

Abstract
The aryl hydrocarbon receptor (AHR) is a PAS-family protein that mediates the toxicity of 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) in vertebrates. Frogs are remarkably insensitive to TCDD, and AHRs from Xenopus laevis bind TCDD with low affinity. We sought to identify structural features of X. laevis AHR1β associated with low TCDD sensitivity. Substitution of the entire ligand-binding domain (LBD) with the corresponding sequence from mouse AHRb-1 dramatically increased TCDD responsiveness in transactivation assays. To identify amino acid residues responsible, we constructed a comparative model of the AHR1β LBD using homologous domains of PAS proteins HIF2α and ARNT. The model revealed an internal cavity of similar dimensions to the putative binding cavity of mouse AHRb-1, suggesting the importance of side-chain interactions over cavity size. Of residues with side chains clearly pointing into the cavity, only two differed from the mouse sequence. When A354, located within a conserved β-strand, was changed to serine, the corresponding mouse residue, the EC50 for TCDD decreased more than 15-fold. When N325 was changed to serine, EC50 declined 3-fold. When the mutations were combined, the EC50 declined from 18.6 nM to 0.8 nM, nearly matching mouse AHR for TCDD sensitivity. Velocity sedimentation analysis confirmed that mutant frog AHRs exhibited correspondingly increased TCDD binding. We also assayed mutant AHRs for responsiveness to a candidate endogenous ligand, 6-formylindolo[3,2b]carbazole (FICZ). Mutations that increased TCDD sensitivity also increased sensitivity to FICZ. This comparative study represents a novel approach to discerning fundamental information about the structure of AHR and its interactions with biologically important agonists.
Keywords: AH receptor, TCDD, homology model, ligand binding domain, Xenopus laevis
The aryl hydrocarbon receptor is a member of the Per-ARNT-Sim (PAS) family of proteins (1). A ligand activated transcription factor, the AHR binds a wide range of structurally related xenobiotics, including halogenated aromatic hydrocarbons (chlorinated dioxins, furans, and biphenyls) and polynuclear aromatic hydrocarbons, as well as several natural products, including some high affinity agonists considered candidate endogenous ligands (2). AHR exists in the cytoplasm in a complex of molecular chaperones (3). Following ligand binding, the protein enters the nucleus, forms a heterodimer with the ARNT protein, and binds specific, cis-acting regulatory elements associated with target genes (4). Subsequent changes in gene expression are thought to underlie the biological activity of AHR agonists, including the toxicity of xenobiotics. Target genes include members of the cytochrome P450 I family of Phase I detoxification enzymes (5) as well as dozens of other genes (e.g., 6, 7). Interactions with additional nuclear proteins also contribute to the biological activity of the AHR (8, 9).
Specific structural and functional properties of the AHR can play important roles in the sensitivity of animal groups to the toxic effects of xenobiotics. Low binding affinity underlies reduced TCDD toxicity in mouse strains and humans (10-12) as well as a range of bird species (13-16). These differences have been attributed to specific amino acid residues within the ligand-binding domain.
Frogs demonstrate extreme insensitivity to TCDD toxicity (17). AHR1α and AHR1β, the two AHR paralogs in Xenopus laevis (African clawed frog), bind TCDD with more than 25- fold lower affinity than mouse AHR (18). Residues previously associated with high affinity binding by mouse AHR are conserved in X. laevis, yet alternative sequence elements that confer low ligand-binding affinity to frog AHRs have not been determined. The goal of this study was to identify the sequence and structural determinants of low TCDD affinity in X. laevis AHRs. To this end, we constructed a homology model of AHR1α and AHR1β ligand binding domains using the NMR structures of PAS-B domains of Hypoxia-inducible factor (HIF) 2α and ARNT as templates (19). Comparison of these models with that of AHRb-1, encoded by the high affinity allele from mouse (11), as well as with models of other mammalian and avian AHRs guided the selection of candidate residues for mutagenesis and subsequent screening for increased TCDD responsiveness and binding. We identified two residues with side chains protruding into the putative ligand-binding cavity and demonstrated that changing them to their mouse-like counterparts substantially enhanced TCDD binding by frog AHR1β. We also demonstrated that a single mutation in a residue that controls the binding cavity characteristics of the high affinity chicken AHR (13) increased TCDD binding by the frog receptor.
EXPERIMENTAL PROCEDURES
AHR Ligands
2,3,7,8-Tetrachlorodibenzo-p-dioxin (TCDD) was obtained from Ultra Scientific (N. Kingstown, RI). 6-formylindolo[3,2b]carbazole (FICZ) was from Enzo Life Sciences (Farmingdale, NY). [1,6-3H]TCDD (33.1 Ci/mmol; >99% radiopurity) was obtained from Chemsyn (Lenexa, KS).
Construction of Chimeric AHR
To determine the general location of the residues responsible for low TCDD responsiveness, a chimeric frog AHR1β containing the mouse LBD was synthesized by Epoch Biolabs (Sugarland, TX). Residues 225-392 of AHR1β were replaced with the homologous residues from mouse AHRb-1 (230-397). The resulting construct was subcloned into pCMVTNT (Promega) at the XhoI and NotI sites. The complete sequence was verified by Sanger sequencing.
Homology Modeling
Three dimensional models of the LBD from X. laevis AHR1α (residues 271-377) and AHR1β (residues 273-379) were constructed as described previously for mouse AHR (19). Briefly, NMR structures of the aligned region of HIF2α (PDB ID 1P97) and ARNT (1X0O) were used as templates in MODELLER version 8v1 (20-22) with spatial restraints obtained from a database of protein structure alignments and CHARMM energy terms (23). Templates were structurally aligned according to DALILite (24). Sequence alignments were obtained by CLUSTALW (25), and the result confirmed by the Align-2D command within MODELLER, using the secondary structures of the AHR LBD predicted by PSIPRED (26). The optimal model among the one hundred candidates that were derived for each target was selected on the basis of the lowest value of the MODELLER objective function. Quality of the models was evaluated using MODELLER’s ENERGY scores and reliability indices obtained using PROCHECK (27) and the ProSA validation method (28). Secondary structures were attributed by DSSPcont (29). Binding cavities within the modeled LBDs were characterized using the CASTp server (30). Visualization of the models was accomplished using PYMOL (31).
Site-Directed Mutagenesis
Frog AHR1β point mutant constructs were generated using PCR-based site-directed mutagenesis (QuikChange XL, Stratagene). HPLC-purified primers (Table S1; Operon, Huntsville, AL) were designed with the Quickchange primer design program (Stratagene). Frog AHR1β in pCMVTNT was used as the template. Some mutant constructs were generated in house, while others were constructed by TopGene Technologies (Montreal, Canada). The entire sequence of all mutated open reading frames was verified by Sanger sequencing (University of Maine DNA Sequencing Facility, Orono ME).
Transactivation assays
COS-7 cells (ATCC, Manassas, VA) were maintained at 37° with 5% CO2 in Dulbecco’s modified Eagle’s medium (DMEM; Sigma) and 10% fetal bovine serum (Invitrogen). Transcriptional activity of each AHR construct was measured in reporter gene assays as described (18). Cells were co-transfected with expression constructs for each AHR, X. laevis ARNT1, the Renilla luciferase transfection control pRL-TK (Promega), and the firefly luciferase reporter pGudLuc6.1, which contains a 480 bp fragment of the upstream enhancer region of mouse CYP1A1 gene, including four XREs (32). AHRs were in the pCMVTNT plasmid (Promega), while mouse AHR (gift from Dr. C. Bradfield) and X. laevis ARNT (Open Biosystems, Huntsville, AL) were in pSPORT, all driven by the CMV promoter. 30,000 cells were plated in each well of a 48-well plate. After 24 hours, 50 ng plasmid expressing AHR1β or mouse AHR, 50 ng ARNT plasmid, 20 ng of reporter construct, and 3 ng pRL-TK were transfected into triplicate wells using Lipofectamine 2000 (Invitrogen). The total amount of transfected DNA was kept constant (300 ng) by addition of pCMVTNT plasmid containing no insert. Five hours following transfection, cells were treated with DMSO vehicle (0.5%) or graded concentrations of agonist. Cells were lysed 18 hours after dosing with TCDD or 3 hours after dosing with FICZ, which is substantially metabolized during long incubations in this cell line (33). The Dual Luciferase Assay kit (Promega, Madison, WI) and a TD 20/20 Luminometer (Turner Designs, Sunnyvale, CA) were used to lyse cells and measure luminescence. Luminescence values are given as the ratio of firefly luciferase units to Renilla luciferase units (relative luciferase units, RLU). The fractional response was then determined for each AHR at each agonist concentration by subtracting the relative luminescence of vehicle-treated cells and determining the ratio of each value to the maximal responsiveness level in the concentration-response curve (34). Graphing, non- linear regression, and statistical analyses were performed using Prism version 6.0b (GraphPad). EC50 values in dose response curves were determined by non-linear regression of the fractional response (34), constraining the background response to 0 and the maximal response to 1.
Velocity sedimentation analysis
Total TCDD binding was detected by velocity sedimentation on sucrose gradients in a vertical tube rotor using [1,6-3H]TCDD as described previously (35). Frog AHRs were synthesized in TNT reactions, diluted 1:1 in MEDMG buffer (25 mM MOPS, pH 7.5, 1 mM EDTA, 5 mM EGTA, 20 mM Na2MoO4, 0.02% NaN3, 10% glycerol 1 mM DTT), and incubated for 18 hr at 4° C with 2 nM [3H]TCDD. Nonspecific binding was determined using reactions containing an empty pCMVTNT vector [unprogrammed lysate (UPL)]. Specific binding was calculated by subtracting the radioactivity of UPL fractions from that of the corresponding fractions comprising the peak of total binding by proteins of the correct sedimentation coefficients.
Western Blotting
AHR quantity in transactivation assays and velocity sedimentation assays was monitored by western blotting. Co-transfected COS-7 cells from single wells of a 48 well plate were lysed in 10 μl of Laemmli buffer and boiled prior to SDS-PAGE. Three microliters of a TNT lysates were prepared in Laemmli buffer and incubated for 30 minutes at room temperature prior to SDS-PAGE. Following electrophoresis, samples were blotted to nitrocellulose. Blots were probed with a 1:1000 dilution of a monoclonal antibody SA210 (Enzo Life Sciences; 1 mg/ml), directed against the N-terminal half of mouse AHR (36).
RESULTS
AHRs from Xenopus laevis bind TCDD with exceptionally low affinity (18). We sought to characterize the structural basis for low TCDD affinity by screening mutants of AHR1β for the ability to activate transcription of a reporter gene driven by the mouse CYP1A1 enhancer following agonist exposure and to bind TCDD in velocity sedimentation assays.
The LBD confers low TCDD responsiveness to AHR1β
We first tested the hypothesis that the amino acid residues that confer low TCDD affinity are located within in the LBD previously characterized by deletion mutagenesis (37, 38). Replacement of the frog AHR1β LBD (a.a. 225-392) with the homologous mouse residues (a.a. 230-397) restored high TCDD responsiveness (Fig. 1). The frog:mouse chimera exhibited an EC50 nearly 100-fold lower than AHR1β (0.20 ± 0.10 nM vs. 18.6 ± 4.7 nM), roughly comparable to the value for mouse AHRb-1 (0.50 ± 0.16 nM). Intriguingly, the chimera also displayed much greater constitutive activity than either frog or mouse proteins (Fig. 1B), suggesting that the unbound mouse LBD affects the dimerization, DNA binding or transactivation domains of the frog protein in a less inhibitory fashion than the native frog sequence.
Figure 1. The Ligand Binding Domain confers low responsiveness of AHR1β to TCDD.

COS-7 cells were co-transfected with expression plasmids for an AHR, X. laevis ARNT1, pGudLuc6.1 (reporter construct), and pRL-TK (transfection control construct) as described in Materials and Methods. Cells were treated with DMSO or the indicated concentrations of TCDD, and luciferase activity was measured after 18 hours. (A) Expression of AHRs determined by western blotting. (B) Transactivation activity of AHR1β, mouse AHRb-1, and chimeric frog AHR containing the mouse LBD. RLUs are given as a ratio of firefly to Renilla luciferase activity. (C) Fractional induction. To compare responsiveness more readily, luciferase activity at each TCDD concentration was normalized to the maximum response of each AHR. Values represent the mean ± standard error for 4-7 replicate analyses.
Frog AHR homology models suggest structural determinants of low TCDD affinity
Having established that the LBD confers low TCDD responsiveness to AHR1β, we sought to more specifically identify amino acid residues that underlie this phenotype. To this end, we constructed homology models of AHR1α and AHR1β based on the solution NMR structures of the PASB regions of HIF2α (PDB ID 1P97) and ARNT (1X0O), an approach previously used to model the LBD of the mouse AHR (19) as well as AHRs from several other species (39, 40). In the modeled region (positions 271-377 for AHR1α, 273-379 for AHRβ), frog sequences share approximately 26% identity with HIF-2α and 21% identity with ARNT. For each sequence, the model with the lowest value of the MODELLER objective function was selected for the analysis and submitted to validation. Both PROCHECK and ProSA analyses indicated a good quality, similar to that obtained for the previous models (19, 39, 40). In fact, the 89% (AHR1α) and 85% (AHRβ) of residues have values of the backbone dihedral angles (ψ and φ) within the range of the most favored areas of the Ramachandran plot, the overall G-factors were -0.14 and -0.11 (for AHR1α and AHR1β respectively) and the ProSA z-scores (-3.65 and -3.98) were within the range observed in native protein structures of similar size.
Comparison of the two frog AHR models with the previously published mouse AHR model revealed striking overall structural similarity (Fig. 2A), with root-mean-square distance (RMSD) values less than 0.5 Å on the Cα atoms. The DSSPcont attribution of secondary structures (Fig. 2A-C) revealed that like the mouse AHR, each frog LBD is composed of a five-strand anti-parallel β-sheet (strands Gβ, Hβ, Iβ, Aβ, Bβ), three relatively short helices (Cα and Dα are α-helices; Eα is a 310-helix), and a long α-helical connector (Fα). Less ordered loops link these secondary structure elements. Except for small discrepancies in the connecting loops, the models suggest a high degree of similarity in the main-chain conformations between the two frog paralogs as well as with the reference mouse AHR. Therefore, basic structural differences cannot underlie the species-specific TCDD-binding phenotypes of the AHRs.
Figure 2. Homology model for the frog AHR1β LBD.

(A) Comparison of cartoon renderings for modeled LBDs of frog and mouse AHR. Green, mouse AHR; cyan, frog AHR1α; orange, frog AHR1β. Black labels indicate the conserved secondary structure elements attributed by DSSPcont. (B) Cartoon representation of the modeled AHR1β LBD. Residues highlighted in red (N325 and A354) represent positions that differ between frog and mouse and protrude into the modeled binding cavity. These were mutated individually and in combination in these studies. Residues highlighted in blue differ between frog and mouse, but they do not point into the binding cavity. The residue highlighted in green (A370) plays an important role in determining the binding affinity of human, mouse, and bird AHRs. The light green shaded area indicates the molecular surface of the binding cavity of frog AHR1β identified by CASTp. The numbering of the three mutated residues in all the indicated AHR sequences is reported in the scheme for comparative purposes. (C) Sequence alignment of the indicated AHR LBDs, obtained by ClustalW. Only the residues that differ from the reference mouse AHR sequence are shown, while dots indicate the conserved positions. Variable residues are boxed, using the same color scheme as in panel B. Secondary structures attributed to the homology models by DSSPcont are indicated below: light grey bars for helices and dark grey bars for β-strands.
Since changes in internal cavity volume of the AHR LBD can dramatically decrease or alter the specificity of AHR ligand binding (40, 41) and responsiveness in transactivation assays (16), we compared the internal cavity space available for binding in frog and mouse AHRs using CASTp analysis. The cavities have similar predicted volumes of about 500Å3. Both the size and the cavity shape (Fig. 2B) are similar to those previously predicted for mouse AHR and for other mammalian and avian AHRs with high affinity for TCDD (19, 39, 40). Thus, the low TCDD affinity of the X. laevis AHRs cannot be explained by a reduced internal space available for the ligand but rather by differences in the interactions of TCDD with specific amino acid residues.
The frog AHRs have 23 (AHR1α) or 22 (AHR1β) residues that differ from the corresponding positions in the mouse AHR LBD (Fig. 2C; blue and red boxes). Of these, only two have side chains that point unambiguously toward the interior of the modeled binding cavity (N325 and A354; Fig. 2B, C; red), while the remainder protrude outward from the external surface (Fig. 2B, C; blue). We hypothesized that altering these two residues to make them more mouse-like would enhance TCDD responsiveness of frog AHRs, presumably through increased affinity.
Also notable in the alignment (Fig. 2C) and model (Fig. 2B) is the presence of alanine at position 370 of AHR1β (368 of AHR1α; green). The residue at the corresponding position belongs to the “TCDD-binding fingerprint” for mammalian AHRs (39) and underlies the difference in TCDD binding between the high affinity mouse AHRb-1 allele (containing alanine at this position) and the lower affinity mouse AHRd and human AHR (10-12) (valine). This position also plays a significant role in the TCDD affinity of bird AHRs. For example, AHR from the common tern (Sterna hirundo, a sea bird) contains alanine and binds TCDD with 7-fold lower affinity than AHR from chicken, which contains a serine (13). We hypothesized (40) that optimal TCDD binding requires the presence of a polar residue (e.g. serine) in the region defined by the faced residue pair S359-A375 of mouse AHR or A364-S380 of chicken AHR (Fig. 2B). TCDD binding with the tern AHR (A365-A381) lacks this stabilizing effect. Like the tern receptor, both frog AHRs have two faced alanine residues at these positions (A354 and A370 in the frog AHR1β, see Fig. 2B). These observations suggest that, in addition to making frog AHR1β more mouse-like by changing A354 to serine, making it more chicken-like by changing A370 to serine would increase its TCDD binding and responsiveness.
TCDD responsiveness of AHR mutants
To test the efficacy of the targeted mutations suggested by our homology models, we inserted point mutations within AHR1β to make it more strongly resemble the high affinity AHRs from mouse or chicken. Each mutant AHR was screened for TCDD responsiveness (i.e. sensitivity) in transient transfection transactivation assays. As predicted, each mutation at least partially restored the high affinity phenotype (Fig. 3; EC50s in Table 1). Comparison of EC50 values from TCDD-concentration response analysis revealed that N325S exhibited 3.2-fold greater TCDD responsiveness, while A354S and A370S mutant AHRs were 15.5- and 18.6-fold more responsive, respectively (Table 1). When both N325 and A354 were mutated to serine to resemble mouse AHRb-1, the resulting mutant AHR was nearly 23-fold more responsive to TCDD. Remarkably, while the LBDs differed in 22 of 168 modeled positions, alteration of only the two residues predicted to protrude into the modeled binding cavity was necessary to dramatically increase the potency of TCDD in transactivation assays. In fact, the internal cavity of the double mutant (N325S/A354S) reproduces almost completely the characteristics of the mouse AHRb-1 cavity, and the A370S mutation reproduces the residues characteristic of the chicken AHR binding cavity (40). Although relative luciferase expression varied somewhat between replicate experiments, we did not observe systematic differences in the efficacy of the response between individual AHRs within an individual experiment (data not shown).
Figure 3. Mutation of specific LBD residues increases TCDD responsiveness of AHR1β.

(A) Expression of each AHR in transfected COS-7 cells determined by western blotting. (B) Fractional induction of reporter gene expression by each AHR following TCDD exposure as described in figure legend 1. Values represent the mean ± standard error for 3-9 replicates.
Table 1.
EC50 (nM) values for reporter gene induction by TCDD and FICZ in COS-7 cells transfected with mouse or frog AHRs bearing the indicated mutations.
| AHR1β | N325S | A354S | A370S | A354S/N325S | chimera | mouse | |
|---|---|---|---|---|---|---|---|
| TCDD | 18.6 ± 4.7a | 5.7 ± 1.6 | 1.2 ± 0.4 | 1.0± 0.4 | 0.82± 0.42 | 0.20 ± 0.10 | 0.50± 0.16 |
| (24 hr) | |||||||
| FICZ | 0.08 ± | 0.07 ± | 0.04 ± | 0.04 ± | 0.03 ± 0.01 | ||
| 0.02 | 0.02 | 0.01 | 0.01 | ||||
| (3 hr) |
Values represent mean ± standard error for 3-9 replicates.
We also tested the effect of amino acid changes at residues with side chains that protrude outside of the modeled binding cavity. While it is possible that the architecture of the pocket could be affected in a functionally important way by complex interactions of these side chains with other parts of the AHR or its associated protein subunits, the simplest hypothesis suggests that these residues would not affect TCDD responsiveness because they do not have the potential to directly alter contact with the ligand. We screened several mutants for the ability to increase TCDD responsiveness, including T256A and S263P (outside the modeled domain); S294Q (Cα); the combination of R345K, D346H, T347S, and G348R (the connecting loop between Gβ and Hβ); and S371T (Iβ). As predicted, none of these mutants exhibited significantly greater TCDD responsiveness than wild type AHR1β (data not shown; EC50s summarized in Table 2).
Table 2.
EC50 (nM) values for reporter gene induction by TCDD. COS-7 cells were transfected with mouse or frog AHRs bearing the indicated mutations. Standard error is indicated for each mean. n=3-4 for each AHR (except where indicated)
| T256A | S263P | T256A/S263P | S371T | S294Q | KHSRb | |
|---|---|---|---|---|---|---|
| TCDD | 23.1 ± | 23.7 ±8.8 | 20.6 ± 8.0 | 13.5 ±3.3 | 25.9 ± 1.1 | 23.0 |
| 8.9a | ||||||
| (24 hr) |
Values represent mean ± standard error for 3-9 replicates; n=1 for KHSR.
KHSR represents a combination of four mutations: R345K, D346H, T347S, and G348R
Implicit in the hypotheses suggested by our models is the idea that residues protruding into the modeled cavity affect TCDD responsiveness by altering the ability of each mutant receptor to bind TCDD. This is consistent with the well-demonstrated low TCDD affinity of frog AHRs (18). To test this hypothesis, we measured TCDD binding by mutant AHRs directly using velocity sedimentation analysis on sucrose density gradients following incubation with 2 nM [3H]-TCDD, a concentration that will saturate TCDD binding by mouse or chimeric AHR but only partially occupy frog AHR1β. As predicted, the chimeric AHR exhibited the greatest TCDD binding, while AHR1β binding was barely detectable (Fig. 4). Mutant AHRs showed increasing ability to bind TCDD, with binding to the A354S/N325S mutant approaching that of the chimera. Somewhat contrary to the prediction of the transactivation assays, N325S AHR demonstrated greater TCDD binding than either A354S or A370S, a relationship that could not be readily explained by the amount of AHR used in each binding assay (Fig. 4A). Overall, however, these results are consistent with the hypothesis that the increased TCDD responsiveness observed in these mutant results primarily from heightened affinity for TCDD.
Figure 4. Mutation of specific LBD residues increases TCDD binding of AHR1β.

(A) Expression of each AHR in TNT reactions determined by western blotting. (B) Velocity sedimentation analysis of TCDD binding. Synthetic AHR proteins or unprogrammed TNT lysates were incubated with 2 nM [3H]TCDD and fractionated on sucrose density gradients. Sedimentation marker [14C]catalase (11.3S) eluted in fractions 14-20 of all gradients. The experiment was replicated three times; a single example is shown. (C) Quantification of TCDD specific binding revealed by sedimentation analysis. Radioactivity (dpm) in fractions comprising each peak in (B) was summed. Specific binding is the difference between total binding (preparations containing an AHR) and non-specific binding (preparation lacking AHR). Bar graph plots specific binding relative to that found for AHR1β. Values represent the mean ± standard error for 3 replicates.
FICZ responsiveness of AHR mutants
FICZ is a candidate endogenous AHR ligand that displays greater potency than TCDD with human and rodent AHRs (42). Our previous studies demonstrated that although frog AHR is less responsive to FICZ than mouse AHR, the difference is much less striking than for TCDD (33). Thus, the molecular interactions of FICZ and TCDD with the LBD may differ. We tested the hypothesis that the mouse AHR residues that restore TCDD responsiveness to the frog receptor also affect FICZ potency. The chimeric AHR bearing the mouse LBD was 2.7-fold more responsive to FICZ than AHR1β (EC50 of 0.03 ± 0.01 nM vs. 0.08 ± 0.02 nM; Fig. 5 and Table 1). N325S and A370S mutations each increased FICZ responsiveness about 2-fold, while A354S restored responsiveness to a level comparable to the chimera (Fig. 5 and Table 1). Once again, systematic differences in the efficacy of the response between individual AHRs within an individual experiment were not evident (data not shown).
Figure 5. Mutation of specific LBD residues increases FICZ responsiveness of AHR1β.
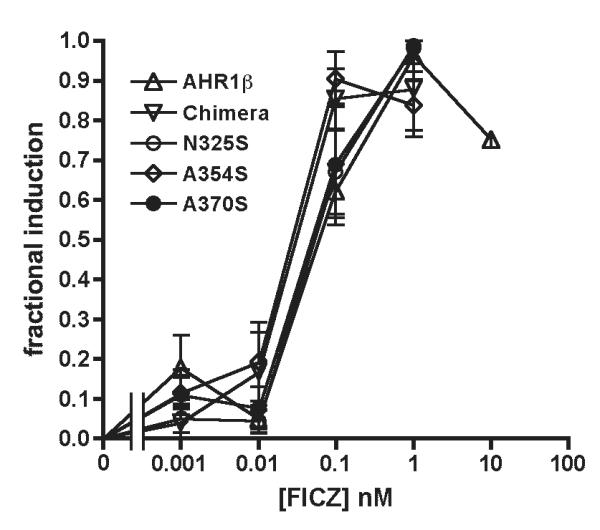
Transactivation assays with the indicated AHRs were performed as described in the legend to Figure 1 following 3-hr exposure to indicated concentrations of FICZ. Graph depicts fractional induction of luciferase reporter gene activity. The response at each FICZ concentration was normalized to the maximum response for each AHR. Values are expressed as the mean ± standard error of 3-4 replicate analyses.
DISCUSSION
In this study we used a homology model to guide identification of candidate amino acid residues responsible for low TCDD affinity of X. laevis AHRs. The frog AHR models generated here share substantial structural similarity with the models of mammalian AHRs published previously by some of us (19, 39, 40) as well as other groups. The mouse AHR LBD structure modeled by Bisson et al. (43) on the basis of the same HIF2α template shows good overall agreement with the number, position, and lengths of secondary structure elements. A recently published model by Xing et al. (44) used THS017-bound HIF2α (3H7W) to model the same region by using an iterative threading, TCDD docking, and energy minimization protocol. This model describes a “flexible belt” structure corresponding to the region that includes the Dα and Eα helices common to other AHR LBD models (residues 307-322 of mouse AHR) and a bifurcation of Gβ into two smaller, successive β-strands.
Of the 22-23 LBD residues that differ between mouse and frog AHRs, our model predicts that only two protrude into the putative ligand binding cavity to potentially affect ligand binding directly: N325 on the Fα helix and A354 on the Hβ strand (Fig. 2B). We show here that both of these positions affect the ligand binding affinity of frog AHR1β. N325 corresponds to S330 in the mouse AHR. A variable position, its role in ligand binding has not been extensively investigated previously. The importance of position 354 is highlighted in recently published AHR LBD models, although different ideas are proposed for the role of this residue in conjunction with AHR agonists. Motto et al. (45) demonstrated that S359A in mouse AHR, the reciprocal substitution of our experiments (A354S in frog AHR1β), reduced TCDD binding by 40%. Xing et al. (44) proposed that this serine is involved in a structurally important hydrogen bond network with Q377 and the medial oxygen of TCDD. This model implies that the absence of hydrogen bonding potential between A354 and Q372 of frog AHR1β would be consistent with low TCDD affinity. However, Xing et al.’s model also predicts a relative increase in benzo[a]pyrene affinity to result from this configuration. That prediction does not fit observations of PAH insensitivity of frogs (46) and the extremely low potency of benzo[a]pyrene for CYP1A6 induction in X. laevis cell line XLK-WG (E. Engelbrecht and W. Powell, unpublished observation). The mouse AHR model of Bisson et al. (43) emphasizes the importance of this position to FICZ binding, predicting that the hydroxyl group of S359 makes direct contact with FICZ as a hydrogen bond donor to the carbonyl oxygen. In the present study, the A354S mutation in frog AHR1β enhanced FICZ sensitivity to the level of the frog:mouse chimeric AHR, confirming the importance of this residue in FICZ agonism. An additional hydrogen bonding interaction was predicted to involve the Q377 side chain of mouse AHR and the FICZ carbazole group (43). Glutamine (Q372) is conserved at the corresponding position in frog AHR1β.
The position homologous to 370 of frog AHR1β (an alanine) is well documented to underlie strain and species differences in TCDD binding by AHRs and TCDD sensitivity in both mammals and birds. While the mouse AHRb-1 allele and other high affinity mammalian AHRs contain alanine at this position (A375 in mouse AHR), the lower affinity mouse AHRd and human AHR contain valine (10-12). The bulkier valine side chain is thought to preclude high affinity TCDD binding by affecting ligand interactions with other closely associated amino acids (19, 39, 40). The presence of alanine at this position in the frog AHRs suggests that the structural basis for low TCDD affinity differs from the classical examples involving mouse and human AHRs. On the other hand, the positive effect of our more chicken-like mutation (A370S) on TCDD binding of AHR1β confirms the Val<Ala<Ser series of increasing TCDD interactions with AHR at this position first hypothesized by Karchner et al. (13).
Taken together, our results and previously published models suggest that the presence of a serine at either position 354 (in Hβ; resembling S359 in mouse AHR) or 370 (in Iβ; resembling S380 in chicken AHR) is important for high affinity ligand binding. This effect is consistent with the hypothesis that optimal electrostatic interactions with the TCDD oxygen atoms require the presence of one polar residue in the region defined by this faced residue pair (40). However, the presence of alanines at both positions is not necessarily sufficient to confer low TCDD affinity to all AHRs. Examination of a wide range of AHR sequences reveals that some fish AHRs bearing two alanines [e.g. Fundulus heteroclitus AHR1 (35) and zebrafish AHR2 (47)] can bind TCDD with relatively high affinity that corresponds to much higher sensitivity to TCDD toxicity than frogs. Fraccalvieri et al. (40) recently observed that while cavities of the high affinity mammalian and avian AHRs are very similar in shape and size and share internal conserved residues, the cavities of high affinity fish AHRs are bordered by a larger number of internal residues that define a more elongated shape. This distinct cavity architecture suggests a different TCDD binding mode for fish AHRs that precludes a direct, sequence-based comparison with the tetrapod orthologs.
Pandini et al. (39) identified a “TCDD-binding fingerprint”—a collection of amino acids that is well conserved in several high affinity AHRs. These residues are identical in the low affinity frog receptors. We conclude that in addition to the invariant signature residues, additional positions contribute to the physicochemical nature of the frog AHR binding cavity in ways that affect the interaction with individual ligands. Variability at these positions could ultimately underlie the efficacy of specific agonists and influence the sensitivity of different groups of animals to their biological effects. The comparative approach exemplified by this study offers a logical framework for discerning insights into the architecture of the AHR. Functionally important differences in frog and other diverse AHR sequences represent the natural equivalent of mutations identified in phenotypic screens. Knowledge of the biochemical, developmental, physiological, and toxicological effects of such sequence differences will provide important benchmarks for testing the validity of AHR homology models and crystal structures that will undoubtedly become available in the future.
Supplementary Material
ACKNOWLEDGEMENTS
This work was funded by the National Institute of Environmental Health Sciences: ES011130 (WHP), ES07685 (MSD), ES006272 (MEH), and the Kenyon College Summer Science Scholars program.
Funding Sources This work was funded by the National Institute of Environmental Health Sciences: ES011130 (WHP), ES07685 (MSD), ES006272 (MEH), and the Kenyon College Summer Science Scholars program.
ABBREVIATIONS
- AHR
aryl hydrocarbon receptor
- DMSO
dimethylsulfoxide
- DTT
ditiothreitol
- EDTA
Ethylenediaminetetraacetic acid
- EGTA
ethylene glycol tetraacetic acid
- FICZ
6-formylindolo[3,2b]carbazole
- HIF
hypoxia-inducible factor
- LBD
ligand binding domain
- MEDMG
25 mM MOPS, pH 7.5, 1 mM EDTA, 5 mM EGTA, 20 mM Na2MoO4, 0.02% NaN3, 1
- mM DTT
10% glycerol
- PAS
Per-ARNT-Sim
- TCDD
2,3,7,8-tetrachlorodibenzo-p-dioxin
- UPL
unprogrammed lysate
Footnotes
SUPPLEMENTAL INFORMATION Supplemental materials may be accessed free of charge online at http://pubs.acs.org.
REFERENCES
- 1.McIntosh BE, Hogenesch JB, Bradfield CA. Mammalian Per-Arnt-Sim proteins in environmental adaptation. Annu Rev Physiol. 2010;72:625–645. doi: 10.1146/annurev-physiol-021909-135922. [DOI] [PubMed] [Google Scholar]
- 2.Denison MS, Nagy SR. Activation of the aryl hydrocarbon receptor by structurally diverse exogenous and endogenous chemicals. Annu Rev Pharmacol Toxicol. 2003;43:309–334. doi: 10.1146/annurev.pharmtox.43.100901.135828. [DOI] [PubMed] [Google Scholar]
- 3.Petrulis JR, Perdew GH. The role of chaperone proteins in the aryl hydrocarbon receptor core complex. Chem Biol Interact. 2002;141:25–40. doi: 10.1016/s0009-2797(02)00064-9. [DOI] [PubMed] [Google Scholar]
- 4.Beischlag TV, Luis Morales J, Hollingshead BD, Perdew GH. The aryl hydrocarbon receptor complex and the control of gene expression. Crit Rev Eukaryot Gene Expr. 2008;18:207–250. doi: 10.1615/critreveukargeneexpr.v18.i3.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nebert DW, Roe AL, Dieter MZ, Solis WA, Yang Y, Dalton TP. Role of the aromatic hydrocarbon receptor and [Ah] gene battery in the oxidative stress response, cell cycle control, and apoptosis. Biochem Pharmacol. 2000;59:65–85. doi: 10.1016/s0006-2952(99)00310-x. [DOI] [PubMed] [Google Scholar]
- 6.Frueh FW, Hayashibara KC, Brown PO, Whitlock JPJ. Use of cDNA microarrays to analyze dioxin-induced changes in human liver gene expression. Toxicol Lett. 2001;122:189–203. doi: 10.1016/s0378-4274(01)00364-2. [DOI] [PubMed] [Google Scholar]
- 7.Puga A, Maier A, Medvedovic M. The transcriptional signature of dioxin in human hepatoma HepG2 cells. Biochem Pharmacol. 2000;60:1129–1142. doi: 10.1016/s0006-2952(00)00403-2. [DOI] [PubMed] [Google Scholar]
- 8.Carlson DB, Perdew GH. A dynamic role for the Ah receptor in cell signaling? Insights from a diverse group of Ah receptor interacting proteins. J Biochem Mol Toxicol. 2002;16:317–325. doi: 10.1002/jbt.10051. [DOI] [PubMed] [Google Scholar]
- 9.Puga A, Tomlinson CR, Xia Y. Ah receptor signals cross-talk with multiple developmental pathways. Biochem Pharmacol. 2005;69:199–207. doi: 10.1016/j.bcp.2004.06.043. [DOI] [PubMed] [Google Scholar]
- 10.Ema M, Ohe N, Suzuki M, Mimura J, Sogawa K, Ikawa S, Fujii-Kuriyama Y. Dioxin binding activities of polymorphic forms of mouse and human aryl hydrocarbon receptors. J Bioll Chem. 1994;269:27337–27343. [PubMed] [Google Scholar]
- 11.Poland A, Palen D, Glover E. Analysis of the four alleles of the murine aryl hydrocarbon receptor. Mol Pharmacol. 1994;46:915–921. [PubMed] [Google Scholar]
- 12.Ramadoss P, Perdew GH. Use of 2-azido-3-[125I]iodo-7,8-dibromodibenzo-p-dioxin as a probe to determine the relative ligand affinity of human versus mouse aryl hydrocarbon receptor in cultured cells. Mol Pharmacol. 2004;66:129–136. doi: 10.1124/mol.66.1.129. [DOI] [PubMed] [Google Scholar]
- 13.Karchner SI, Franks DG, Kennedy SW, Hahn ME. The molecular basis for differential dioxin sensitivity in birds: role of the aryl hydrocarbon receptor. Proc Natl Acad Sci USA. 2006;103:6252–6257. doi: 10.1073/pnas.0509950103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Head JA, Hahn ME, Kennedy SW. Key amino acids in the aryl hydrocarbon receptor predict dioxin sensitivity in avian species. Environ Sci Technol. 2008;42:7535–7541. doi: 10.1021/es801082a. [DOI] [PubMed] [Google Scholar]
- 15.Farmahin R, Wu D, Crump D, Herve JC, Jones SP, Hahn ME, Karchner SI, Giesy JP, Bursian SJ, Zwiernik MJ, Kennedy SW. Sequence and in vitro function of chicken, ring-necked pheasant, and Japanese quail AHR1 predict in vivo sensitivity to dioxins. Environ Sci Technol. 2012;46:2967–2975. doi: 10.1021/es2043992. [DOI] [PubMed] [Google Scholar]
- 16.Farmahin R, Manning GE, Crump D, Wu D, Mundy LJ, Jones SP, Hahn ME, Karchner SI, Giesy JP, Bursian SJ, Zwiernik MJ, Fredricks TB, Kennedy SW. Amino Acid sequence of the ligand-binding domain of the aryl hydrocarbon receptor 1 predicts sensitivity of wild birds to effects of dioxin-like compounds. Toxicol Sci. 2012;131:139–152. doi: 10.1093/toxsci/kfs259. [DOI] [PubMed] [Google Scholar]
- 17.Jung RE, Walker MK. Effects of 2,3,7,8-Tetrachlorodibenzo-p-dioxin (TCDD) on development of anuran amphibians. Environ Toxicol Chem. 1997;16:230–240. [Google Scholar]
- 18.Lavine JA, Rowatt AJ, Klimova T, Whitington AJ, Dengler E, Beck C, Powell WH. Aryl hydrocarbon receptors in the frog Xenopus laevis: two AhR1 paralogs exhibit low affinity for 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) Toxicol Sci. 2005;88:60–72. doi: 10.1093/toxsci/kfi228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pandini A, Denison MS, Song Y, Soshilov AA, Bonati L. Structural and functional characterization of the aryl hydrocarbon receptor ligand binding domain by homology modeling and mutational analysis. Biochemistry. 2007;46:696–708. doi: 10.1021/bi061460t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sali A, Blundell TL. Comparative protein modelling by satisfaction of spatial restraints. J Mol Biol. 1993;234:779–815. doi: 10.1006/jmbi.1993.1626. [DOI] [PubMed] [Google Scholar]
- 21.Marti-Renom MA, Stuart AC, Fiser A, Sanchez R, Melo F, Sali A. Comparative protein structure modeling of genes and genomes. Annu Rev Biophys Biomol Struct. 2000;29:291–325. doi: 10.1146/annurev.biophys.29.1.291. [DOI] [PubMed] [Google Scholar]
- 22.Fiser A, Do RK, Sali A. Modeling of loops in protein structures. Protein Sci. 2000;9:1753–1773. doi: 10.1110/ps.9.9.1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.MacKerell AD, Bashford D, Bellott M, Dunbrack RL, Evanseck JD, Field MJ, Fischer S, Gao J, Guo H, Ha S, Joseph-McCarthy D, Kuchnir L, Kuczera K, Lau FTK, Mattos C, Michnick S, Ngo T, Nguyen DT, Prodhom B, Reiher WE, Roux B, Schlenkrich M, Smith JC, Stote R, Straub J, Watanabe M, Wiorkiewicz-Kuczera J, Yin D, Karplus M. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J Phys Chem B. 1998;102:3586–3616. doi: 10.1021/jp973084f. [DOI] [PubMed] [Google Scholar]
- 24.Holm L, Park J. DaliLite workbench for protein structure comparison. Bioinformatics. 2000;16:566–567. doi: 10.1093/bioinformatics/16.6.566. [DOI] [PubMed] [Google Scholar]
- 25.Thompson JD, Higgins DG, Gibson TJ. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignments through sequence weighting, position-specific gap penalties and weight matrix choice. Nucl Acids Res. 1994;22:4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jones DT. Protein secondary structure prediction based on position-specific scoring matrices. J Mol Biol. 1999;292:195–202. doi: 10.1006/jmbi.1999.3091. [DOI] [PubMed] [Google Scholar]
- 27.Laskowski RA, Macarthur MW, Moss DS, Thornton JM. Procheck - a Program to Check the Stereochemical Quality of Protein Structures. J Appl Crystallogr. 1993;26:283–291. [Google Scholar]
- 28.Sippl MJ. Recognition of Errors in 3-Dimensional Structures of Proteins. Proteins. 1993;17:355–362. doi: 10.1002/prot.340170404. [DOI] [PubMed] [Google Scholar]
- 29.Andersen CA, Palmer AG, Brunak S, Rost B. Continuum secondary structure captures protein flexibility. Structure. 2002;10:175–184. doi: 10.1016/s0969-2126(02)00700-1. [DOI] [PubMed] [Google Scholar]
- 30.Dundas J, Ouyang Z, Tseng J, Binkowski A, Turpaz Y, Liang J. CASTp: computed atlas of surface topography of proteins with structural and topographical mapping of functionally annotated residues. Nucl Acids Res. 2006;34:W116–W118. doi: 10.1093/nar/gkl282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.The PyMOL Molecular Graphics System Version 1.3. LLC; Schrodinger: [Google Scholar]
- 32.Long WP, Pray-Grant M, Tsai JC, Perdew GH. Protein kinase C activity is required for aryl hydrocarbon receptor pathway-mediated signal transduction. Mol Pharmacol. 1998;53:691–700. doi: 10.1124/mol.53.4.691. [DOI] [PubMed] [Google Scholar]
- 33.Laub LB, Jones BD, Powell WH. Responsiveness of a Xenopus laevis cell line to the aryl hydrocarbon receptor ligands 6-formylindolo[3,2-b]carbazole (FICZ) and 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) Chem Biol Interact. 2010;183:202–211. doi: 10.1016/j.cbi.2009.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Poland A, Glover E. Genetic expression of aryl hydrocarbon hydroxylase by 2,3,7,8-tetrachlorodibenzo-p-dioxin: evidence for a receptor mutation in genetically non-responsive mice. Mol Pharmacol. 1975;11:389–398. [Google Scholar]
- 35.Karchner SI, Powell WH, Hahn ME. Structural and Functional Characterization of Two Highly Divergent Aryl Hydrocarbon Receptors (AHR1 and AHR2) in the teleost Fundulus heteroclitus. Evidence for a novel class of ligand-binding basic helix-loop-helix Per-ARNT-Sim (bHLH-PAS) factors. J Biol Chem. 1999;274:33814–33824. doi: 10.1074/jbc.274.47.33814. [DOI] [PubMed] [Google Scholar]
- 36.Pollenz RS, Sattler CA, Poland A. The aryl hydrocarbon receptor and aryl hydrocarbon receptor nuclear translocator protein show distinct subcellular localizations in Hepa 1c1c7 cells by immunofluorescence. Mol Pharmacol. 1994;45:428–438. [PubMed] [Google Scholar]
- 37.Fukunaga BN, Probst MR, Reiszporszasz S, Hankinson O. Identification of functional domains of the aryl hydrocarbon receptor. J Biol Chem. 1995;270:29270–29278. doi: 10.1074/jbc.270.49.29270. [DOI] [PubMed] [Google Scholar]
- 38.Coumailleau P, Poellinger L, Gustafsson J-A, Whitelaw ML. Definition of a minimal domain of the dioxin receptor that is associated with hsp90 and maintains wild type ligand binding affinity and specificity. J Biol Chem. 1995;270:25291–25300. doi: 10.1074/jbc.270.42.25291. [DOI] [PubMed] [Google Scholar]
- 39.Pandini A, Soshilov AA, Song Y, Zhao J, Bonati L, Denison MS. Detection of the TCDD binding-fingerprint within the Ah receptor ligand binding domain by structurally driven mutagenesis and functional analysis. Biochemistry. 2009;48:5972–5983. doi: 10.1021/bi900259z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fraccalvieri D, Soshilov AA, Karchner SI, Franks DG, Pandini A, Bonati L, Hahn ME, Denison MS. Comparative analysis of homology models of the Ah receptor ligand binding domain: verification of structure-function predictions by site-directed mutagenesis of a non-functional AHR. Biochemistry. 2013;52:714–425. doi: 10.1021/bi301457f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Goodale BC, La Du JK, Bisson WH, Janszen DB, Waters KM, Tanguay RL. AHR2 mutant reveals functional diversity of aryl hydrocarbon receptors in zebrafish. PLoS One. 2012;7:e29346. doi: 10.1371/journal.pone.0029346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wincent E, Amini N, Luecke S, Glatt H, Bergman J, Crescenzi C, Rannug A, Rannug U. The suggested physiologic aryl hydrocarbon receptor activator and cytochrome P4501 substrate 6-formylindolo[3,2-b]carbazole is present in humans. J Biol Chem. 2009;284:2690–2696. doi: 10.1074/jbc.M808321200. [DOI] [PubMed] [Google Scholar]
- 43.Bisson WH, Koch DC, O’Donnell EF, Khalil SM, Kerkvliet NI, Tanguay RL, Abagyan R, Kolluri SK. Modeling of the aryl hydrocarbon receptor (AhR) ligand binding domain and its utility in virtual ligand screening to predict new AhR ligands. J Med Chem. 2009;52:5635–5641. doi: 10.1021/jm900199u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Xing Y, Nukaya M, Satyshur KA, Jiang L, Stanevich V, Korkmaz EN, Burdette L, Kennedy GD, Cui Q, Bradfield CA. Identification of the ah-receptor structural determinants for ligand preferences. Toxicol Sci. 2012;129:86–97. doi: 10.1093/toxsci/kfs194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Motto I, Bordogna A, Soshilov AA, Denison MS, Bonati L. New aryl hydrocarbon receptor homology model targeted to improve docking reliability. J Chem Inf Model. 2011;51:2868–2881. doi: 10.1021/ci2001617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bantle JA. FETAX--A developmental toxicity assay using frog embryos. In: Rand GM, editor. Fundamentals of aquatic toxicology. Taylor and Francis; Washington, DC: 1996. pp. 207–230. [Google Scholar]
- 47.Tanguay RL, Abnet CC, Heideman W, Peterson RE. Cloning and characterization of the zebrafish (Danio rerio) aryl hydrocarbon receptor. Biochim Biophys Acta (Gene Struct Express) 1999;1444:35–48. doi: 10.1016/s0167-4781(98)00252-8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.