

Abstract
The delta opioid receptor (DOR) agonist DPDPE decreases ethanol (EtOH) consumption when injected into the ventral tegmental area (VTA). We previously showed that DPDPE inhibition of GABAA receptor-mediated inhibitory postsynaptic currents (GABAAR IPSCs) is associated with reduced EtOH consumption. To determine whether self-administration of EtOH is required to change VTA DOR function, we compared the effects of passively administered (gavaged) and self-administered (two-bottle choice) EtOH. Because rats showed variability in DOR regulation of drinking and inhibition of GABAAR IPSCs, we examined whether these changes can be predicted before EtOH exposure by behavioral measures of anxiety or intoxication. Functional DORs were seen with both gavaged and self-administered EtOH, although the magnitude of DPDPE-induced inhibition correlated with behavioral measures only when EtOH was self-administered. Specifically, DPDPE-induced inhibition correlated with predrinking measures of open arm time in the plus maze (n = 19, R = .69, p = .001), with change in maximum fall latency on the rotarod (n = 17, R = .89, p = .000001), and with blood corticosterone (n = 17, R = .66, p = .004) in drinking animals. This DOR-mediated inhibition persisted for at least 14 days after EtOH access was terminated. Together, these findings indicate that anxious animals and those with the greatest EtOH-induced motor impairment have the most robust DPDPE-induced inhibition of GABAAR IPSCs in VTA neurons. These data also extend our understanding of the possible therapeutic value of the DOR for treatment of alcoholism by showing that its relevant synaptic action persists during abstinence.
Keywords: ethanol, self-administration, gavage, delta opioid receptor, opioid
The ventral tegmental area (VTA) regulates alcohol consumption in rodents. Alcohol-preferring rats self-administer ethanol (EtOH) (McBride et al., 1993) and its primary metabolite acetaldehyde (Rodd-Henricks et al., 2002) directly into the VTA and VTA cFOS expression increases after exposure to an EtOH-associated context (Hill, Ryabinin, & Cunningham, 2007). Opioid actions in the VTA are implicated in the regulation of EtOH consumption (McBride et al., 1991), and we have previously reported that both intra-VTA naltrexone (Mitchell, Bergren, Chen, Rowbotham, & Fields, 2009) and the mu opioid receptor (MOR) selective antagonist CTOP can reduce alcohol consumption in Lewis rats (Margolis, Fields, Hjelmstad, & Mitchell, 2008). Furthermore, intra-VTA administration of an opioid antagonist attenuates EtOH place preference in the mouse (Bechtholt & Cunningham, 2005). In contrast to the promotion of EtOH consumption by mu opioids, VTA microinjection of the delta opioid receptor (DOR) selective agonist DPDPE decreases EtOH consumption (two-bottle choice; 10% vol/vol) in chronically drinking animals (Margolis et al., 2008). Importantly, DPDPE-induced inhibition of GABA release onto VTA neurons is inversely correlated with levels of EtOH consumption in chronically drinking animals. Conversely, VTA infusion of the DOR selective antagonist TIPP-Ψ increases drinking, and this effect is blocked by VTA pretreatment with the GABAA receptor (GABAAR) antagonist bicuculline (Margolis et al., 2008), indicating that DOR agonists in the VTA reduce EtOH consumption by inhibiting GABAergic terminals.
To extend our understanding of the role of the DOR in inhibiting EtOH consumption, we studied the time course of the functional expression of the DOR during initial exposure to EtOH. We tested the hypothesis that passively administered EtOH through intraoral gavage would show effects similar to EtOH self-administration, that is, that EtOH exposure rather than the act of drinking was the critical factor in changing DOR function. We used slice electrophysiology to measure the function of DOR on GABAAR-mediated inhibitory postsynaptic currents (IPSCs) at different time points after EtOH exposure. We also prescreened rats for anxiety (plus maze) and EtOH-induced motor impairment (rotarod), two behaviors that are thought to correlate with potential EtOH consumption (Bacopoulos, Bize, Levine, & Van Orden, 1979; Margolis et al., 2008), in an attempt to determine whether it was possible to predict future EtOH consumption levels and amount of DPDPE-induced inhibition of GABAAR IPSCs in VTA neurons.
Method
Animals
Forty-nine male Lewis rats (Harlan Laboratories, Hayward, CA) weighing between 275 and 300 g on arrival were housed individually in a temperature controlled colony room (21°C) on a 12-hr reversed light/dark cycle (lights off at 10 a.m.). All experiments were performed during the dark portion of the cycle. Rat chow and water were available ad libitum. During EtOH self-administration periods (see below) 10% EtOH (vol/vol Gold Shield, Hayward, CA) was also available ad libitum. Experiments were conducted in accordance with the Guide for the Care and Use of Laboratory Animals (NIH) and were approved by the EGCRC Committee on Animal Research. Animals were never food- or water-deprived.
EtOH Self-Administration
EtOH was administered via a two-bottle continuous access, free-choice paradigm in which one bottle contained 10% EtOH (vol/vol) and the other bottle contained water. Sucrose was never added to the EtOH solution. Animals were weighed daily, and the amount of EtOH and water consumed was measured at the same time daily (10 a.m.). Bottles were identical and their positions were counterbalanced and rotated daily. Self-administration was permitted for either 3, 7, or 14 days before electrophysiological recordings. For abstinence studies, animals were continued on the two-bottle choice paradigm until consumption had stabilized (at least 4 months) and EtOH bottles were then removed for 3, 7, or 14 days before electrophysiological recordings.
EtOH Gavage
Animals were lightly anesthetized with isoflurane gas and then intraorally gavaged with 1 mL of 30% vol/vol (approximately 1.1 g/kg) EtOH daily for either 3, 7, or 14 days. Gavaging was conducted at the same time each day (10 a.m.). The gavage concentration was chosen to mimic the average g/kg consumed per day in our two-bottle choice continuous access animals, and time courses were chosen to match the EtOH self-administering groups.
Elevated Plus-Maze
Before EtOH exposure, all animals were tested in an elevated plus-maze (Med Associates, St Albans, VT) for 5 minutes. The platform was elevated 75 cm from the ground and consisted of four arms (50 × 10 cm each) joined by a central platform (10 × 10 cm) with floor lines delineating the entrance to each arm. Closed arm walls were 40 cm high. Amount of time in open and closed arms and number of rearings in open and closed arms were recorded for analysis. Testing commenced when animals were placed in the central hub facing toward an open arm.
Rotarod
Before initial EtOH exposure, animals were screened for motor response to a challenge dose of EtOH using a rotarod. The rotarod apparatus consisted of a textured stainless steel cylinder (7 cm diameter) divided into four lanes (9 cm wide) and suspended 30.5 cm above the table surface. On the training day, animals were trained to stay on the stationary rotarod for 2 minutes without falling (12 trials maximum). Immediately before beginning EtOH self-administration or gavage, animals were tested on the rotarod with a challenge dose of EtOH. The animals were administered 3 g/kg of EtOH (30%) s.c. five minutes before being placed upon the rotarod. Latency to fall was measured for 10 trials with the rotarod rotating at 2.5 rpm.
Corticosterone (CORT)
To analyze CORT levels, trunk blood was collected at the time of sacrifice (1.5 mL), spun for 10 minutes (7000 RPM), and plasma was removed and frozen until the end of the study. Once all samples were collected, they were thawed, diluted 1:40, and plasma was run through a commercial assay (Corticosterone EIA kit, Assay Designs, Ann Arbor, MI) and read on a Spectra Max 190 plate reader (Molecular Devices, Sunnyvale, CA) at 405 nm.
Slice Preparation and Electrophysiology
Electrophysiological experiments were completed blind to EtOH treatment and consumption levels. Rats were anesthetized with isoflurane and their brains were removed. Horizontal brain slices (200 μm thick) containing the VTA were prepared using a vibratome (Leica Microsystems, Berlin, Germany). Slices were submerged in artificial cerebrospinal fluid solution containing (in mM): 126 NaCl, 2.5 KCl, 1.2 MgCl, 1.4 NaH2PO4, 2.5 CaCl2, 25 NaHCO3, and 11 glucose saturated with 95% O2 – 5% CO2 and allowed to recover at 32 °C for at least 1 hr. Individual slices were visualized using a Zeiss Axioskop microscope with differential interference contrast optics and infrared illumination. Recordings were made throughout the VTA. Whole cell patch-clamp recordings were made at 32 °C using 2.5-5 MΩ pipettes containing (in mM): 128 KCl, 20 NaCl, 1 MgCl2, 1 EGTA, 0.3 CaCl2, 10 HEPES, 2 MgATP, and 0.3 Na3GTP (pH 7.2, osmolarity adjusted to 275), plus 0.1% biocytin to label the recorded neuron. Signals were amplified using an Axopatch 1-D amplifier (Molecular Devices, Sunnyvale, CA), filtered at 2 kHz, and collected at 5 kHz using IGOR Pro (Wavemetric, Lake Oswego, OR). Cells were recorded in voltage-clamp mode (V = −70 mV). Series resistance and input resistance were sampled throughout the experiment with 4 mV, 200-ms depolarizing steps. For all experiments, neurons in which there was a change in series resistance of more than 5 MΩ, or 15% of baseline, were excluded from analysis. GABAAR mediated IPSCs were pharmacologically isolated by including 6,7-dinitroquinoxaline-2,3 (1H,4H)-dione (DNQX: 10 μmol/L), strychnine (1 μmol/L), and sulpiride (10 μmol/L) in the bath solution. We previously confirmed that this approach isolates GABAAR signaling using both picrotoxin (Margolis et al., 2008; Margolis, Mitchell, Hjelmstad, & Fields, 2011) and gabazine (Margolis et al., 2011). To measure drug effects on evoked IPSCs, paired electrical pulses (50 ms interval) were delivered once every 10 s through stimulating electrodes placed 80–250 μm away from the patched soma. The IPSC amplitude was calculated by comparing a 2-ms period around the peak to a 2-ms interval just before stimulation. Recordings included, but were not limited to, confirmed dopaminergic neurons (Margolis et al., 2008).
VTA Cannulations
Animals were anesthetized and maintained on isoflurane (0.5 L/min) as needed for the duration of surgery. Animals were placed in a stereotaxic frame and were implanted with bilateral 26-gauge stainless steel chronic guide cannulas (Plastics One, Roanoke, VA) into the VTA (AP, −5.8; ML, ± 0.75; DV, −7.5) based on the atlas of Paxinos and Watson (1997). Cannulas were secured to the skull with dental cement. At the end of the surgical procedure, animals were treated with penicillin and topical antibiotics. A stainless steel dummy cannula (Plastics One, Roanoke, VA) was inserted into each guide cannula and remained in place when the guide cannulas were not in use. Animals were allowed a 2-week recovery period before behavioral testing.
VTA Microinjections
Each injection was made using a 1-μl syringe (Hamilton, Reno, NV) attached to 20 cm of PE 50 tubing connected to a 33-gauge injection cannula (Plastics One, Roanoke, VA). Microinjections of 0.5 μl volumes were given at a rate of 0.5 μl/min using a syringe pump (KD Scientific, Holliston, MA) into each side of the VTA. Injection cannulas extended 2 mm beyond guide cannula and were left in place for 1 min after microinjections to minimize the backflow of drug solution. Animals were divided into two matched groups based on baseline drinking (days 1–7); each group received daily saline or TIPP-Ψ for 5 days (days 8–12).
Drugs and Doses
For electrophysiology, DPDPE (1 mmol/L stock solution; Sigma, St. Louis, MO) was applied by bath perfusion (1 μmol/L). Stock solution was diluted in artificial cerebrospinal fluid immediately before application. EtOH (100%; Gold Shield, Hayward, CA) was diluted to 10% (vol/vol) for self-administration and to 30% (vol/vol) for intraoral gavage and rotarod challenge before use. TIPP-Ψ (5 μmol/L) was dissolved in physiological saline for microinjections into the VTA.
Data Analysis
Summary data are presented as mean ± SEM. For consumption data, drinking was analyzed using 24-hr time points (10 a.m. to 10 a.m.). Comparisons and regression analyses were completed in Excel (v.11.4.1). To assess level of motor impairment on the rotarod, change in the maximum latency to fall was compared before and after the challenge dose of EtOH using the following formula:
The number of seconds the animal spent in the open arm during the 5 min plus maze test was used to assess basal anxiety level.
IPSC data were first analyzed for each time point (3, 7, and 14 days of EtOH self-administration or gavage). As no significant differences were found between the time points for either self-administration or gavaged IPSCs, data were collapsed for analyses.
For electrophysiology, the analyzed data was composed of the 4 min of baseline just preceding drug application and minutes 4–7 of drug application. Paired pulse ratio (PPR) was calculated as the amplitude of the second pulse over the amplitude of the first pulse and was measured both at baseline and after bath application of DPDPE.
One cell displayed a robust increase in GABAAR signaling in the presence of DPDPE; this response was greater than two standard deviations away from the mean and was therefore excluded from data analysis. For CORT analysis, blood samples from two rats were lost and could not be included in the analysis. Additionally, one rat was accidentally overlooked at the time of rotarod testing and therefore could not be included in the analysis. For the TIPP-Ψ injections, one animal consumed EtOH at a rate greater than three standard deviations away from the mean for the first 4 days of baseline drinking and was therefore excluded from data analysis. p < .05 was required for significance in all experiments.
Previously published IPSC data from naïve and chronically drinking animals are shown in Figure 1 for comparison with current self-administration and gavage data (Margolis et al., 2008).
Figure 1.
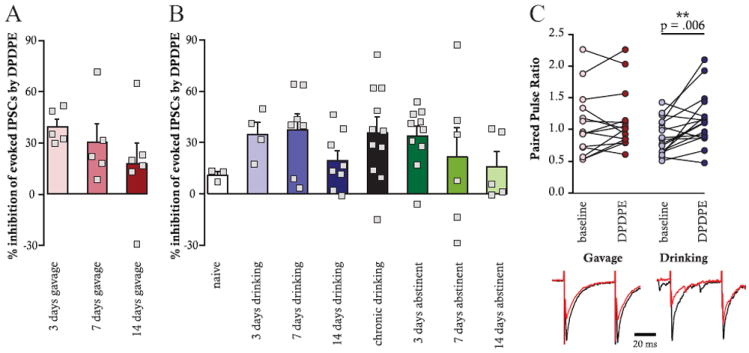
Comparison of DPDPE-induced inhibition of evoked GABAAR-mediated IPSCs after either EtOH gavage (A) or EtOH self-administration (B). The range of inhibitions is similar across time points and exposure procedures and is not significantly different after up to 14 days of EtOH abstinence. PPR (C) was significantly increased by DPDPE in EtOH self-administering animals only (p = .006, n = 19). Representative traces are shown for gavaged and self-administering animals; black = baseline, red = DPDPE. Data from naïve and chronic drinking animals were reported previously (Margolis et al., 2008).
Results
DOR Function in EtOH Gavaged and Self-Administering Animals
We previously found that EtOH consumption leads to increased DOR inhibition of GABAAR IPSCs in VTA neurons. However, it is uncertain how rapidly this inhibition arises and whether exposure to EtOH is sufficient to cause this change or whether active self-administration is required. In contrast to the small inhibition of evoked IPSCs produced by DPDPE in VTA neurons from EtOH naïve animals (Margolis et al., 2008; Margolis et al., 2011), robust inhibitions of IPSC magnitude in response to bath application of the DOR selective agonist DPDPE (1 μmol/L) were observed after 3, 7, and 14 days of either EtOH gavage (t = 4.81, p = .0002, n = 16 cells; Figure 1A) or EtOH self-administration (t = 6.35, p < .00001, n = 19 cells; Figure 1B). This enhanced inhibition persisted after 3, 7, and 14 days of EtOH abstinence in chronic drinking animals (t = 4.47, p = .0002, n = 21 cells; Figure 1B), indicating that this DOR functionality does not require ongoing EtOH exposure.
We recently reported that DPDPE can decrease GABAAR IPSCs in VTA neurons of EtOH naïve rats through a postsynaptic mechanism (Margolis et al., 2011), whereas the mechanism of inhibition is presynaptic in EtOH consuming rats (Margolis et al., 2008). A drug-induced increase in PPR suggests a decrease in probability of neurotransmitter release, whereas no change in PPR is more consistent with a postsynaptic effect. Here we found that PPR significantly increased with DPDPE application in VTA neurons from self-administering animals (t = −3.13, p = .006, n = 17 cells) but not from gavaged animals (t = −.66, p = .52, n = 15 cells; Figure 1C), suggesting that the mechanisms underlying inhibitions of GABAAR IPSCs in these two groups of rats are qualitatively different. Specifically, in drinking animals presynaptic DORs may be inducing inhibitions, whereas in gavaged animals postsynaptic DORs are more likely responsible for this effect.
Elevated Plus Maze and DOR Function in the VTA of EtOH Gavaged and Self-Administering Animals
Time in the open arm of the elevated plus maze before EtOH exposure was negatively correlated with DPDPE-induced inhibition of GABAAR IPSCs in self-administering animals (R = .69, p = .001, n = 19 cells; Figure 2B) but was not significantly correlated with inhibition of IPSCs in gavaged animals (R = .20, p = .46, n = 16 cells; Figure 2A). This pattern appears as early as 3 days after commencing EtOH consumption. These data indicate that the degree of anxiety before initiation of EtOH consumption predicts increased DPDPE induced inhibition of GABAAR IPSCs but only in animals that actively consume EtOH.
Figure 2.
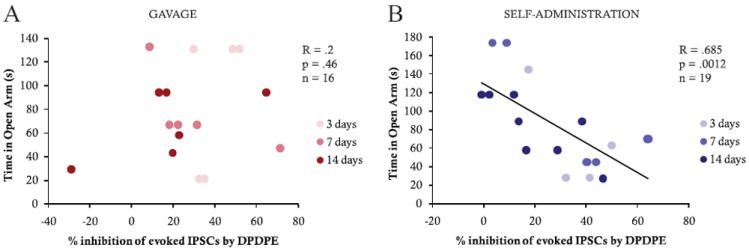
Correlation of plus maze (time in the open arm) with DPDPE-induced inhibition of GABAAR evoked IPSCs after either EtOH gavage (A) or EtOH self-administration (B) after 3, 7, and 14 days of EtOH exposure. Time in the open arm was significantly correlated with the DPDPE effect only in EtOH self-administering animals (R =.685, p = .0012, n = 19).
Rotarod and DOR Function in the VTA of EtOH Gavaged and Self-Administering Animals
The change in maximum fall latency on the rotarod was positively correlated with the effect of DPDPE on GABAAR IPSCs in neurons from self-administering animals (R = .89, p = .000001, n = 17 cells; Figure 3B). However, no such relationship was observed in gavaged animals (R = .11, p = .68, n = 16 cells; Figure 3A). Therefore, the self-administering animals with the greatest motor impairment after an EtOH challenge were also those with the greatest DPDPE-induced inhibition of IPSCs after EtOH consumption. Additionally, only when animals are allowed to freely consume EtOH is it possible to use their initial EtOH-induced motor impairment to predict the subsequent development of DOR-mediated inhibition of GABA release.
Figure 3.

Correlation of rotarod (change in latency to fall) with DPDPE-induced change in GABAAR evoked IPSCs after either EtOH gavage (A) or EtOH self-administration (B) after 3, 7, and 14 days of EtOH exposure. Change in latency to fall was significantly correlated with the DPDPE effects only in EtOH self-administering animals (R =.893, p ≤ .00001, n = 17).
CORT Levels and DOR Function in the VTA of EtOH Gavaged and Self-Administering Animals
Blood CORT sampled at the time of sacrifice, after animals self-administered EtOH, was positively correlated with DPDPE induced inhibition of GABAAR IPSCs (R = .66, p = .0038, n = 17 cells) such that higher CORT levels predicted greater inhibition of GABA (Figure 4B). There was, however, no correlation between CORT and DPDPE effect on evoked IPSCs in gavaged animals (R = .08, p = .77, n = 15 cells; Figure 4A). Interestingly, CORT also did not correlate with average daily EtOH consumed before sacrifice (R = .267, p = .48, n = 9; Figure 5C). These data indicate that CORT levels can be used to predict DPDPE-induced inhibition of GABAAR IPSCs in animals actively consuming EtOH but not in gavaged animals, even though EtOH consumption per se does not correlate with CORT levels at the time of sacrifice.
Figure 4.

Correlation of blood CORT levels with DPDPE effect on evoked GABAAR-mediated IPSCs after either EtOH gavage (A) or EtOH self-administration (B) after 3, 7, and 14 days of EtOH exposure. Blood CORT was significantly correlated with the DPDPE effect only in EtOH self-administering animals (R = .663, p = .0038, n = 17).
Figure 5.
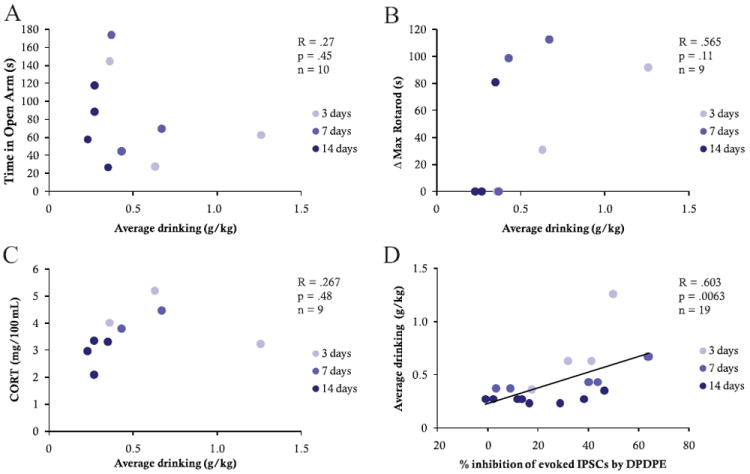
There was no correlation between EtOH self-administration and either time in the open arm of the plus maze (A), latency to fall on the rotarod (B), or CORT at time of sacrifice (C). However, there was a significant positive correlation between EtOH self-administration and DPDPE inhibition of evoked GABAAR mediated IPSCs (R = .603, p = .0063, n = 19; D).
EtOH Consumption and Behavioral Measures in Self-Administering Animals
EtOH consumption did not itself correlate with either behavioral measure presented here (plus maze: R = .267, p = .45, n = 10; Figure 5A; rotarod: R = .565, p = .11, n = 9; Figure 5B) but did correlate directly with DPDPE modulation of VTA IPSCs (R = .603, p = .0063, n = 19 cells; Figure 5D) such that the greater the drinking, the greater the GABAAR IPSC inhibition.
The Role of DOR in EtOH Consumption During Acquisition
While our previous findings show a negative correlation between EtOH consumption and DOR inhibition of GABAAR IPSCs in chronic drinking animals, such that low drinkers have greater DOR inhibition than high drinkers, here we show a positive correlation during acquisition, suggesting that the contribution of DOR inhibition to the control of EtOH consumption changes over the course of EtOH exposure. To determine whether DORs in the VTA control drinking in a similar manner in both acquisition and chronic drinking, we tested the effect of the DOR selective antagonist, TIPP-Ψ, microinjected for 5 days into the VTA during acquisition of EtOH consumption (injections on Days 8–12 of EtOH self-administration). Although we previously found that DOR antagonists increase drinking in chronic drinking animals (Margolis et al., 2008), here we find no significant difference in acquisition of EtOH consumption between animals that received VTA TIPP-Ψ microinjections compared with those that received saline injections (5-day average saline group = .66 g/kg, 5-day average TIPP-Ψ group = .81 g/kg; t = 2.12, p = .48, n = 18; Figure 6).
Figure 6.
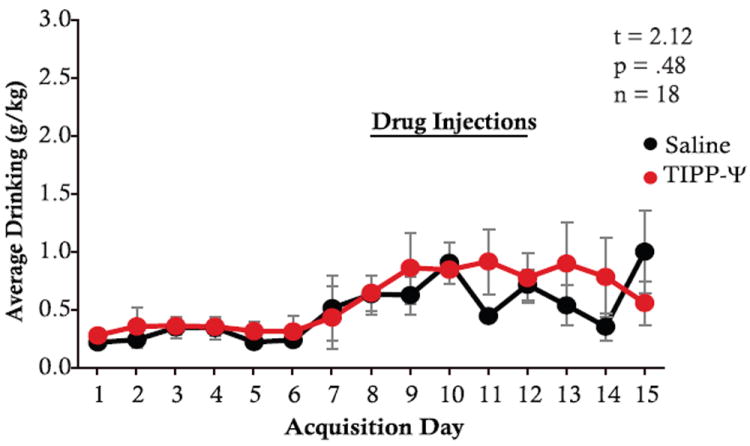
DORs in the VTA do not control drinking during acquisition of EtOH consumption; black = saline injections, red = TIPP-Ψ injections (t = 2.12, p = .48, n = 18).
Discussion
Our data show that an increase in DOR function in the VTA is apparent as early as 3 days after initial EtOH exposure and persists for at least 14 days after EtOH exposure is terminated. In contrast to our previous finding that DOR inhibition of GABA release is greater in chronic low drinking animals than in high drinking animals (Margolis et al., 2008), we show here that during acquisition DOR inhibition is actually greater in higher drinking animals. This suggests that, although the VTA DOR on GABAergic terminals is functional after initial exposure to EtOH, DOR functionality subsides in animals that continue on to become high drinkers. The lack of an effect of DOR antagonists on acquisition suggests that endogenous ligands acting at the DOR do not control EtOH consumption until animals have had chronic exposure to EtOH (see Figure 6). Additional experiments are necessary to determine which factors contribute to the downregulation or inactivation of DOR over time in high drinking animals and to determine how and when the DOR begins to modulate EtOH consumption. However, one parsimonious possibility is that overactivation of DOR, perhaps resulting from robust endogenous ligand release, could lead to internalization of DOR in high drinking animals, and that those DOR would then be degraded or would fail to be appropriately recycled (see Figure 7).
Figure 7.

Schematic diagram indicating hypothetical change in DOR function over time and EtOH exposure in high- and low-drinking animals.
A number of previous studies also indicate that passive administration of EtOH induces a different range of neural and behavioral effects from those seen in animals actively consuming EtOH. For example, EtOH affects the threshold of brain stimulation reward in EtOH self-administering rats but not in EtOH yoked controls (Moolten & Kornetsky, 1990), and EtOH-induced ataxia is attenuated in EtOH self-administering rats but not in EtOH yoked controls (Weise-Kelly & Siegel, 2001). Further, rats permitted to control their EtOH exposure show differences in subsequent locomotor activity in response to an EtOH challenge and operant responding for EtOH compared with EtOH-yoked controls (Pautassi, Truxell, Molina, & Spear, 2008). Together these results demonstrate that current approaches to passive EtOH administration do not mimic the effects of active EtOH consumption. Although it remains possible that an alternative approach may better mimic EtOH consumption, when considered with our present data, these studies suggest that active consumption is a necessary experimental component when studying the neural effects of alcohol or drug abuse.
Human data suggest that initial motor response to alcohol is an important predictor of future propensity to develop problem drinking. Specifically, individuals who show less motor impairment (body sway) in response to an alcohol challenge are more likely to become alcoholic (Schuckit, 1993, 1994). To our knowledge, ours is the first demonstration of the extension of these findings to an animal model and suggest that motor performance on the rotarod test may be a useful means of assessing how differences in low and high level of response to alcohol in animals affect subsequent EtOH intake. Furthermore, our current results are consistent with the idea that one of the factors contributing to the relation between initial response to EtOH and subsequent consumption levels is the development of DOR functionality with repeated exposure.
The anxiolytic properties of DOR selective agonists are well established (Jutkiewicz, 2007; Perrine, Hoshaw, & Unterwald, 2006; Randall-Thompson, Pescatore, & Unterwald, 2010; Saitoh et al., 2004; Solati, Zarrindast, & Salari, 2010; van Rijn, Brissett, & Whistler, 2010). DOR knock-out mice display greater measures of anxiety than matched controls (Filliol et al., 2000), and selective DOR antagonists can attenuate the anxiolytic actions of benzodiazepines (Primeaux, Wilson, McDonald, Mascagni, & Wilson, 2006). Consistent with this idea, we recently demonstrated that EtOH-consuming animals exhibiting lower levels of anxiety show greater DOR inhibition of GABA release in the VTA, suggesting that endogenous DOR activation in the VTA could be anxiolytic (Margolis et al., 2008). The present data extend these findings and indicate that the interaction between DOR and anxiety can change in response to EtOH exposure: the negative correlation between time in the open arm of the plus maze and DPDPE-induced inhibition of GABA release presented here indicates that the previously noted relationship was most likely the result of chronic EtOH consumption. Additional studies are necessary to understand the relationship between anxiety, duration of EtOH consumption, and change in DOR function.
EtOH stimulates hypothalamic production of corticotrophin releasing factor, leading to an increase in the stress hormone CORT (Rivier, 1996; Rivier, Bruhn, & Vale, 1984). Previous data also indicate that EtOH and stress can interact synergistically to increase CORT levels (Patterson-Buckendahl, Kubovcakova, Krizanova, Pohorecky, & Kvetnansky, 2005). In the present study we did not see a difference in CORT levels between EtOH-gavaged and EtOH self-administering animals. Because we attempted to minimize stress by lightly anesthetizing animals before gavage, these data may be explained by lack of a gavage stress–induced increase in CORT level. However, because a stressful stimulus (footshock) can elicit a change in the DPDPE effect on GABAAR signaling in the VTA (Margolis et al., 2011), gavage stress, though less extreme than footshock stress, may have still influenced the DPDPE effect on GABAAR signaling.
In summary, we find that anxious animals and those with the greatest EtOH-induced motor impairment have the most robust DPDPE-induced inhibition of GABAAR IPSCs in VTA neurons over the first two weeks of EtOH self-administration. Importantly, the DOR effect on synaptic transmission persists for at least two weeks after onset of abstinence, raising the possibility that VTA DORs are a viable treatment target in relapse prevention. As anxiety is a major factor promoting EtOH intake, these data also emphasize the potential therapeutic importance of DOR agonists in the treatment of alcohol abuse in conjunction with anxiety.
Acknowledgments
We thank the National Institute on Drug Abuse for the generous gift of TIPP-Ψ. This work was supported by California State Funds for the Study of Drug and Alcohol Abuse, by the Wheeler Center for the Neurobiology of Addiction, and by NIAAA AA017072 and NIDA DA016782.
References
- Bacopoulos NG, Bize I, Levine J, Van Orden LS. Modification of ethanol intoxication by dopamine agonists and antagonists. Psychopharmacology. 1979;60:195–201. doi: 10.1007/BF00432293. [DOI] [PubMed] [Google Scholar]
- Bechtholt A, Cunningham C. Ethanol-induced conditioned place preference is expressed through a ventral tegmental area dependent mechanism. Behavioral Neuroscience. 2005;119:213–223. doi: 10.1037/0735-7044.119.1.213. [DOI] [PubMed] [Google Scholar]
- Filliol D, Ghozland S, Chluba J, Martin M, Matthes H, Simonin F, Kieffer B, et al. Mice deficient for delta- and mu-opioid receptors exhibit opposing alterations of emotional responses. Nature Genetics. 2000;25:195–200. doi: 10.1038/76061. [DOI] [PubMed] [Google Scholar]
- Hill KG, Ryabinin AE, Cunningham CL. FOS expression induced by an ethanol-paired conditioned stimulus. Pharmacology, Biochemistry, and Behavior. 2007;87:208–221. doi: 10.1016/j.pbb.2007.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jutkiewicz E. RB101-mediated protection of endogenous opioids: Potential therapeutic utility? CNS Drug Reviews. 2007;13:192–205. doi: 10.1111/j.1527-3458.2007.00011.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margolis EB, Fields HJ, Hjelmstad GO, Mitchell JM. Delta-opioid receptor expression in the ventral tegmental area protects against elevated alcohol consumption. The Journal of Neuroscience. 2008;28:12672–12681. doi: 10.1523/JNEUROSCI.4569-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margolis EB, Mitchell JM, Hjelmstad GO, Fields HL. A novel delta opioid receptor-mediated enhancement of GABAA receptor function induced by stress in ventral tegmental area neurons. Journal of Physiology. 2011 doi: 10.1113/jphysiol.2011.209023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McBride W, Murphy J, Gatto G, Levy A, Lumeng L, Li T. Serotonin and dopamine systems regulating alcohol intake. Alcohol and Alcoholism Supplement. 1991;1:411–416. [PubMed] [Google Scholar]
- McBride WJ, Murphy JM, Gatto GJ, Levy AD, Yoshimoto K, Lumeng L, Li TK. CNS mechanisms of alcohol self-administration. Alcohol and Alcoholism Supplement. 1993;2:463–467. [PubMed] [Google Scholar]
- Mitchell JM, Bergren LJ, Chen KS, Rowbotham MC, Fields HL. Naltrexone aversion and treatment efficacy are greatest in humans and rats that actively consume high levels of alcohol. Neurobiology of Disease. 2009;33:72–80. doi: 10.1016/j.nbd.2008.09.018. [DOI] [PubMed] [Google Scholar]
- Moolten M, Kornetsky C. Oral self-administration of ethanol and not experimenter-administered ethanol facilitates rewarding electrical brain stimulation. Alcohol. 1990;7:221–225. doi: 10.1016/0741-8329(90)90008-z. [DOI] [PubMed] [Google Scholar]
- Patterson-Buckendahl P, Kubovcakova L, Krizanova O, Pohorecky LA, Kvetnansky R. Ethanol consumption increases rat stress hormones and adrenomedullary gene expression. Alcohol. 2005;37:157–166. doi: 10.1016/j.alcohol.2005.09.007. [DOI] [PubMed] [Google Scholar]
- Pautassi RM, Truxell E, Molina JC, Spear NE. Motivational effects of intraorally-infused ethanol in rat pups in an operant self-administration task. Physiology & Behavior. 2008;93:118–129. doi: 10.1016/j.physbeh.2007.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paxinos G, Watson C. The rat brain in stereotaxic coordinates. 3. San Diego, CA: Academic Press; 1997. Compact, CD-Rom. [DOI] [PubMed] [Google Scholar]
- Perrine S, Hoshaw B, Unterwald E. Delta opioid receptor ligands modulate anxiety-like behaviors in the rat. British Journal of Pharmacology. 2006;147:864–872. doi: 10.1038/sj.bjp.0706686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Primeaux S, Wilson S, McDonald A, Mascagni F, Wilson M. The role of delta opioid receptors in the anxiolytic actions of benzodiazepines. Pharmacology, Biochemistry, and Behavior. 2006;85:545–554. doi: 10.1016/j.pbb.2006.09.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Randall-Thompson JF, Pescatore KA, Unterwald EM. A role for delta opioid receptors in the central nucleus of the amygdala in anxiety-like behaviors. Psychopharmacology. 2010;212:585–595. doi: 10.1007/s00213-010-1980-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivier C, Bruhn T, Vale W. Effect of ethanol on the hypothalamic-pituitary-adrenal axis in the rat: Role of corticotropinreleasing factor (CRF) The Journal of Pharmacology and Experimental Therapeutics. 1984;229:127–131. [PubMed] [Google Scholar]
- Rivier C. Alcohol stimulates ACTH secretion in the rat: Mechanisms of action and interactions with other stimuli. Alcoholism, Clinical and Experimental Research. 1996;20:240–254. doi: 10.1111/j.1530-0277.1996.tb01636.x. [DOI] [PubMed] [Google Scholar]
- Rodd-Henricks ZA, Melendez RI, Zaffaroni A, Goldstein A, McBride WJ, Li TK. The reinforcing effects of acetaldehyde in the posterior ventral tegmental area of alcohol-preferring rats. Pharmacology, Biochemistry, and Behavior. 2002;72:55–64. doi: 10.1016/s0091-3057(01)00733-x. [DOI] [PubMed] [Google Scholar]
- Saitoh A, Kimura Y, Suzuki T, Kawai K, Nagase H, Kamei J. Potential anxiolytic and antidepressant-like activities of SNC80, a selective delta-opioid agonist, in behavioral models in rodents. Journal of Pharmacological Sciences. 2004;95:374–380. doi: 10.1254/jphs.fpj04014x. [DOI] [PubMed] [Google Scholar]
- Schuckit MA. Reaction to alcohol as a predictor of alcoholism. Alcohol and Alcoholism Supplement. 1993;2:91–94. [PubMed] [Google Scholar]
- Schuckit MA. Low level of response to alcohol as a predictor of future alcoholism. American Journal of Psychiatry. 1994;151:184–189. doi: 10.1176/ajp.151.2.184. [DOI] [PubMed] [Google Scholar]
- Solati J, Zarrindast MR, Salari AA. Dorsal hippocampal opioidergic system modulates anxiety-like behaviors in adult male Wistar rats. Psychiatry and Clinical Neurosciences. 2010;64:634–641. doi: 10.1111/j.1440-1819.2010.02143.x. [DOI] [PubMed] [Google Scholar]
- van Rijn RM, Brissett DI, Whistler JL. Dual efficacy of delta opioid receptor-selective ligands for ethanol drinking and anxiety. The Journal of Pharmacology and Experimental Therapeutics. 2010;335:133–139. doi: 10.1124/jpet.110.170969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weise-Kelly L, Siegel S. Self-administration cues as signals: Drug self-administration and tolerance. Journal of Experimental Psychology: Animal Behavior Processes. 2001;27:125–136. [PubMed] [Google Scholar]