [PubMed] [Google Scholar]
- 24.Xu A, Yao M, Xu G, Ying J, Ma W, Li B, Jin Y. A physical model for the size-dependent cellular uptake of nanoparticles modified with cationic surfactants. Int J Nanomedicine. 2012;7:3547–3554. doi: 10.2147/IJN.S32188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Heilbronn R, Weger S. Viral vectors for gene transfer: current status of gene therapeutics. Handb Exp Pharmacol. 2010:143–170. doi: 10.1007/978-3-642-00477-3_5. [DOI] [PubMed] [Google Scholar]
- 26.Hatakeyama H, Akita H, Kogure K, Oishi M, Nagasaki Y, Kihira Y, Ueno M, Kobayashi H, Kikuchi H, Harashima H. Development of a novel systemic gene delivery system for cancer therapy with a tumor-specific cleavable PEG-lipid. Gene Ther. 2007;14:68–77. doi: 10.1038/sj.gt.3302843. [DOI] [PubMed] [Google Scholar]
- 27.Xu L, Wempe MF, Anchordoquy TJ. The effect of cholesterol domains on PEGylated liposomal gene delivery in vitro. Ther Deliv. 2011;2:451–460. doi: 10.4155/tde.11.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kusumoto K, Ishikawa T. Didodecyldimethylammonium bromide (DDAB) induces caspase-mediated apoptosis in human leukemia HL-60 cells. J Control Release. 2010;147:246–252. doi: 10.1016/j.jconrel.2010.07.114. [DOI] [PubMed] [Google Scholar]
- 29.Peer D. Immunotoxicity derived from manipulating leukocytes with lipid-based nanoparticles. Adv Drug Deliv Rev. 2012;64:1738–1748. doi: 10.1016/j.addr.2012.06.013. [DOI] [PubMed] [Google Scholar]
- 30.Kedmi R, Ben-Arie N, Peer D. The systemic toxicity of positively charged lipid nanoparticles and the role of Toll-like receptor 4 in immune activation. Biomaterials. 2010;31:6867–6875. doi: 10.1016/j.biomaterials.2010.05.027. [DOI] [PubMed] [Google Scholar]
- 31.Chen QR, Mixson AJ. Systemic gene therapy with p53 inhibits breast cancer: recent advances and therapeutic implications. Front Biosci. 1998;3:D997–D1004. doi: 10.2741/a340. [DOI] [PubMed] [Google Scholar]
- 32.Brown CJ, Lain S, Verma CS, Fersht AR, Lane DP. Awakening guardian angels: drugging the p53 pathway. Nat Rev Cancer. 2009;9:862–873. doi: 10.1038/nrc2763. [DOI] [PubMed] [Google Scholar]
- 33.Pisters LL, Pettaway CA, Troncoso P, McDonnell TJ, Stephens LC, Wood CG, Do KA, Brisbay SM, Wang X, Hossan EA, Evans RB, Soto C, Jacobson MG, Parker K, Merritt JA, Steiner MS, Logothetis CJ. Evidence that transfer of functional p53 protein results in increased apoptosis in prostate cancer. Clin Cancer Res. 2004;10:2587–2593. doi: 10.1158/1078-0432.ccr-03-0388. [DOI] [PubMed] [Google Scholar]
- 34.Cristofanilli M, Krishnamurthy S, Guerra L, Broglio K, Arun B, Booser DJ, Menander K, Van Wart Hood J, Valero V, Hortobagyi GN. A nonreplicating adenoviral vector that contains the wild-type p53 transgene combined with chemotherapy for primary breast cancer: safety, efficacy, and biologic activity of a novel gene-therapy approach. Cancer. 2006;107:935–944. doi: 10.1002/cncr.22080. [DOI] [PubMed] [Google Scholar]
- 35.Zhang S, Li Y, Li L, Zhang Y, Gao N, Zhang Z, Zhao H. Phase I study of repeated intraepithelial delivery of adenoviral p53 in patients with dysplastic oral leukoplakia. J Oral Maxillofac Surg. 2009;67:1074–1082. doi: 10.1016/j.joms.2008.06.079. [DOI] [PubMed] [Google Scholar]