

Abstract
Fibrous dysplasia (FD) is a skeletal disorder caused by activating mutations in Gsα that result in elevations in cAMP. A feature of FD is elevated blood levels of the bone cell-derived phosphaturic hormone, fibroblast growth factor-23 (FGF23). FGF23 regulates serum phosphorus and active vitamin D levels by action on proximal renal tubule cells. An essential step in the production of biologically active FGF23 is glycosylation by the UDP-N-acetyl-α-D-galactosamine:polypeptide N-acetylgalactosaminyl transferase (ppGalNAc-T3). In the absence of glycosylation, FGF23 is processed into inactive N- and C-terminal proteins by a subtilisin proprotein convertase, probably furin. Normally, most if not all circulating FGF23 is intact. In FD, C-terminal levels are elevated, suggesting altered FGF23 processing. Altered processing in FD is the result of a cAMP-dependent, coordinated decrease in ppGalNAc-T3 and an increase in furin enzyme activity. These findings, and emerging data from other diseases, suggest regulation of FGF23 processing may be a physiologically important process.
Keywords: FGF23, fibrous dysplasia, phosphorus, vitamin D, mineral metabolism, bone
Fibrous Dysplasia Background
Fibrous dysplasia (FD; OMIM 174800) is an uncommon and debilitating skeletal disorder resulting in fracture, deformity, functional impairment, and pain1–3. FD occurs along a broad clinical spectrum, and may affect one bone (monostotic FD) or many bones (polyostotic FD). FD may occur in association with extraskeletal features, most commonly café-au-lait macules and hyperfunctioning endocrinopathies, including precocious puberty, hyperthyroidism, growth hormone excess, hypercortisolism, and FGF23-mediated hypophosphatemia. FD in combination with one or more of these extraskeletal features is termed McCune-Albright syndrome 4–7.
FD arises from .somatic activating mutations in the GNAS gene, which encodes the α-subunit of the Gs-stimulatory protein 8,9. Disease burden depends on the stage of embryogenesis at which the mutation occurs, and the specific clone in which the mutation occurs; this determines the locations to where mutated progenitor cells subsequently migrate 10,11. The vast majority of the mutations that occur in Gsα in FD arise at the R201 position, which includes the intrinsic GTPase domain of the molecule 12,13. This results in prolonged interaction of activated Gs with the effector molecule adenylyl cyclase, and increased production of the second messenger cyclic AMP (cAMP)14. In bone, constitutive Gsα activity results in proliferation of undifferentiated bone marrow stromal cells, which accumulate in marrow spaces leading to local loss of hematopoiesis and marrow fibrosis 15. Osteogenic cells derived from these progenitors are functionally impaired, leading to abnormal matrix deposition and formation of bone that is morphologically and structurally unsound. FD demonstrates site-specific histologic features; in the craniofacial area it appears as dense with uninterrupted trabecular networks, while in the axial and appendicular skeleton the trabecular pattern is discontinuous (often referred to as having a ‘Chinese writing’ appearance) 16. Typical features recognized at all skeletal sites include abnormal collagen formation as manifested by so-called Sharpey’s fibers and loss of typical lamellation patterns, increased vascularization, and abnormal persistence of woven bone 16. Undermineralized matrix and accumulation of osteoid is also a common and clinically important feature of FD 17. It may be the lesional osteomalacia in FD tissue that lends plasticity to bones affected by FD and their tendency to bow and fracture 18,19.
Areas of skeletal involvement are established early, with 90% of craniofacial lesions evident by age 5 years, and 75% of all FD lesions established by age 15 years 20. Although any area of the skeleton may be affected, the most commonly involved sites include the proximal femora and skull base 21,22. Femoral disease may present with a classic coxa vara, or shepherd’s crook deformity. Skull disease may result in facial asymmetry, and rarely damage to the cranial nerves leading to loss of vision and/or hearing. Complications of craniofacial FD are more common in patients with uncontrolled growth hormone excess 23,24. The radiographic appearance of FD varies depending on the location and activity of the affected bone. During childhood, FD in the appendicular and craniofacial skeleton typically demonstrates a homogeneous ‘ground glass’ appearance. Disease activity may wane during adulthood, at which point FD often adopts a more sclerotic and less homogeneous appearance. X-ray findings in the axial skeleton (including the ribs, pelvis, and spine) may be subtle, and FD in these areas is often more easily detected using bone scintigraphy 22.
The Role of FGF23 in Mineral Metabolism in Fibrous Dysplasia
Andrea Prader was the first to propose the existence of a phosphaturic factor. In 1959 he reported the case of an 11-year old girl who developed severe rickets over the course of a year that resolved after resection of a benign connective tissue tumor. Prader speculated that the tumor produced a circulating factor responsible for her phosphate wasting 25, a condition later described as tumor-induced osteomalacia (reviewed in 26,27). Since then, FGF23 overproduction has been identified as the underlying etiology of tumor-induced osteomalacia and other hypophosphatemic disorders, including X-linked, autosomal dominant, and autosomal recessive hypophosphatemic rickets, and FD 28–31.
Although frank hypophosphatemia in patients with FD is infrequent, a renal tubulopathy including some degree of phosphate wasting is one of the most commonly associated extraskeletal manifestations 32,33. In 2003 it was recognized that FD-associated phosphate wasting arises from overproduction of FGF23 by abnormal osteogenic precursors, and that in normal bone FGF23 is produced by both osteoblasts and osteocytes 31. This provided the first evidence that bone is the physiologic source of FGF23 production, a finding that was subsequently supported by other investigators 34–36. The degree of FGF23 overproduction in FD is correlated with disease severity and tissue burden; thus significant hypophosphatemia is seen only in patients with a substantial FD burden 31,32. In contrast to most other features of McCune-Albright syndrome, hypophosphatemia may resolve as patients become older 7,37. This likely reflects intrinsic changes in FD that occur with age, including decline in the number of mutated progenitor cells and the tendency of histologic features to improve over time 37.
FGF23 reduces serum phosphate levels by three mechanisms: 1) directly by down-regulation of the NaPi 2a and 2c transporters in the proximal tubule, leading to reduced renal phosphate reabsorption at the level of the kidney, 2) directly by down-regulation of renal 1α-hydroxylase, which decreases production of active 1,25-dihydroxyvitamin D and as a result decreases intestinal phosphate absorption, and 3) indirectly by the compensatory increase in PTH that develops in order to maintain eucalcemia in response to relatively or frankly low 1,25-dihydroxyvitamin D 38–40. Elevated PTH exacerbates the phosphate wasting effect of FGF23 and worsens the disease.
Clinical sequelae of FGF23-mediated hypophosphatemia in FD include earlier onset and increased risk of fractures relative to patients with hypophosphatemia, and FD-related bone pain 19. Treatment is similar to other forms of FGF23-mediated hypophosphatemia, and includes supplementation with phosphorus and vitamin D analogs 6,7.
The regulation of FGF23
The physiologic mechanisms by which FGF23 transcription and translation, and ultimately blood levels are regulated have not yet been clearly defined. Clinically, changes in FGF23 levels likely involve feedback from serum phosphorus and 1,25-dihydroxyvitamin D 41–45. Molecularly, the fact that mutations in phosphate regulating endopeptidase homolog, X-linked (PHEX)28 and dentin matrix protein-1 (DMP-1)36, (the genes mutated in X-linked hypophosphatemic rickets (XLH) and autosomal recessive hypophosphatemic rickets, respectively), cause dysregulated elevations in blood FGF23 and hypophosphatemia/rickets, implicates PHEX and DMP-1 in FGF23 regulation. There is also evidence that pathologically elevated levels of α-Klotho (αKL) are able to directly elevate FGF23 levels 46,47. In addition, mutations in the FAM20C gene that encodes the phosphorylation enzyme Golgi-enriched fraction casein kinase (GEF-CK) have recently been reported in several families with FGF23-mediated hypophosphatemic rickets 48.
Human FGF23 is a 251 amino acid protein with a high degree of homology across many species that includes a 24 amino acid signal peptide, indicating it is a secretory hormone 28,38,49. The FGF23 protein has two major functional domains: the N-terminal portion, which includes homology to other FGFs, and a unique C-terminal domain. FGF23 exerts its action via interaction with various FGF cell-surface receptors including FGFR1, 3c, and 4. FGF23 signaling is dependent upon its interaction not only with an FGFR, but also with its co-receptor, αKL. Evidence indicates that the most potent signal transduction is exhibited when FGF23 binds to FGFR1 50,51. However, recent evidence also suggests that in certain pathophysiologic states, such as renal failure where FGF23 levels may be elevated by two to three orders of magnitude, FGF23 may also interact with other FGFRs such as FGFR4 to mediate pathophysiologic effects 52. While FGFRs including FGFR1 are widely expressed, it may be the relatively narrow tissue expression profile of αKL that lends specificity to FGF23 action under normal physiologic conditions. FGF23 receptor binding induces various cell signaling events, including activation of extracellular signal-regulatory kinases (ERK) and mitogen activated kinases (MAPK), that lead to activation of the early growth response protein 1 (EGR1) 50.
Recent evidence suggests the mineral metabolism effects of FGF23 may be PTH-dependent 53, with PTH acting synergistically as part of a coordinated physiological process to induce phosphate excretion via the action of sodium-hydrogen exchanger regulatory factor-1 (NHERF-1), a PDZ domain containing scaffolding protein 54. Upon addition of PTH and/or FGF23, specific receptor molecules induce signal transduction pathways by activating either PKC or MAPK pathways, causing the dissociation of NHERF-1 from the Npt2a complexes, which in turn induces Npt2a endocytosis and reduces renal phosphate reabsorption55.
FGF23 processing
FGF23 circulates in several forms: biologically active intact FGF23 (iFGF23), and inactive C-terminal (cFGF23) and N-terminal FGF23 (nFGF23) fragments (56, reviewed in 57). There are two assays commonly used for the measurement of FGF23. One assay detects only iFGF23 (Kainos, Tokyo, Japan). The other assay uses capture and detection antibodies that are both directed to the C-terminus, and as a result detects both iFGF23 and cFGF23. Currently there are no nFGF23-specific assays.
FGF23 processing appears to be primarily regulated by two major steps: 1) post-translational O-linked glycosylation by UDP-N-acetyl-α-D-galactosamine:polypeptide N-acetylgalactosaminyl transferase (ppGalNAc-T3) at Ser or Thr residues, and 2) protein cleavage around amino acids 176–179 (RXXR, 176RHTR179) (58, reviewed in 59). Studies from cell culture models suggest that production and secretion of active iFGF23 requires glycosylation, and that under-glycosylated FGF23 is prone to cleavage to inactive fragments 56,58,60. In the absence of glycosylation, the FGF23 cleavage site is recognized by subtilisin-like proprotein convertase enzymes such as furin. In support of this, FGF23 has been co-localized with ppGalNAc-T3 and furin by confocal microscopy61. In addition, when LoVo cells, a cell line that lacks furin, are transfected with FGF23 there is no FGF23 processing, in spite of the fact that the other subtilisin-like proprotein convertases are expressed (unpublished data, NB and MTC). However recent evidence suggests other proprotein convertases, such as PC2, may also play a role 62. Mass spectrometric analyses of purified FGF23 indicates that several Ser and Thr residues may be O-glycosylated and therefore important for protection against cleavage of FGF23 by furin 56,60. However, demonstration of this in clinical specimens remains to be seen.
The most dramatic example of altered FGF23 processing is the disease of hyperphosphatemic familial tumoral calcinosis (HFTC). These patients present with reduced urinary phosphate excretion, hyperphosphatemia, and inappropriately normal or elevated 1,25-dihydroxyvitamin D. HFTC can result from mutations in three different genes. The most common form is due to loss of function mutations in ppGalNAc-T3, and is sometimes referred to as HFTC type 1 63. The second most common form is HFTC type 2, which is due to mutations in FGF23 64. The least common (to date only one case has been reported) is HFTC type 3 due to mutations in αKL, the FGF23 co-receptor, which results in a form of FGF23 resistance 65. In types 1 and 2 FGF23 processing is altered, resulting in elevation of nFGF23 and cFGF23 and low levels of iFGF23. In Type 1 loss of function mutations in ppGalNAc-T3 presumably leads to decreased glycosylation and increased FGF23 cleavage. FGF23 mutations in type 2 HFTC typically arise at Ser or Thr residues, which are potential sites for O-glycosylation by ppGalNAc-T3. However, mutations at sites other than Ser or Thr have also been reported (H41Q, Q54K and M96T). It has been speculated that disrupted protein folding may also play important roles in FGF23 stability and processing 66.
FGF23 Processing in Fibrous Dysplasia
Studies in FD have provided novel insights into both production and processing of FGF23. The first insight into differential regulation of FGF23 in FD was the recognition that in spite of high levels of circulating FGF23, frank hypophosphatemia and/or rickets was relatively uncommon 31,32. Analysis of FD patient serum confirmed that while total FGF23 levels (a combination of iFGF23 and cFGF23) were elevated, there was a proportionally greater elevation in cFGF23 levels relative to iFGF23, consistent with increased FGF23 processing 61.
The mechanism of FGF23 processing in FD was studied in detail in both human bone marrow stromal cells from FD patients, as well as an FGF23 producing cell line that was transiently transfected with either mutant or wild type Gsα61. Patient bone marrow stromal cells, which had elevated cAMP levels due to the activating mutation in Gsα, showed a cAMP-dependent coordinated increase in furin enzyme activity and a decrease in ppGalNAC-T3 enzyme activity. These data are consistent with increased cleavage of active iFGF23 into its inactive fragments. Also consistent with this was the demonstration of perinuclear co-localization by confocal microscopy of FGF23, furin, and ppGalNAC-T3 in patient bone marrow stromal cells. These studies were validated and extended by studies in FGF23 processing in HEKF cells, a cell line that overexpresses FGF23. When HEKF cells were treated with either cAMP-inducing doses of forskolin or co-transfected with mutant Gsα, there was an increase in cFGF23 relative to iFGF23, as well as a coordinated increase in furin activity and decrease ppGalNAC-T3 enzyme activity. These data explain the biochemical phenotype observed in patients with FD.
There is precedent for the sort of coordinated O-glycosylation and furin protein processing that is seen in FD. Semenov and colleagues demonstrated that the mechanism of inhibition of pro-brain natriuretic peptide processing is a coordinated inhibition of O-glycosylation and increase in furin activity 67.
Other examples of altered FGF23 processing
A pattern similar to what has been observed in FD can be seen in Jansen metaphyseal chondrodysplasia. Jansen metaphyseal chondrodysplasia is a skeletal dysplasia caused by activating mutations in the PTH/PTHrP receptor. Constitutive receptor activation in Jansen metaphyseal chondrodysplasia leads to elevations in cAMP, as is seen in FD. In a report of one patient, levels of total FGF23 (cFGF3 + iFGF23) were elevated out of proportion to iFGF23, supporting a model of cAMP-mediated FGF23 overproduction with a compensatory increase in FGF23 processing 68.
An important new finding is a potential role for serum iron levels in regulation of FGF23 synthesis and processing. Durham et al demonstrated elevated cFGF23 but normal iFGF23 levels in a group of anemic patients, suggesting low iron levels caused an increase in both FGF23 synthesis and processing 69. Imel et al studied the relationship between serum iron concentrations and plasma cFGF23 and iFGF23 levels in ADHR subjects and healthy controls 70. In individuals with ADHR, serum iron levels were negatively correlated with both iFGF23 and cFGF23, while in controls low serum iron was associated with elevated cFGF23, but not iFGF23. These findings suggest that in spite of increased transcription and translation of FGF23 in normal subjects, serum iFGF23 levels are maintained in the normal range, possibly by increased FGF23 processing. It appears that FGF23 mutations in subjects with ADHR (most of which are at the 176RHTR179 site) do not allow for the increased processing seen in normal subjects. These findings were supported in studies by Farrow et al 71. In a mouse model of ADHR they showed that on a low iron diet, both ADHR and wild type mice had increased FGF23 transcription and translation, but only the ADHR mice had elevated iFGF23 and were hypophosphatemic. The wild type mice were able to compensate by processing iFGF23 to cFGF23, which was elevated in the wild type mice. They further showed that, as might be expected in an iron-regulated pathway, the effects on FGF23 transcription, translation and processing may be mediated by the HIF-1α pathway.
It has been speculated that excess levels of C-terminal FGF23 may exert an inhibitory effect on the action of iFGF23 72. This would suggest increased FGF23 processing may serve a physiologic role in diseases such as FD by inhibiting the effects of excess FGF23. However, evidence from the phex and galnt3 knockout mouse suggests otherwise 73. This double knockout mouse is frankly hypophosphatemic in the setting of extremely elevated cFGF23 and only moderately elevated iFGF23 levels, suggesting the excess cFGF23 was unable to inhibit the effects of iFGF23. The answer to the question as to whether or not excess cFGF23 can inhibit iFGF23 action is important, as it will determine whether or not FGF23 replacement would be an effective treatment for patients with iFGF23-deficent HFTC.
Taken together, these findings support a model in which FGF23 processing is a regulated process mediated by the relative activities of ppGalNAcT3 glycosylation and furin degradation; a process in which FGF23 that is fully glycosylated by ppGalNAcT3 is protected from degradation by furin. In FD and Jansen metaphyseal dysplasia cAMP pathways are operative, and in ADHR the HIF-1α pathway may play an important role. Further elucidation of this exciting area of investigation is awaited.
Conclusions
The overall levels of intact biologically active FGF23 reflect the balance between the activity of a subtilisin-like proprotein convertase (most likely furin) that degrades iFGF23 and the galactosyl transferase enzyme, ppGalNAcT3, which by glycosylating FGF23, protects it from degradation (Figure). The processing of iFGF23 to its inactive degradation products is a regulated process that likely has physiological significance. In FD, there is a coordinated, cAMP-mediated regulation of both furin and ppGalNAcT3 enzyme activity that results in lower ppGalNAcT3 activity and elevated furin activity, with the net result of increased FGF23 processing and a proportionally elevated increase in cFGF23 relative to iFGF23. Evidence of altered FGF23 processing has also been observed in other disease states, such as ADHR, and suggest that FGF23 processing may represent a possible target for therapeutic intervention in FGF23-mediated diseases.
Figure.
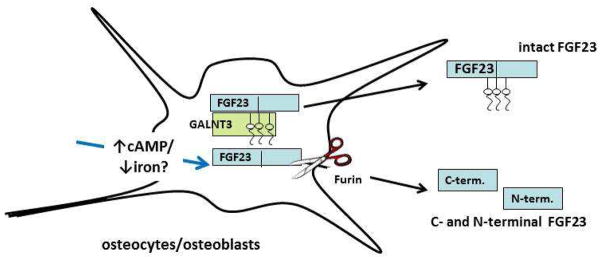
Bone cells (osteocytes and osteoblasts) are responsible for production of FGF23 (blue boxes). The galactosyl transferase UDP-N-acetyl-α-D-galactosamine:polypeptide N-acetylgalactosaminyl transferase (GALNT3, green box) glycosylates FGF23 and by doing so protects it from enzymatic degradation by a subtilisin proprotein convertase, probably furin (scissors). Unglycosylated, unprotected FGF23 (lower blue box) is degraded and C- and N-terminal degradation products (probably biologically inactive) are secreted into the circulation. In fibrous dysplasia, under the regulation of cAMP, the process of degradation is enhanced (blue arrow). This degradation process may also be increased in low iron states, possibly mediated by the HIF-1 pathway.
Acknowledgments
This manuscript was supported by Division of Intramural Research of the National Institute of Dental and Craniofacial Research, National Institutes of Health, Department of Health and Human services (AMB, NB, and MTC), and the Bone Health Program at Children’s National Medical Center (AMB).
Footnotes
Disclosure
AM Boyce declares no conflicts of interest; N Bhattacharyya declares no conflicts of interest; and MT Collins declares no conflicts of interest .
References
Papers of particular interest, published recently, have been highlighted as:
•• Of major importance
• Of importance
- 1.Lichtenstein L. Poloyostotic fibrous dysplasia. Arch Surg. 1938;36:874–898. [Google Scholar]
- 2.Lichtenstein L, Jaffe HL. Fibrous dysplasia of bone: a condition affecting one, several or many bones, the graver cases of which may present abnormal pigmentation of skin, premature sexual development, hyperthyroidism or still other extraskeletal abnormalities. Arch Pathol. 1942;33:777–816. [Google Scholar]
- 3.Collins MT, Bianco P. In: Primer on the Metabolic Bone Diseases and Disorders of Mineral Metabolism. Favus Murray J., editor. Ch 72. American Society for Bone and Mineral Research; 2006. pp. 415–418. [Google Scholar]
- 4.McCune D. Osteitis fibrosa cystica; the case of a nine year old girl who also exhibits precocious puberty, multiple pigmentation of the skin and hyperthyroidism. American journal of diseases of children (1960) 1936;52:743–744. [Google Scholar]
- 5.Albright FBA, Hampton AO, Smith P. Syndrome characterized by osteitis fibrosa disseminata areas of pigmentation and endocrine dysfunction with precocious puberty in females: report of 5 cases. The New England journal of medicine. 1937;216:727–746. [Google Scholar]
- 6•.Dumitrescu CE, Collins MT. McCune-Albright syndrome. Orphanet journal of rare diseases. 2008;3:12. doi: 10.1186/1750-1172-3-12. This reference provides a thorough overview of McCune-Albright syndrome that includes recommendations for the evaluation and management of specific aspects of the disease. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Collins MT, Singer FR, Eugster E. McCune-Albright syndrome and the extraskeletal manifestations of fibrous dysplasia. Orphanet journal of rare diseases. 2012;7(Suppl 1):S4. doi: 10.1186/1750-1172-7-S1-S4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Weinstein LS, et al. Activating mutations of the stimulatory G protein in the McCune-Albright syndrome. N Engl J Med. 1991;325:1688–1695. doi: 10.1056/NEJM199112123252403. [DOI] [PubMed] [Google Scholar]
- 9.Schwindinger WF, Francomano CA, Levine MA. Identification of a mutation in the gene encoding the α subunit of the stimulatory G protein of adenylyl cyclase in McCune-Albright syndrome. Proc Natl Acad Sci USA. 1992;89:5152–5156. doi: 10.1073/pnas.89.11.5152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Happle R. The McCune-Albright syndrome: a lethal gene surviving by mosaicism. Clinical genetics. 1986;29:321–324. doi: 10.1111/j.1399-0004.1986.tb01261.x. [DOI] [PubMed] [Google Scholar]
- 11.Riminucci M, Saggio I, Robey PG, Bianco P. Fibrous dysplasia as a stem cell disease. J Bone Miner Res. 2006;21 (Suppl 2):P125–131. doi: 10.1359/jbmr.06s224. [DOI] [PubMed] [Google Scholar]
- 12.Weinstein LS. G(s)alpha mutations in fibrous dysplasia and McCune-Albright syndrome. J Bone Miner Res. 2006;21 (Suppl 2):P120–124. doi: 10.1359/jbmr.06s223. [DOI] [PubMed] [Google Scholar]
- 13.Landis CA, et al. GTPase inhibiting mutations activate the α chain of Gs and stimulate adenylyl cyclase in human pituitary tumours. Nature. 1989;340:692–696. doi: 10.1038/340692a0. [DOI] [PubMed] [Google Scholar]
- 14.Iiri T, Herzmark P, Nakamoto JM, van Dop C, Bourne HR. Rapid GDP release from Gs alpha in patients with gain and loss of endocrine function. Nature. 1994;371:164–168. doi: 10.1038/371164a0. [DOI] [PubMed] [Google Scholar]
- 15.Riminucci M, et al. Fibrous dysplasia of bone in the McCune-Albright syndrome: abnormalities in bone formation. The American journal of pathology. 1997;151:1587–1600. [PMC free article] [PubMed] [Google Scholar]
- 16.Riminucci M, et al. The histopathology of fibrous dysplasia of bone in patients with activating mutations of the Gs alpha gene: site-specific patterns and recurrent histological hallmarks. J Pathol. 1999;187:249–258. doi: 10.1002/(SICI)1096-9896(199901)187:2<249::AID-PATH222>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- 17.Corsi A, et al. Osteomalacic and hyperparathyroid changes in fibrous dysplasia of bone: core biopsy studies and clinical correlations. J Bone Miner Res. 2003;18:1235–1246. doi: 10.1359/jbmr.2003.18.7.1235. [DOI] [PubMed] [Google Scholar]
- 18.Bianco P, et al. Mutations of the GNAS1 gene, stromal cell dysfunction, and osteomalacic changes in non-McCune-Albright fibrous dysplasia of bone. J Bone Miner Res. 2000;15:120–128. doi: 10.1359/jbmr.2000.15.1.120. [DOI] [PubMed] [Google Scholar]
- 19.Leet AI, et al. Fracture incidence in polyostotic fibrous dysplasia and the McCune-Albright syndrome. J Bone Miner Res. 2004;19:571–577. doi: 10.1359/JBMR.0301262. [DOI] [PubMed] [Google Scholar]
- 20.Hart ES, et al. Onset, progression, and plateau of skeletal lesions in fibrous dysplasia and the relationship to functional outcome. J Bone Miner Res. 2007;22:1468–1474. doi: 10.1359/jbmr.070511. [DOI] [PubMed] [Google Scholar]
- 21.Kelly MH, Brillante B, Collins MT. Pain in fibrous dysplasia of bone: age-related changes and the anatomical distribution of skeletal lesions. Osteoporos Int. 2008;19:57–63. doi: 10.1007/s00198-007-0425-x. [DOI] [PubMed] [Google Scholar]
- 22.Collins MT, et al. An instrument to measure skeletal burden and predict functional outcome in fibrous dysplasia of bone. J Bone Miner Res. 2005;20:219–226. doi: 10.1359/JBMR.041111. [DOI] [PubMed] [Google Scholar]
- 23.Akintoye SO, et al. Characterization of gsp-mediated growth hormone excess in the context of McCune-Albright syndrome. J Clin Endocrinol Metab. 2002;87:5104–5112. doi: 10.1210/jc.2001-012022. [DOI] [PubMed] [Google Scholar]
- 24.Boyce AM, et al. Optic Neuropathy in McCune-Albright Syndrome: Effects of Early Diagnosis and Treatment of Growth Hormone Excess. The Journal of clinical endocrinology and metabolism. 2012 doi: 10.1210/jc.2012-2111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Prader A, Illig R, Uehlinger E, Stalder G. Rickets following bone tumor. Helvetica paediatrica acta. 1959;14:554–565. [PubMed] [Google Scholar]
- 26••.Chong WH, Molinolo AA, Chen CC, Collins MT. Tumor-induced osteomalacia. Endocrine-related cancer. 2011;18:R53–77. doi: 10.1530/ERC-11-0006. This article is a thorough review of tumor-induced osteomalacia (TIO) that includes guidance on the diagnosis, tumor localization, and when necessary, medical treatment of TIO. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jan de Beur SM. Tumor-induced osteomalacia. Jama. 2005;294:1260–1267. doi: 10.1001/jama.294.10.1260. [DOI] [PubMed] [Google Scholar]
- 28.ADHR C. Autosomal dominant hypophosphataemic rickets is associated with mutations in FGF23. The ADHR Consortium. Nat Genet. 2000;26:345–348. doi: 10.1038/81664. [DOI] [PubMed] [Google Scholar]
- 29.White KE, et al. The autosomal dominant hypophosphatemic rickets (ADHR) gene is a secreted polypeptide overexpressed by tumors that cause phosphate wasting. J Clin Endocrinol Metab. 2001;86:497–500. doi: 10.1210/jcem.86.2.7408. [DOI] [PubMed] [Google Scholar]
- 30.Jonsson KB, et al. Fibroblast growth factor 23 in oncogenic osteomalacia and X-linked hypophosphatemia. N Engl J Med. 2003;348:1656–1663. doi: 10.1056/NEJMoa020881. [DOI] [PubMed] [Google Scholar]
- 31.Riminucci M, et al. FGF-23 in fibrous dysplasia of bone and its relationship to renal phosphate wasting. J Clin Invest. 2003;112:683–692. doi: 10.1172/JCI18399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Collins MT, et al. Renal phosphate wasting in fibrous dysplasia of bone is part of a generalized renal tubular dysfunction similar to that seen in tumor-induced osteomalacia. J Bone Miner Res. 2001;16:806–813. doi: 10.1359/jbmr.2001.16.5.806. [DOI] [PubMed] [Google Scholar]
- 33.Dent CE, Gertner JM. Hypophosphataemic osteomalacia in fibrous dysplasia. Quart J Med. 1976;45:411–420. [PubMed] [Google Scholar]
- 34.Mirams M, Robinson BG, Mason RS, Nelson AE. Bone as a source of FGF23: regulation by phosphate? Bone. 2004;35:1192–1199. doi: 10.1016/j.bone.2004.06.014. [DOI] [PubMed] [Google Scholar]
- 35.Sitara D, et al. Homozygous ablation of fibroblast growth factor-23 results in hyperphosphatemia and impaired skeletogenesis, and reverses hypophosphatemia in Phex-deficient mice. Matrix Biol. 2004;23:421–432. doi: 10.1016/j.matbio.2004.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Feng JQ, et al. Loss of DMP1 causes rickets and osteomalacia and identifies a role for osteocytes in mineral metabolism. Nat Genet. 2006;38:1310–1315. doi: 10.1038/ng1905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kuznetsov SA, et al. Age-dependent demise of GNAS-mutated skeletal stem cells and “normalization” of fibrous dysplasia of bone. J Bone Miner Res. 2008;23:1731–1740. doi: 10.1359/jbmr.080609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shimada T, et al. Cloning and characterization of FGF23 as a causative factor of tumor-induced osteomalacia. Proc Natl Acad Sci U S A. 2001;98:6500–6505. doi: 10.1073/pnas.101545198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shimada T, et al. FGF-23 is a potent regulator of vitamin D metabolism and phosphate homeostasis. J Bone Miner Res. 2004;19:429–435. doi: 10.1359/JBMR.0301264. [DOI] [PubMed] [Google Scholar]
- 40.Shimada T, et al. Targeted ablation of Ffg23 demonstrates an essential physiological role of FGF23 in pohsphate and vitamin D metabolism. J Clin Invest. 2004;113:561–568. doi: 10.1172/JCI19081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Larsson T, Nisbeth U, Ljunggren O, Juppner H, Jonsson KB. Circulating concentration of FGF-23 increases as renal function declines in patients with chronic kidney disease, but does not change in response to variation in phosphate intake in healthy volunteers. Kidney Int. 2003;64:2272–2279. doi: 10.1046/j.1523-1755.2003.00328.x. [DOI] [PubMed] [Google Scholar]
- 42.Ferrari SL, Bonjour JP, Rizzoli R. Fibroblast growth factor-23 relationship to dietary phosphate and renal phosphate handling in healthy young men. J Clin Endocrinol Metab. 2005;90:1519–1524. doi: 10.1210/jc.2004-1039. [DOI] [PubMed] [Google Scholar]
- 43.Nishida Y, et al. Acute effect of oral phosphate loading on serum fibroblast growth factor 23 levels in healthy men. Kidney international. 2006;70:2141–2147. doi: 10.1038/sj.ki.5002000. [DOI] [PubMed] [Google Scholar]
- 44.Ito N, et al. Effect of acute changes of serum phosphate on fibroblast growth factor (FGF)23 levels in humans. J Bone Miner Metab. 2007;25:419–422. doi: 10.1007/s00774-007-0779-3. [DOI] [PubMed] [Google Scholar]
- 45.Collins MT, et al. Fibroblast growth factor-23 is regulated by 1alpha,25-dihydroxyvitamin D. J Bone Miner Res. 2005;20:1944–1950. doi: 10.1359/JBMR.050718. [DOI] [PubMed] [Google Scholar]
- 46.Brownstein CA, et al. A translocation causing increased alpha-klotho level results in hypophosphatemic rickets and hyperparathyroidism. Proc Natl Acad Sci U S A. 2008;105:3455–3460. doi: 10.1073/pnas.0712361105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47•.Smith RC, et al. Circulating alphaKlotho influences phosphate handling by controlling FGF23 production. J Clin Invest. 2012;122:4710–4715. doi: 10.1172/JCI64986. The preceding two articles (Brownsetin et al, and Smith et al) provide important evidence for a direct role for circulating α-Klotho in the regulation of FGF23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rafaelsen SH, et al. Exome sequencing reveals FAM20c mutations associated with FGF23-related hypophosphatemia, dental anomalies and ectopic calcification. J Bone Miner Res. 2013 doi: 10.1002/jbmr.1850. [DOI] [PubMed] [Google Scholar]
- 49.Yamashita T, Yoshioka M, Itoh N. Identification of a novel fibroblast growth factor, FGF-23, preferentially expressed in the ventrolateral thalamic nucleus of the brain. Biochem Biophys Res Commun. 2000;277:494–498. doi: 10.1006/bbrc.2000.3696. [DOI] [PubMed] [Google Scholar]
- 50.Urakawa I, et al. Klotho converts canonical FGF receptor into a specific receptor for FGF23. Nature. 2006 doi: 10.1038/nature05315. [DOI] [PubMed] [Google Scholar]
- 51.Kurosu H, et al. Regulation of fibroblast growth factor-23 signaling by klotho. J Biol Chem. 2006;281:6120–6123. doi: 10.1074/jbc.C500457200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52••.Faul C, et al. FGF23 induces left ventricular hypertrophy. J Clin Invest. 2011;121:4393–4408. doi: 10.1172/JCI46122. This is an excellent article that provides both clinical and experimental evidence for the “off-target,” i.e. non-mineral metabolism effects of FGF23 in the pathophysiology of FGF23-mediated cardiovascular disease in renal failure. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gupta A, Winer K, Econs MJ, Marx SJ, Collins MT. FGF-23 is elevated by chronic hyperphosphatemia. J Clin Endocrinol Metab. 2004;89:4489–4492. doi: 10.1210/jc.2004-0724. [DOI] [PubMed] [Google Scholar]
- 54.Weinman EJ, Steplock D, Shenolikar S, Biswas R. Fibroblast growth factor-23-mediated inhibition of renal phosphate transport in mice requires sodium-hydrogen exchanger regulatory factor-1 (NHERF-1) and synergizes with parathyroid hormone. J Biol Chem. 2011;286:37216–37221. doi: 10.1074/jbc.M111.288357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Weinman EJ, Lederer ED. NHERF-1 and the regulation of renal phosphate reabsoption: a tale of three hormones. American journal of physiology Renal physiology. 2012;303:F321–327. doi: 10.1152/ajprenal.00093.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kato K, et al. Polypeptide GalNAc-transferase T3 and familial tumoral calcinosis. Secretion of fibroblast growth factor 23 requires O-glycosylation. J Biol Chem. 2006;281:18370–18377. doi: 10.1074/jbc.M602469200. [DOI] [PubMed] [Google Scholar]
- 57••.Bhattacharyya N, Chong WH, Gafni RI, Collins MT. Fibroblast growth factor 23: state of the field and future directions. Trends in endocrinology and metabolism: TEM. 2012;23:610–618. doi: 10.1016/j.tem.2012.07.002. This article provides an excellent overview of the subject of FGF23, highlighting key papers in the development of our current understanding of the physiology and pathophysiology of FGF23 and pointing to the future direction to which the current research is leading. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gram Schjoldager KT, et al. A systematic study of site-specific GalNAc-type O-glycosylation modulating proprotein convertase processing. J Biol Chem. 2011;286:40122–40132. doi: 10.1074/jbc.M111.287912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Fukumoto S. Post-translational modification of Fibroblast Growth Factor 23. Ther Apher Dial. 2005;9:319–322. doi: 10.1111/j.1744-9987.2005.00289.x. [DOI] [PubMed] [Google Scholar]
- 60.Frishberg Y, et al. Hyperostosis-hyperphosphatemia syndrome: a congenital disorder of O-glycosylation associated with augmented processing of fibroblast growth factor 23. J Bone Miner Res. 2007;22:235–242. doi: 10.1359/jbmr.061105. [DOI] [PubMed] [Google Scholar]
- 61•.Bhattacharyya N, et al. Mechanism of FGF23 processing in fibrous dysplasia. J Bone Miner Res. 2012;27:1132–1141. doi: 10.1002/jbmr.1546. This paper is an important paper that details the mechanism of regulation of FGF23 processing in fibrous dysplasia. It points to the potentially important role FGF23 processing may play in mineral metaoblism physiology. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Yuan B, et al. Hexa-D-arginine treatment increases 7B2*PC2 activity in hyp-mouse osteoblasts and rescues the HYP phenotype. J Bone Miner Res. 2013;28:56–72. doi: 10.1002/jbmr.1738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Topaz O, et al. Mutations in GALNT3, encoding a protein involved in O-linked glycosylation, cause familial tumoral calcinosis. Nat Genet. 2004;36:579–581. doi: 10.1038/ng1358. [DOI] [PubMed] [Google Scholar]
- 64.Benet-Pages A, Orlik P, Strom TM, Lorenz-Depiereux B. An FGF23 missense mutation causes familial tumoral calcinosis with hyperphosphatemia. Hum Mol Genet. 2004 doi: 10.1093/hmg/ddi034. [DOI] [PubMed] [Google Scholar]
- 65.Ichikawa S, et al. A homozygous missense mutation in human KLOTHO causes severe tumoral calcinosis. J Clin Invest. 2007;117:2684–2691. doi: 10.1172/JCI31330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Bergwitz C, et al. Defective O-glycosylation due to a novel homozygous S129P mutation is associated with lack of fibroblast growth factor 23 secretion and tumoral calcinosis. J Clin Endocrinol Metab. 2009;94:4267–4274. doi: 10.1210/jc.2009-0961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Semenov AG, et al. Processing of pro-brain natriuretic peptide is suppressed by O-glycosylation in the region close to the cleavage site. Clinical chemistry. 2009;55:489–498. doi: 10.1373/clinchem.2008.113373. [DOI] [PubMed] [Google Scholar]
- 68.Brown WW, et al. Hypophosphatemia with elevations in serum fibroblast growth factor 23 in a child with Jansen’s metaphyseal chondrodysplasia. J Clin Endocrinol Metab. 2009;94:17–20. doi: 10.1210/jc.2008-0220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Durham BH, Joseph F, Bailey LM, Fraser WD. The association of circulating ferritin with serum concentrations of fibroblast growth factor-23 measured by three commercial assays. Annals of clinical biochemistry. 2007;44:463–466. doi: 10.1258/000456307781646102. [DOI] [PubMed] [Google Scholar]
- 70.Imel EA, et al. Iron modifies plasma FGF23 differently in autosomal dominant hypophosphatemic rickets and healthy humans. The Journal of clinical endocrinology and metabolism. 2011;96:3541–3549. doi: 10.1210/jc.2011-1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Farrow EG, et al. Iron deficiency drives an autosomal dominant hypophosphatemic rickets (ADHR) phenotype in fibroblast growth factor-23 (Fgf23) knock-in mice. Proc Natl Acad Sci U S A. 2011;108:E1146–1155. doi: 10.1073/pnas.1110905108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Goetz R, et al. Isolated C-terminal tail of FGF23 alleviates hypophosphatemia by inhibiting FGF23-FGFR-Klotho complex formation. Proc Natl Acad Sci U S A. 2009 doi: 10.1073/pnas.0902006107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ichikawa S, Austin AM, Gray AK, Econs MJ. A Phex mutation in a murine model of X-linked hypophosphatemia alters phosphate responsiveness of bone cells. J Bone Miner Res. 2012;27:453–460. doi: 10.1002/jbmr.544. [DOI] [PMC free article] [PubMed] [Google Scholar]