

Abstract
The role of cannabinoid receptors in inflammation has been the topic of many research endeavors. Despite this effort, to date the involvement of the endocannabinoid system (ECS) in inflammation remains obscure. The ambiguity of cannabinoid involvement may be explained by the existence of cannabinoid receptors, other than CB1 and CB2, or a consequence of interaction of endocannabinoids with other signaling systems. GPR55 has been proposed to be a cannabinoid receptor; however the interaction of the endocannabinoid system with GPR55 remains elusive. Consequently this study set about to examine the effects of the endocannabinoids, anandamide (AEA) and virodhamine, on GPR55 mediated signaling. Specifically, we assessed changes in β-arrestin2 (βarr2) distribution and GPR55 receptor internalization following activation by lysophosphatidylinositol (LPI), the synthetic cannabinoid ligand SR141716A, and new selective synthetic GPR55 agonists. Data obtained from the experiments presented herein demonstrate that AEA and virodhamine modulate agonist-mediated recruitment of βarr2. AEA and virodhamine act as partial agonists; enhancing the agonist effect at low concentrations and inhibiting it at high concentrations. Furthermore, both virodhamine and AEA significantly attenuated agonist-induced internalization of GPR55. These effects are attributed to the expression of GPR55, and not CB1 and CB2 receptors, as we have established negligible expression of CB1 and CB2 in these GPR55-transfected U2OS cells. The identification of select endocannabinoids as GPR55 modulators will aide in elucidating the function of GPR55 in the ECS.
Discoveries of the CB1 receptor, a G-protein coupled receptor (GPCR), and of an endogenous ligand which activates it, arachidonic acid ethanolamide, named “anandamide” (AEA) for the Sanskrit word for “bliss (Devane et al. 1992b), have led to subsequent breakthroughs elucidating an endocannabinoid system (ECS) (Di Marzo et al. 1998). Within the ECS, six endogenous compounds (Scotter et al., 2010) have been isolated thus far, and two receptors (CB1 and CB2) have been definitively characterized (Pertwee et al. 2010). In the quest to understand the ECS, and to ascribe biological functions to one or both of these receptors, many different reporter assays and model systems have been employed. Results from these research efforts are often contradictory, suggesting that signal transduction via CB1 and CB2 is dependent upon the assay system. Furthermore, studies employing cannabinoid receptor knockout mice suggest the existence of additional cannabinoid receptors. Previous reports have proposed the existence of a third cannabinoid receptor, GPR55 (Ryberg et al., 2007; Lauckner et al., 2008). However, the reported profiles of ligands that activate and inhibit GPR55 are contradictory. For example, activation of GPR55 by the endocannabinoids AEA and virodhamine yielded inconsistent results. AEA was equipotent in activating GPR55, CB1 and CB2 receptors according to Ryberg et al (2007), but much less potent in stimulating increases in intracellular calcium (Lauckner et al. 2008). Previous findings from our laboratory indicated that AEA did not activate GPR55 (Kapur et al., 2009). Agonist activity of virodhamine at GPR55 with potency much higher than that observed for CB1 and CB2 (12 nM vs 2.9 μM and 381 nM, respectively) has been demonstrated (Ryberg et al., 2007). In contrast, other reports indicated that virodhamine was a low potency (3 μM) GPR55 agonist (Lauckner et al. 2008). Virodhamine has also been reported to act as a partial agonist/antagonist at the CB1 receptor and a full agonist at the CB2 receptor (Porter et al., 2002).
Tissue levels of virodhamine and AEA have been measured in peripheral and brain regions (Porter et al., 2002). Quantities of these two endocannabinoids were similar in most brain regions, except for brainstem and striatum where AEA was 2- to 4- fold higher than virodhamine. In peripheral tissues, the concentration of virodhamine was found to be 2- to 9 fold higher than AEA. Immune tissue such as the spleen exhibited almost 8-fold higher levels of virodhamine as compared to AEA. This difference in tissue levels may reflect the affinity of degradative enzymes, or uptake transporter mechanisms for these two ligands. Porter et al. (2002) have demonstrated that virodhamine inhibits AEA transport, indicating that these two endocannabinoids share common uptake mechanisms.
Localization of GPR55 to human neutrophils (PMNs) has recently been reported (Balenga et al. 2011). Furthermore, these investigators established that activation of GPR55 by LPI augmented the migratory response of PMNs towards the endocannabinoid, 2-arachidonyl glycerol (2-AG). Since 2-AG is regarded as the primary endocannabinoid at the CB2 receptor (Sugiura et al., 2000), the findings by Balenga et al (2011) supported an interaction of GPR55 and CB2. In addition to the presence of GPR55 in PMN’s, it has also been localized to primary cultures of microglia, as well as to the murine microglial cell line, BV2 (Pietr et al., 2009). Furthermore, these investigators reported that lipopolysaccharide (LPS) and interferon gamma (IFN-γ), established activators of microglia, differentially modulated GPR55 and CB2 expression levels. Both GPR55 and CB2 mRNAs were found to be downregulated by LPS in both primary microglia and BV-2 cells (Pietr et al. 2009). However, IFN-γ activation resulted in the up regulation of GPR55 and CB2 in BV2 cells and a down regulation in primary microglia. To elucidate the involvement of GPR55 in the inflammatory response, identification of endogenous compounds which antagonize known GPR55 mediated inflammatory events, such as migration, are imperative.
Our previous studies have demonstrated that the distribution of β-arrestin2 (βarr2) is an indication of GPR55 responsiveness. In the presence of agonist, GPR55 recruits βarr2 to the membrane. Consequently, this study set about to examine the effects of the endocannabinoids AEA and virodhamine on GPR55 mediated signaling. Specifically, we assessed the effects of AEA and virodhamine on changes in β-arrestin distribution and GPR55 receptor internalization following activation by LPI, the synthetic cannabinoid ligand SR141716A, and new selective synthetic GPR55 agonists which we identified from a non-biased screen for GPR55 activity.
Materials and Methods
Reagents
Dulbecco’s modified Eagle’s medium (DMEM), Hanks’ balanced salt solution (HBSS), fetal bovine serum, (FBS) and phosphate buffered saline (PBS, with or without Ca2+ and Mg2+) were purchased from Cellgro, Mediatech, Inc and Hyclone (Fisher). Triton X-100 was purchased from Fisher.G418 was purchased from A..G. Scientific, (San Diego, CA). Anti-HA mouse monoclonal antibody was purchased from Covance (Emeryville, CA). Alexa Fluor 568 goat anti-mouse antibody and zeocin were purchased from Invitrogen (Carlsbad,CA). LPI (L7635) and bovine serum albumin (BSA) were purchased from Sigma-Aldrich Corp, (St. Louis, MO). Virodhamine, (O-arachidonyl ethanol amine) and anandamide (N-arachidonylethanolamide) were purchased from Tocris Bioscience, (Ellisville, MO). SR141716A was obtained from the National Institute on Drug Abuse (NIDA) Research Resources Drug Supply System (Research Triangle Park, NC, USA). GPR55 selective agonists ML184 (CID 2440433), ML185 (CID1374043) and ML186 (CID15945391) were obtained from the Sanford-Burnham Center for Chemical Genomics & John C. Reed (Kotsikorou et al. 2011).
Cell lines
U2OS cells permanently expressing HA-GPR55E and βarr2-GFP (green fluorescent protein) were obtained from Drs. Larry Barak and Marc Caron (Duke University) and have previously been described (Kapur et al. 2009). Development of the CB1-HEK293 cell line is described in Tao and Abood (1998) and CB2-CHO cells in Showalter et al. (1996).
RNA extraction, RT-PCR and quantitative real time PCR (qRT-PCR)
Total RNA was isolated from HA-GPR55E/βarr2-GFP U2OS, U2OS, CB1-HEK293 and CB2-CHO cell lines using TRIzol reagent (Invitrogen) and DNase treated using Rnase-DNA free kit (Qiagen,MD). RNA was quantified using spectrophotometry and quality was confirmed by formaldehyde-agarose gel electrophoresis. For cDNA synthesis, 1 μg total RNA was reverse transcribed using Superscript First Strand Synthesis System (Invitrogen). cDNA samples was amplified using the cycling program: 94°C for 7 min and 30 cycles of 94°C for 1 min, 55°C for 1 min, and 72°C for 1 min. The reaction products were analyzed on a 3% agarose gel for CB1 and CB2 expression.
Gene Expression for CB1 and CB2 in the cell lines was determined by qRT-PCR analysis using SYBR Green Master Mix (Applied Biosystems). Reactions were run on a 7,500 Real-Time PCR System (Applied Biosystems). Primers for qRT-PCR as as follows: CB1 (forward: 5′-GACCATAGCCATTGTGATCG-3′ and reverse: 5′GGTTTCATCAATGTGTGGGA 3′, 99 bp transcript), and CB2 (forward: 5′-GACCGCCATTGACCGATACC-3′and reverse: 5′-GGACCCACATGATGCCCAG-3; 100bp transcript). The cycling program was as follows: 50°C for 2 min, 95°C for 10 min, 40 cycles of 95°C for 15 sec and 60°C for 1 min. Three independent experiments were performed for each gene and each experiment was performed in triplicate. Data are expressed as the average Ct value.
LDH Cytotoxicity assay
Lactate dehydrogenase (LDH) release was used as an indicator of cell membrane damage as a consequence of exposure to agonist and antagonist compounds. Supernatants from HA-GPR55E/βarr2-GFP U2OS cells were evaluated using the LDH Cytotoxicity Assay Kit (Cayman Chemical Company, MI). This colorimetric assay is based on the cleavage of a tetrazolium salt when LDH activity is present in the cell culture supernatant.
Briefly, cells were seeded at 50,000 cells/well onto a 96-well plate (BD Falcon ™) and grown to confluency. Confluent cells were deprived of serum for 24 h, at which time the assay was performed. Aliquots (100 μl) of supernatant and reaction mixture were transferred into corresponding wells of an optically clear 96- well flat -bottom microtiter plate and incubated for up to 30 min at RT. The increase in enzyme activity directly correlates to the amount of formazan produced by the reduction of the tetrazolium salt. The absorbacnce was measured at 490 nm using a spectrophotometric microtiter plate reader (Wallac Victor 2, Perkin Elmer, USA). Background and negative controls were obtained by LDH measurement of assay medium and wells containing no cells. Unexposed cells represented the negative control whereas total cellular LDH release was obtained by treatment with 1% Triton X-100. Data from control and treated cells were normalized to total LDH release and calculated as percentage cytotoxicity. The data represent the mean of two independent experiments, each using triplicate wells per drug treatment.
β-arrestin Assessment of GPR55 Activation
U2OS cells permanently expressing HA-GPR55E and βarr2-GFP were seeded onto glass coverslips at 80–85% confluence and placed in 24-well plates (BD Falcon ™). Cells were maintained at 37°C in 5% CO2 overnight. Cells were washed briefly with Hanks’ balanced salt solution (HBSS) before drug application. Experiments were performed using HBSS. Agonist-stimulated redistribution of βarr2-GFP was assessed following 40 minute (min) drug treatment at room temperature (RT). Cells were then fixed with 4% paraformaldehyde for 25 min at room temperature followed by three washes with PBS and one wash with double distilled water. The antagonism protocol included 15 minutes of pre-incubation with the antagonist, followed by a 40 min co-incubation of antagonist and agonist. Glass coverslips were mounted onto slides and imaged using a fluorescence microscope (Nikon E1000; Tokyo, Japan) using a 40X oil objective and 488 nm excitation for GFP.
Internalization Assay, Immunocytochemistry
HA-GPR55E/βarr2-GFP U2OS cells grown on coverslips were incubated over ice for 40 min with a mouse monoclonal anti-HA antibody (1:500) diluted in PBS containing 3% bovine serum albumin (blocking buffer). This was followed by three washes (3 times at RT) and 40 min incubation with the secondary antibody Alexa Fluor 568 goat anti-mouse (1:1500 in blocking buffer). Antibody-labeled cells were treated with compounds for 40 min (RT) in HBSS. The antagonism protocol included a pre-incubation of 15 min (RT) with the antagonist compounds. Cells were fixed and imaged with fluorescence microscopy (as described above) using a 40X oil objective and 568-nm excitation for Alexa Fluor 568 antibody.
Confocal Microscopy
Representative images of βarr2 recruitment and the internalization assays were visualized with a Leica DM IRE2 confocal microscope with a TCS SL system. For GFP visualization, the excitation wavelength was 488 nm and emission was captured at wavelengths between 475 and 575 nm; YFP was excited at 488 nm, and emission was captured at wavelengths between 500 and 625 nm.
Data Quantification and Statistical analyses
The RGB color Images captured from the fluorescent microscope were transformed into 8-bit gray-scale images using the Automate-Batch function in Adobe Photoshop CS5. To quantify βarr2-GFP aggregates, Gray-scale images were processed through ImageJ software, (http://rsbweb.nih.gov/ij/), using a custom written plug-in provided by Pingwei Zhao, Temple University). Briefly, the plug-in extracts those pixels that generate objects of interest that fall within a predetermined range of sizes and intensities after varying local backgrounds are subtracted from the images. The assay’s readout was based on the number of detected fluorescent spots representing pits of vesicles. Inhibition curves were analyzed by nonlinear regression using Graph-Pad Prism 5.0 software (GraphPad, San Diego, CA), and data was fitted to sigmoidal concentration-response curves to obtain IC50 values. Activity values were normalized to the agonist’s response (3μM LPI or 30 μM SR141716A was considered as 100%). Statistical analysis was performed using one-way analysis of variance (ANOVA) followed by a Dunnett’s post-test. The data presented represents mean values ±SEM. *<0.05; **<0.01, ***<0.001 are considered as statistically different.
Results
Virodhamine and AEA inhibit GPR55-mediated βarr2-GFP re-distribution
Our previous study demonstrated that the distribution pattern of βarr2-GFP is indicative of GPR55 responsiveness (Kapur et al. 2009). Using this assay, three agonists at GPR55 were determined; LPI, SR141716A and AM251 with EC50 values of 1.2 μM, 3.9 μM and 9.6 μM respectively. Importantly, the ability of these compounds to activate GPR55 was further validated in other downstream assays by our group and others (Kapur et al. 2009; Henstridge et al. 2009; Oka et al. 2007; Yin et al. 2009; Balenga et al. 2011). Both AEA and virodhamine have been previously reported to activate GPR55 (Ryberg et al. 2007; Lauckner et al. 2008). Another previous report utilizing a β-arrestin-based assay for readout demonstrated a weak response mediated by endocannabinoids at GPR55 (Yin et al. 2009). In our hands, however, AEA alone at varying concentrations of 0.1–30 μM did not result in recruitment of βarr2-GFP (Fig. 1c). In contrast, virodhamine resulted in a weak recruitment of βarr2-GFP (Fig. 2c). Since virodhamine appeared to function as a partial agonist, we examined the ability of both virodhamine and AEA to antagonize recruitment of GPR55-mediated βarr2-GFP trafficking. For the antagonist assay, cells were pre-incubated with AEA (0.01–30 μM) or with virodhamine (0.01–30 μM) 15 min prior to the application of the agonist (LPI or SR141716A) at the EC90 concentration. As illustrated in Figure 1(a–c), application of AEA significantly inhibited the βarr2-GFP response mediated by the agonists. AEA elicited a concentration–dependent inhibition of LPI response with an IC50 value of 5.91 (1.14–30.8) μM and a maximal inhibition of 20% (Fig 1a). AEA produced a concentration–dependent inhibition of SR141716A response with an IC50 value of 12.0 (4.6–34.7) μM and a maximal inhibition of 65% (Fig 1b). Notably, at low concentrations, AEA enhanced the recruitment of LPI, and to a lesser extent, that of SR14716A (Fig 1a and b). Similar to AEA, pre-incubation with virodhamine resulted in a concentration-dependent inhibition of LPI and SR141716A with IC50 values of 6.57 (1.58–27.4) μM and 9.44 (3.07–29.07) μM and maximal inhibition of 46% and 68%, respectively (Fig 2a–c). As seen with AEA, lower concentrations of virodhamine enhanced the recruitment mediated by LPI and SR141716A.
Fig. 1. AEA inhibits agonist mediated βarr2-GFP distribution in U2OS cells co-expressing GPR55E and βarr2-GFP.

(A) (i–iv) Cells were pre-incubated with increasing concentrations of the antagonist (AEA) for 15 min prior to agonist application (3 μM LPI or 30 μM SR141716A). The response was normalized to the averaged response obtained by the agonist. Data are mean ± SEM from three independent experiments performed in duplicate (*P<0.05, **P<0.01, ***P<0.001). (a) AEA inhibition of LPI. (b) AEA inhibition of SR141716A. (c) Representative images, captured with confocal microscopy at 40x magnification depicting βarr2-GFP recruitment pattern are illustrated. Cells treated with vehicle and 30 μM AEA show that βarr2-GFP is homogenously distributed in the cytoplasm. Treatment with 3 μM LPI and 30 μM SR141716A resulted in cytosolic distribution βarr2-GFP which was largely attenuated following treatment with AEA.
Fig. 2. Virodhamine inhibits agonist mediated βarr2-GFP distribution in U2OS cells co-expressing GPR55E and βarr2-GFP.

Cells were pre-incubated with increasing concentrations of virodhamine for 15 min prior to agonist application (3 μM LPI or 30 μM SR141716A). The response was normalized to the averaged response obtained by the agonist. Data are mean ± SEM from three independent experiments performed in duplicate (*P<0.05, **P<0.01, ***P<0.001). (a) Virodhamine inhibition of LPI. (b) Virodhamine inhibition of SR141716A. (c) Representative images, captured with confocal microscopy at 40x magnification depicting βarr2-GFP recruitment pattern are illustrated. Cells treated with vehicle and 30 μM virodhamine show that βarr2-GFP is homogenously distributed in the cytoplasm. Treatment with 3 μM LPI and 30 μM SR141716A resulted in cytosolic distribution of β-arr2-GFP which was largely attenuated following treatment with virodhamine.
Virodhamine and AEA inhibit GPR55’s internalization
Receptor internalization upon ligand stimulation is a key component of a cell’s response and allows a cell to correctly sense its environment (Grimsey et al. 2011). We and others have previously demonstrated the internalization pattern of GPR55 following agonist binding (Kapur et al., 2009, Henstridge et al., 2010). In our studies we utilized an HA-epitope tagged variant of GPR55 which permits visualization of accumulated HA fluorescence within centralized intracellular locations. To confirm the ability of virodhamine and AEA to inhibit GPR55 agonist mediated signaling, we evaluated their effects on the internalization process mediated by agonist-receptor binding. Cells were pre-incubated for 15 min with 10 μM AEA or 10 μM virodhamine prior to agonist application. The GPR55 internalization process was subsequently monitored. Fig 3a depicts membrane bound receptors upon treatment with vehicle (DMSO plus EtOH). As expected, application of either 3 μM LPI or 30 μM SR141716A resulted in a loss of membrane staining and the appearance of cytosolic fluorescent aggregates corresponding to internalized receptors (Fig 3b,c). While AEA alone revealed the presence of membrane bound receptors (Fig 3g), in the presence of agonist LPI (Fig 3h) or SR141716A (Fig 3i), AEA markedly decreased receptor internalization. Similar to AEA, receptors were localized to the membrane following treatment with virodhamine alone (Fig 3m), whereas in the presence of LPI (Fig 3n) or SR141716A (Fig 3o), virodhamine strikingly decreased receptor internalization. We have further tested the ability of AEA and virodhamine to decrease receptor internalization following application of newly identified selective agonists at GPR55. In a recent screen run in collaboration with the Molecular Libraries Probe Production Centers Network program, we have identified potent GPR55-selective agonists ML184, ML 185 and ML186 with EC50 values of 263 nM, 658 nM and 305 nM respectively; β-arrestin trafficking, phospholipase C recruitment and phosphorylation of ERK1/2 were used as measures of agonist activity (Kotsikorou et al. 2011; Heynen-Genel et al. 2010). As illustrated in Fig 3(d–f), application of 5 μM of each of the ML compounds resulted in a significant increase of cytosolic fluorescent aggregates corresponding to internalized receptors. This response was largely attenuated following pre-incubation with either 10 μM AEA (Fig 3j–l) or virodhamine (Fig 3p–r).
Fig. 3. AEA and Virodhamine inhibit agonist-mediated GPR55E internalization.
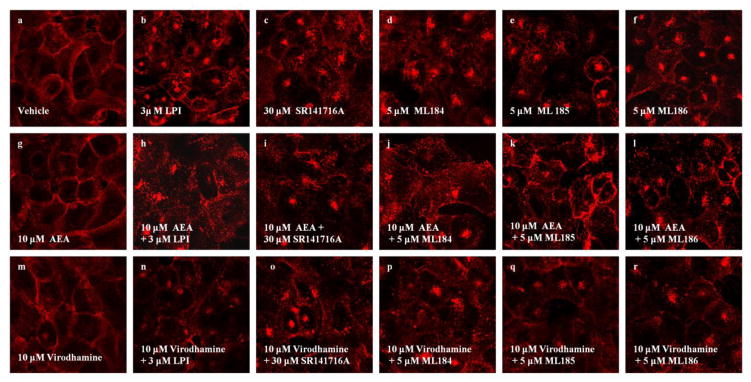
U2OS cells expressing HAGPR55E were pre-labeled with anti-HA antibody and Alexa Fluor 568 antibody. Cells were then pre-incubated with virodhamine (3 μM) for 15 min, followed by additional of 40 min incubation along with the agonist. Membrane staining was captured by confocal microscopy at 63x magnification. Upon treatment with vehicle (a) 10 μM AEA (g) and 10 μM virodhamine (m), cells show primarily membrane localization of GPR55. Treatment with 3 μM LPI (b), 30 μM SR141716A (c), 5 μM ML184 (d), 5 μM ML185 (e) and 5 μM ML186 (f), resulted in receptor internalization, evident by loss of plasma membrane receptor staining. Treatment with 10 μM AEA and 10 μM virodhamine largely attenuated receptor internalization induced by 3 μM LPI (h,n), 30 μM SR141716A (i,o), 5 μM ML184 (j,p), 5 μM ML185 (k,q) and 5 μM ML186 (l,r). Data are representative examples of at least three individual experiments.
U2OS HAGPR55E/βarr2-GFP cells do not express CB1 or CB2 receptors
Since both AEA and virodhamine have been previously demonstrated to activate the well characterized CB1 and CB2 receptors (Pertwee et al. 2010; Porter et al. 2002), it was important to rule out their involvement in the observed effects. In other words, we sought to determine that the inhibition of the agonist mediated signaling was attributed to AEA and virodhamine acting solely at GPR55. Using qRT-PCR we measured mRNA levels of CB1 and CB2 receptors in U2OS HA-GPR55E/βarr2-GFP cells (Fig 4a–c). Fig 4a depicts cDNA transcripts of eukaryotic 18s rRNA in wild-type U2OS, HAGPR55E/βarr2-GFP U2OS, CB1-HEK293 and CB2-CHO cell lines. The negative control for the PCR reaction contained no template mRNA (NTC). As illustrated in Fig 4b, CB1 and CB2 receptor expression was not present in U2OS or HAGPR55E/βarr2-GFP U2OS cell lines. A graph of the average Ct value corresponding to levels of CB1 and CB2 expression in their respective cell lines as compared to levels of expression in U2OS HAGPR55E/βarr2-GFP cells is shown in Fig 4c. The relationship of Ct value is inversely proportional to the level of expression. As expected, low average Ct values were obtained for CB1 in HEKCB1 cells and CB2 in CHOCB2 cells. In HAGPR55E/βarr2-GFP U2OS cells, however, the average Ct values for CB1 and CB2 were 30 and 32 respectively, indicating that they do not express any significant levels of CB1 or CB2 receptors.
Fig. 4. Expression levels of CB1 and CB2 in U2OS and GPR55E U2OS cells.
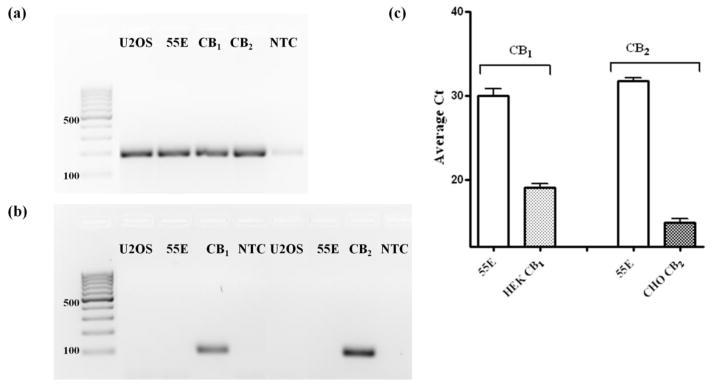
Levels of CB1 and CB2 mRNA expression were assessed by RT-PCR and qRT-PCR. (a) Representative 3% agarose gel showing PCR products for eukaryotic 18S RNA after 30 cycles of RT-PCR. (b) Representative 3% agarose gel showing PCR products for CB1 and CB2 after 30 cycles of RT-PCR. (c) Average Ct values for CB1 and CB2 from qRT-PCR analysis. Data are means from three independent experiments performed in triplicate. 55E, GPR55E U2OS cells; CB1, HEKCB1-293 cells; CB2, CB2-CHO cells, NTC, no template control.
AEA and Virodhamine do not induce cell death
In this study we demonstrate that both AEA and virodhamine (10 μM and 30 μM), significantly attenuate agonist mediated β-arr2 recruitment and receptor internalization. One might argue that the high concentrations of antagonist compounds used may affect cell viability which would account for the reduction in GPR55’s responsiveness. We therefore assessed cell viability using the lactate dehydrogenase (LDH) assay. LDH is a soluble enzyme located in the cytosol which is released into the surrounding culture medium upon cell damage or lysis, processes that commonly correlate with both apoptosis and necrosis (Bonfoco et al. 1995). The levels of LDH released into the culture media can therefore serve as a reliable measurement for cytotoxicity. Results of the LDH assay are summarized in Table 1. Cell death was induced by 1% Triton X-100 (Abood et al. 2001), and was set to be total LDH activity (100%). As depicted in Table 1, no significance difference in LDH activity was observed in control and treated cells. This indicates that inhibition of the agonist-mediated response rather than cell death underlies the reduction in both agonist mediated βarr2 recruitment and receptor internalization.
Table 1. Cytotoxicity assessed by LDH assay.
Values represent means, expressed as a percentage of total LDH activity induced by 1% Triton X-100. Supernatants of vehicle- and ligand- treated GPR55E U2OS cells are similar to each other in that they demonstrate little to no cellular damage, 7% and 6–9% of LDH activity, respectively.
| Treatment | Cell death, % |
|---|---|
|
| |
| HBSS | 5.97 |
| Vehicle | 7.12 |
| 3μM LPI | 7.08 |
| 30 μM SR141716A | 7.12 |
| 10 μM Virodhamine | 7.27 |
| 10 μM Virodhamine +3 μM LPI | 6.94 |
| 10 μM Virodhamine + 30 μM SR141716A | 6.44 |
| 30 μM Virodhamine | 8.08 |
| 30 μM Virodhamine + 3μM LPI | 8.73 |
| 30 μM Virodhamine +30 μM SR141716A | 6.93 |
| 10 μM AEA | 5.90 |
| 10 μM AEA+ 3μM LPI | 6.12 |
| 10 μM AEA + 30 μM SR141716A | 6.79 |
| 30 μM AEA | 5.93 |
| 30 μM AEA + 3 μM LPI | 5.86 |
| 30 μM AEA + 30 μM SR141716A | 8.02 |
Discussion
Increasing amount of evidence suggests an important physiological role for GPR55 (Andradas et al. 2011) (Staton et al. 2008; Whyte et al. 2009; Huang et al. 2011; Pineiro et al. 2011). Recently GPR55 has been demonstrated to play significant role in inflammation where it interacts with the well characterized CB2 receptor (Balenga et al. 2011). While lysophosphatidylinositol, LPI, has been repeatedly reported as an endogenous ligand at GPR55 (Bondarenko et al. 2011; Kapur et al. 2009; Henstridge et al. 2009; Henstridge et al. 2010) the role of endocannabinoids, known activators of the well characterized cannabinoid receptors (CB1 and CB2), in regulating GPR55 is poorly understood. Moreover, no specific and selective antagonists have been identified at GPR55, thus preventing a more thorough understanding of GPR55’s physiological role. The availability of high potency antagonists will facilitate the study of the biological roles of this receptor. In this study we employed the previously characterized βarr2 recruitment assay as readout for receptor responsiveness (Yin et al. 2009; Kapur et al. 2009; Vrecl et al. 2009). Briefly, upon activation by agonist binding receptors undergo deactivation or “desensitization” by binding of the βarr2 protein to the activated receptor. The GPCR-β-arrestin complex internalizes, the ligand is removed and the receptor is recycled back to the cell membrane. Localization of the fluorescently labeled βarr2 can be monitored by image analysis. It is important to note that the calculated IC50’s of tested compounds are probably 5–8 fold higher than the actual binding affinities Ki of the compounds. Nonetheless, in the absence of an established radioligand binding assay for GPR55, the βarr2 recruitment assay provides a reproducible readout of receptor responsiveness.
Data obtained from the experiments presented herein demonstrate that AEA and virodhamine modulate agonist-mediated recruitment of β-arrestin. AEA and virodhamine act as partial agonists; having no detectable response (AEA) or a weak response (virodhamine) on their own, enhancing the agonist effect at low concentrations yet inhibiting it at high concentrations. The binding site of LPI at GPR55 has recently been proposed, whereby agonists adopt an L-shaped conformation for receptor activation (Kotsikorou et al. 2011). It is possible that AEA and virodhamine may interact with some of the same residues required for receptor activation, thus producing partial agonism; but at higher concentrations of these ligands, they interfere with LPI binding, inhibiting receptor activation. Since virodhamine and AEA have been shown to activate CB1 and CB2 receptors (Pertwee et al. 2010; Porter et al. 2002), one might argue that AEA and virodhamine responses reported here are a consequence of CB1 and/or CB2 activation. Therefore we performed qRT-PCR analysis of HAGPR55E/βarr2-GFP cells. The qRT-PCR data clearly indicate that HAGPR55E cells do not express CB1 nor CB2 receptors. Furthermore, to ensure that AEA and virodhamine are not cytotoxic, an LDH assay was performed. The lack of detectable levels of released LDH from cells exposed to AEA and virodhamine indicate that neither AEA nor virodhamine are toxic to the cells at concentrations employed in this study. Together, the results from qRT-PCR analysis and the LDH assay support receptor mediated antagonism of GPR55E/βarr2-GFP cells by both AEA and virodhamine via GPR55 receptors. It appears that the modulation of GPR55 by cannabinoid compounds extends to other structural classes of ligands. Previously, we and others have demonstrated that the synthetic CB1/CB2 agonist CP55,940 acts as an antagonist/partial agonist at GPR55 (Henstridge et al. 2009; Kapur et al. 2009; Pineiro et al. 2011). CP55,940 blocks GPR55 internalization and the formation of β-arrestin-GPR55 complexes; CP55,940 produces only a slight amount of protein kinase CII membrane recruitment but does not stimulate membrane remodeling like LPI, AM251 or SR141716A (Kapur et al. 2009). Recently, a number of cannabinoids were evaluated in the AlphaScreen SureFire ERK1/2 phosphorylation assay, where they were found to modulate the effect of LPI (Anavi-Goffer et al. 2012). AEA and virodhamine were not evaluated by these authors.
Previous reports of virodhamine as a partial agonist at CB1 and full agonist at CB2 support the role of virodhamine in modulating cannabinoid receptor function (Porter et al. 2002). Our finding of partial agonist/antagonist activity at GPR55 in β-arrestin trafficking further support a role of GPR55 in modulating cannabinoid function. In so doing, GPR55 activity may be the key in balancing the involvement of the ECS in the inflammatory process. During inflammation, the levels of LPI and the endocannabinoids are increased (Walter and Stella 2004; Ford et al. 2010). In two independent studies with a neuropathic pain model, increases in both AEA and 2-AG were demonstrated (Mitrirattanakul S et al., 2006, Petrosino et al., 2007); further supporting the involvement of endocannabinoids in inflammation. Hence, it is possible that in the presence of AEA or virodhamine, the overall response of LPI in inflammation could be muted via GPR55. AEA inhibition of SDF-1 induced migration of CD8+ T-lymphocyte was demonstrated by Joseph et al., 2004 (Joseph et al. 2004). These investigators found that AEA-induced inhibition was CB2-mediated as the CB2 agonist, JWH133, also prevented migration of CD8+ T lymphocytes. Our finding that AEA acts as an antagonist at GPR55 lends support for the involvement of GPR55 in inflammation. Perhaps AEA acting at GPR55, and not at CB2, will permit migration of lymphocytes aiding in the development of an inflammatory response, and/or a balance between pro-inflammatory and anti-inflammatory effects of the ECS. This is supported by a previous report that activation of GPR55 by LPI diminishes both de-granulation of PMN’s and reactive oxygen species production via suppression of Rac2 whereas CB2 activation results in activation of Rac2 (Balenga et al. 2011). Thus, crosstalk between GPR55 and CB2 has been suggested to be important in fine-tuning the immune response to prevent excessive tissue injury (Irving 2011).
Previous studies have documented that GPR55−/− mice were protected in models of neuropathic and inflammatory pain (Staton et al., 2008). In the partial nerve ligation model of neuropathic hypersensitivity, GPR55−/− mice of both sexes failed to develop mechanical hyperalgesia up to 28 days post-ligation. In mechanical hyperalgesia induced by intraplantar administration of Freund’s complete adjuvant (FCA), inflammatory mechanical hyperalgesia was completely absent in male GPR55 −/− mice up to 14 days post-injection, while female GPR55−/− mice were protected for one day. Cytokine profiling experiments showed increased levels of IL-4, IL-10, IFN γ and GM-CSF in paws from the FCA-injected GPR55−/− mice when compared with the FCA-injected GPR55 wild-type mice (at 1 and 14 days post-injection for the female and male mice, respectively). This suggests that GPR55 signaling can influence the regulation of certain cytokines and this may contribute to the lack of inflammatory mechanical hyperalgesia in the GPR55 −/− mice. These data suggest that GPR55 antagonists may have therapeutic potential in the treatment of both inflammatory and neuropathic pain.
Further studies are required to determine more precisely the role of GPR55 in inflammation, but the identification of select endocannabinoids as GPR55 modulators will aid in elucidating the function of GPR55 in the ECS, as well as the role of the ECS in the process of inflammation.
Acknowledgments
This research was supported by the National Institutes of Health National Institute on Drug Abuse [Grants DA023204, DA005274]. We would like to thank Dr. Pingwei Zhao for providing the plug-in for data quantification, Drs. Lawrence Barak and Marc Caron for providing the GPR55/βarr2-GFP U2OS cells and express gratitude to Mrs. Genevieve V. Regan for her valuable assistance with the confocal imaging.
Abbreviations
- AEA
Anandamide; N-arachidonyl ethanolamine
- 2-AG
2-arachidonyl glycerol
- βarr2
β-arrestin2
- BV2
murine microglial cell line
- BSA
bovine serum albumin
- CB1
cannabinoid receptor type 1
- CB2
cannabinoid receptor type 2
- CHO
Chinese hamster ovary cell line
- DMEM
Dulbecco’s modified Eagle’s medium
- ECS
endocannabinoid system
- FBS
fetal bovine serum
- FCA
Freund’s complete adjuvant
- GFP
Green Fluorescent Protein
- GPCR
G-protein coupled receptor
- HBSS
Hanks’ balanced salt solution
- HA
hemagglutinin
- HEK293
human embryonic kidney cell line
- IFN-γ
interferon gamma
- LDH
lactate dehydrogenase
- LPI
lysophosphatidylinositol
- LPS
lipopolysaccharide
- ML184 (CID2440433)
3-[4-(2,3-dimethylphenyl)piperazine-1-carbonyl]-N,N-dimethyl-4-pyrrolidin-1-ylbenzenesulfonamide
- ML185 (CID1374043)
N-[4-(4-methoxyphenyl)-1,3-thiazol-2-yl]-2-[(4-methyl-[1,2,4]triazolo[4,3-a]quinolin-1-yl)sulfanyl]acetamide
- ML186 (CID15945391)
N-(2-methoxy-5-morpholin-4-ylsulfonylphenyl)-4,5,6,7-tetrahydro-1-benzothiophene-2-carboxamide
- PBS
phosphate-buffered saline
- PMN
human neutrophils
- qRT-PCR
quantitative real time PCR
- RT-PCR
reverse transcriptase-polymerase chain reaction
- SR141716A
N-(piperidin-1-yl)-5-(4-chlorophenyl)-1-(2,4-dichlorophenyl)-4-methyl-1H-pyrazole-3-carboxamide
- U20S
osteosarcoma cell line
- Virodhamine
O-arachidonoyl ethanolamine
Footnotes
Conflict of interest
The authors declare no conflict of interest.
References
- Abood ME, Rizvi G, Sallapudi N, McAllister SD. Activation of the CB1 cannabinoid receptor protects cultured mouse spinal neurons against excitotoxicity. Neuroscience letters. 2001;309 (3):197–201. doi: 10.1016/s0304-3940(01)02065-1. [DOI] [PubMed] [Google Scholar]
- Anavi-Goffer S, Baillie G, Irving AJ, Gertsch J, Greig IR, Pertwee RG, Ross RA. Modulation of L-alpha-Lysophosphatidylinositol/GPR55 Mitogen-activated Protein Kinase (MAPK) Signaling by Cannabinoids. J Biol Chem. 2012;287(1):91–104. doi: 10.1074/jbc.M111.296020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andradas C, Caffarel MM, Perez-Gomez E, Salazar M, Lorente M, Velasco G, Guzman M, Sanchez C. The orphan G protein-coupled receptor GPR55 promotes cancer cell proliferation via ERK. Oncogene. 2011;30(2):245–252. doi: 10.1038/onc.2010.402. [DOI] [PubMed] [Google Scholar]
- Balenga NA, Aflaki E, Kargl J, Platzer W, Schroder R, Blattermann S, Kostenis E, Brown AJ, Heinemann A, Waldhoer M. GPR55 regulates cannabinoid 2 receptor-mediated responses in human neutrophils. Cell Res. 2011 doi: 10.1038/cr.2011.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bondarenko AI, Malli R, Graier WF. The GPR55 agonist lysophosphatidylinositol acts as an intracellular messenger and bidirectionally modulates Ca2+-activated large-conductance K+ channels in endothelial cells. Pflugers Arch. 2011;461(1):177–189. doi: 10.1007/s00424-010-0898-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonfoco E, Krainc D, Ankarcrona M, Nicotera P, Lipton SA. Apoptosis and necrosis: two distinct events induced, respectively, by mild and intense insults with N-methyl-D-aspartate or nitric oxide/superoxide in cortical cell cultures. Proc Natl Acad Sci U S A. 1995;92 (16):7162–7166. doi: 10.1073/pnas.92.16.7162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Marzo V, Melck D, Bisogno T, De Petrocellis L. Endocannabinoids: endogenous cannabinoid receptor ligands with neuromodulatory action. Trends Neurosci. 1998;21 (12):521–528. doi: 10.1016/s0166-2236(98)01283-1. [DOI] [PubMed] [Google Scholar]
- Ford LA, Roelofs AJ, Anavi-Goffer S, Mowat L, Simpson DG, Irving AJ, Rogers MJ, Rajnicek AM, Ross RA. A role for L-alpha-lysophosphatidylinositol and GPR55 in the modulation of migration, orientation and polarization of human breast cancer cells. Br J Pharmacol. 2010;160(3):762–771. doi: 10.1111/j.1476-5381.2010.00743.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimsey NL, Goodfellow CE, Dragunow M, Glass M. Cannabinoid receptor 2 undergoes Rab5-mediated internalization and recycles via a Rab11-dependent pathway. Biochim Biophys Acta. 2011;1813(8):1554–1560. doi: 10.1016/j.bbamcr.2011.05.010. [DOI] [PubMed] [Google Scholar]
- Henstridge CM, Balenga NA, Ford LA, Ross RA, Waldhoer M, Irving AJ. The GPR55 ligand L-alpha-lysophosphatidylinositol promotes RhoA-dependent Ca2+ signaling and NFAT activation. FASEB J. 2009;23(1):183–193. doi: 10.1096/fj.08-108670. [DOI] [PubMed] [Google Scholar]
- Henstridge CM, Balenga NA, Schroder R, Kargl JK, Platzer W, Martini L, Arthur S, Penman J, Whistler JL, Kostenis E, Waldhoer M, Irving AJ. GPR55 ligands promote receptor coupling to multiple signalling pathways. Br J Pharmacol. 2010;160(3):604–614. doi: 10.1111/j.1476-5381.2009.00625.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heynen-Genel S, Dahl R, Shi S, Milan L, Hariharan S, Bravo Y, Sergienko E, Hedrick M, Dad S, Stonich D, Su Y, Vicchiarelli M, Mangravita-Novo A, Smith LH, Chung TDY, Sharir H, Barak LS, Abood ME. Probe Reports from the Molecular Libraries Program. 2011/11/18. Bethesda, MD: 2010. Screening for Selective Ligands for GPR55 - Agonists. NBK66152 [bookaccession] [PubMed] [Google Scholar]
- Huang L, Ramirez JC, Frampton GA, Golden LE, Quinn MA, Pae HY, Horvat D, Liang LJ, DeMorrow S. Anandamide exerts its antiproliferative actions on cholangiocarcinoma by activation of the GPR55 receptor. Lab Invest. 2011;91(7):1007–1017. doi: 10.1038/labinvest.2011.62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irving A. New blood brothers: the GPR55 and CB(2) partnership. Cell Res. 2011 doi: 10.1038/cr.2011.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joseph J, Niggemann B, Zaenker KS, Entschladen F. Anandamide is an endogenous inhibitor for the migration of tumor cells and T lymphocytes. Cancer Immunol Immunother. 2004;53(8):723–728. doi: 10.1007/s00262-004-0509-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapur A, Zhao P, Sharir H, Bai Y, Caron MG, Barak LS, Abood ME. Atypical responsiveness of the orphan receptor GPR55 to cannabinoid ligands. J Biol Chem. 2009;284(43):29817–29827. doi: 10.1074/jbc.M109.050187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotsikorou E, Madrigal KE, Hurst DP, Sharir H, Lynch DL, Heynen-Genel S, Milan LB, Chung TD, Seltzman HH, Bai Y, Caron MG, Barak L, Abood ME, Reggio PH. Identification of the GPR55 agonist binding site using a novel set of high-potency GPR55 selective ligands. Biochemistry. 2011;50(25):5633–5647. doi: 10.1021/bi200010k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauckner JE, Jensen JB, Chen HY, Lu HC, Hille B, Mackie K. GPR55 is a cannabinoid receptor that increases intracellular calcium and inhibits M current. Proc Natl Acad Sci U S A. 2008;105(7):2699–2704. doi: 10.1073/pnas.0711278105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oka S, Nakajima K, Yamashita A, Kishimoto S, Sugiura T. Identification of GPR55 as a lysophosphatidylinositol receptor. Biochem Biophys Res Commun. 2007;362(4):928–934. doi: 10.1016/j.bbrc.2007.08.078. [DOI] [PubMed] [Google Scholar]
- Pertwee RG, Howlett AC, Abood ME, Alexander SP, Di Marzo V, Elphick MR, Greasley PJ, Hansen HS, Kunos G, Mackie K, Mechoulam R, Ross RA. International Union of Basic and Clinical Pharmacology. LXXIX. Cannabinoid receptors and their ligands: beyond CB and CB. Pharmacol Rev. 2010;62(4):588–631. doi: 10.1124/pr.110.003004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pietr M, Kozela E, Levy R, Rimmerman N, Lin YH, Stella N, Vogel Z, Juknat A. Differential changes in GPR55 during microglial cell activation. FEBS Lett. 2009;583(12):2071–2076. doi: 10.1016/j.febslet.2009.05.028. [DOI] [PubMed] [Google Scholar]
- Pineiro R, Maffucci T, Falasca M. The putative cannabinoid receptor GPR55 defines a novel autocrine loop in cancer cell proliferation. Oncogene. 2011;30(2):142–152. doi: 10.1038/onc.2010.417. [DOI] [PubMed] [Google Scholar]
- Porter AC, Sauer JM, Knierman MD, Becker GW, Berna MJ, Bao J, Nomikos GG, Carter P, Bymaster FP, Leese AB, Felder CC. Characterization of a novel endocannabinoid, virodhamine, with antagonist activity at the CB1 receptor. J Pharmacol Exp Ther. 2002;301 (3):1020–1024. doi: 10.1124/jpet.301.3.1020. [DOI] [PubMed] [Google Scholar]
- Ryberg E, Larsson N, Sjogren S, Hjorth S, Hermansson NO, Leonova J, Elebring T, Nilsson K, Drmota T, Greasley PJ. The orphan receptor GPR55 is a novel cannabinoid receptor. Br J Pharmacol. 2007;152(7):1092–1101. doi: 10.1038/sj.bjp.0707460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Showalter VM, Compton DR, Martin BR, Abood ME. Evaluation of binding in a transfected cell line expressing a peripheral cannabinoid receptor (CB2): Identification of cannabinoid receptor subtype selective ligands. J Pharmacol Exp Ther. 1996;278 (3):989–999. [PubMed] [Google Scholar]
- Staton PC, Hatcher JP, Walker DJ, Morrison AD, Shapland EM, Hughes JP, Chong E, Mander PK, Green PJ, Billinton A, Fulleylove M, Lancaster HC, Smith JC, Bailey LT, Wise A, Brown AJ, Richardson JC, Chessell IP. The putative cannabinoid receptor GPR55 plays a role in mechanical hyperalgesia associated with inflammatory and neuropathic pain. Pain. 2008;139(1):225–236. doi: 10.1016/j.pain.2008.04.006. [DOI] [PubMed] [Google Scholar]
- Tao Q, Abood ME. Mutation of a highly conserved aspartate residue in the second transmembrane domain of the cannabinoid receptors, CB1 and CB2, disrupts G-protein coupling. J Pharmacol Exp Ther. 1998;285 (2):651–658. [PubMed] [Google Scholar]
- Vrecl M, Norregaard PK, Almholt DL, Elster L, Pogacnik A, Heding A. Beta-arrestin-based Bret2 screening assay for the “non”-beta-arrestin binding CB1 receptor. J Biomol Screen. 2009;14(4):371–380. doi: 10.1177/1087057109333101. [DOI] [PubMed] [Google Scholar]
- Walter L, Stella N. Cannabinoids and neuroinflammation. Br J Pharmacol. 2004;141(5):775–785. doi: 10.1038/sj.bjp.0705667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whyte LS, Ryberg E, Sims NA, Ridge SA, Mackie K, Greasley PJ, Ross RA, Rogers MJ. The putative cannabinoid receptor GPR55 affects osteoclast function in vitro and bone mass in vivo. Proc Natl Acad Sci U S A. 2009;106(38):16511–16516. doi: 10.1073/pnas.0902743106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin H, Chu A, Li W, Wang B, Shelton F, Otero F, Nguyen DG, Caldwell JS, Chen YA. Lipid G protein-coupled receptor ligand identification using beta-arrestin PathHunter assay. J Biol Chem. 2009;284(18):12328–12338. doi: 10.1074/jbc.M806516200. [DOI] [PMC free article] [PubMed] [Google Scholar]