

Abstract
Inflammatory processes have a fundamental role in the pathophysiology of stroke. A key initial event is the rapid activation of resident immune cells, primarily microglia. This cell population is an important target for new therapeutic approaches to limit stroke damage. Activation of microglia is normally held in check by strictly controlled mechanisms involving neuronal-glial communication. Ischemic stroke is a powerful stimulus that disables the endogenous inhibitory signaling and triggers microglial activation. Once activated, microglia exhibit a spectrum of phenotypes, release both pro- and anti-inflammatory mediators, and function to either exacerbate ischemic injury or help repair depending on different molecular signals the microglial receptors receive. Various ligands and receptors have been identified for microglial activation. Experimental tools to detect these inflammatory signals are being increasingly developed in an effort to define the functional roles of microglia. Fine-tuning immunomodulatory interventions based on the heterogeneous profiles of microglia are urgently needed for ischemic stroke.
Keywords: CD45, chimera, inflammatory response, ischemic stroke, macrophage, microglia
Introduction
Inflammation plays a critical role in mediating post-ischemic injury. The activation of microglia, the major resident immune cells in the brain, is a key element in triggering the innate immune response [1]. When ischemia occurs, cessation of cerebral blood flow leads to deprivation of oxygen and glucose to areas supplied by the occluded vessel. Resultantly, vulnerable neurons are subject to death and the endogenous signaling that inhibits inflammatory responses under physiological conditions compromises, leading to microglial activation. Once activated, microglia develop macrophage-like capabilities including phagocytosis, cytokine production, antigen presentation and the release of matrix metalloproteinases (MMPs) that weaken the blood brain barrier (BBB) [2]. As a result, peripheral leukocytes infiltrate into the brain and the normally immune-privileged brain environment is exposed to systemic responses that further exacerbate inflammation and brain damage.
The inflammatory response has dual effects on ischemic injury [3]. Stroke-induced microglial activation causes release of a variety of inflammatory mediators many of which are cytotoxic and/or cytoprotective [4]. Phagocytosis of cellular debris and harmful substances along with the release of anti-inflammatory cytokines by microglia occurs in an effort to restore tissue homeostasis by clearing pathogens or necrotic cells, and consequently attenuate the detrimental effects of inflammation and aid in tissue repair [5,6]. Because of their critical roles in the immune response to stroke, microglia have become a recent target of interest for many stroke scientists. This review focuses on current findings, providing an update on microglial activation, phenotype identification, and the roles of microglia in the pathophysiology of cerebral ischemia.
Origin and development of microglia
There has been much controversy over the characterization of the cell lineage of microglia. The most significant hypotheses of the development of microglia debate their embryonic neuroectodermal or mesodermal origin. Unlike the ectodermal development of macroglia (astrocytes and oligodendrocytes) and neurons, a consensus in favor of the mesodermal hematopoietic origin of microglia is currently held [7,8]. A subset of primitive myeloid precursors, localized in the extra-embryonic yolk sac (YS) at embryonic day 8 (E8) was shown to contribute to the rise of yolk sac microglia that persists into adulthood [9]. This cellular subset of primitive hematopoiesis contributes little to blood leukocyte development, which is largely specific to definitive hematopoiesis of hematopoetic stem cells (HSCs) [9-11]. Microglia precursors originate in the embryonic yolk sac before the emergence of definitive HSCs from the aorto-gonad-mesenephros (AGM). The direct precursors of microglia that travel to the neural tube at E8 are exclusively the CD45-cKit+ cells. This subpopulation of erythromyeloid cells eventually begins expressing CX3CR1 and CD45 and travels into the neuroectoderm in a matrix metalloproteinase 8 (MMP 8) and MMP 9 dependent manner to develop into microglia [12,13]. These precursor cells are seen seeding the brain rudiment by E10 in rodents [9] and have a full microglial morphology beginning at E14 [12].
The transcription factor Myb is essential for the development of HSCs [14,15] and can be found in the AGM during embryogenesis [8,16]. By using Myb knockout (KO)mice, Schulz et al. [15] found that yolk sac-derived CD45+CX3CR1hiF4/80hi macrophages and microglia still develop in normal numbers and remain independent of Myb into adulthood; however, CD45+CX3CR1+F4/80lowCD11bhi monocytic phagocytes continually replaced by bone marrow (BM) in an adult are unable to develop without Myb. The transcription factor PU.1, on the other hand, is necessary for myelopoiesis of the YS but dispensable for the development of definitive HSCs [11,15]. This further suggests two separate myeloid lineages of peripheral macrophages and resident microglia.
Moreover, colony stimulating factor (CSF) and its receptor CSF-1R are necessary for the differentiation of most macrophages/microglia [17]. Recent studies showed that in CSF-1R KO mice, yolk sac-derived microglia do not develop and are deficient throughout life, but HSC-derived monocytes are able to differentiate and circulate without dependence on CSF-1R [8,9,18]. Fate mapping analysis of Flt3-Cre x RosaLSL-YFP mice also shows that yolk sac-derived microglia are independent of Flt3, a chemokine present on multipotent hematopoetic progenitors in the blood and brain [15]. Taken together, the separation between HSCs and yolk sac derivations is evident and allows insight into microglial ontogeny.
Markers and methods for identification
To date, no single microglial-specific marker has been identified, keeping the cell type indistinguishable from macrophages and various myeloid-derived cells that infiltrate the brain during pathological states [19]. Changes in microglial activation in response to central nervous system (CNS) injury are illustrated by diverse phenotypes and unique expression of cell surface proteins. Microglial activation stages can be detected by characterizations of these changes, but the similarities with other cell types pose a major hurdle for their definitive characterization and detection.
Ionized calcium binding adaptor molecule-1 (Iba-1) expression by immunohistochemistry (IHC)
Iba-1 is amongst the most useful proteins for distinguishing microglia through IHC and immunocytochemistry (ICC) staining [20,21], especially for studies of cerebral ischemia where the expression of Iba-1 is upregulated [22] (Figure 1). However, Iba-1 has also been shown to bind various cells of monocytic lineage [20], and thus its specificity for microglia staining is limited in injured brain tissue where peripheral macrophages may infiltrate. Other markers, such as CD11b, Isolectin (IB4), and F4/80, have also been used for in vivo and in vitro microglial staining. Although the benefits of IHC include the ability to create a spatiotemporal and morphological profile of microglia within the CNS, it lacks in specificity and may be inconsistent depending on the type of histological preparation.
Figure 1.
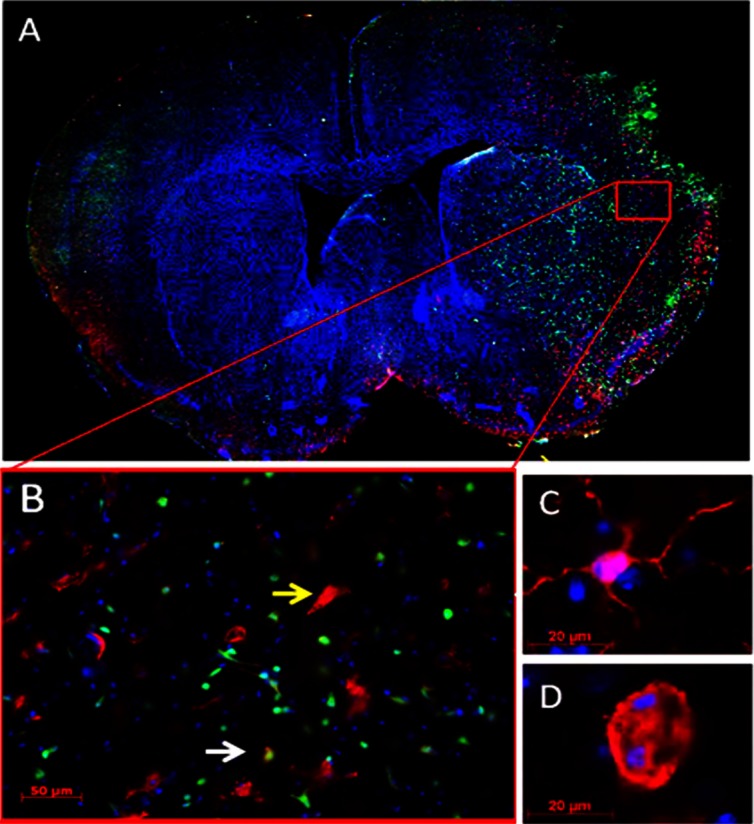
Microglia activation and leukocyte infiltration in the ischemic brain. A. Representative brain slice stained with Iba-1 (red), GFP (green) and DAPI (blue) from a chimeric mouse after stroke (10x magnification). The chimeric mouse model was made by transferring GFP+ bone marrow of a donor mouse to an irradiated WT mouse. Eight weeks after reconstitution, the chimeric mouse was subjected to 90 min MCAO. The mouse was reperfused for 72 hours before sacrifice. GFP+ cells represent bone marrow-derived peripheral leukocyte infiltrates. Green fluorescence is localized to the region of injured tissue in the striatum and cortex. B. 20x magnification of the box area in (A). The yellow arrow indicates a large Iba1+ ameboid microglia that does not colocalize with GFP+ bone marrow-derived cells; the white arrow indicates a cell co-labeled with Iba-1 and GFP. C. Resting microglia morphology with thin, ramified processes in the non-injured brain hemisphere. 63x. D. Activated microglia with large, ameboid cell body in the ischemic cortex. 63x.
CD45/CD11b expression by flow cytometry
Flow cytometry provides a sensitive means to detect various markers and create a multiparameter characterization of different cell types. In 1991, Sedgewick et al [23] observed differences in the expression of the hematopoietic cell surface marker CD45 on resident microglia and infiltrating peripheral leukocytes. To date, the most common characterization profile derived from flow cytometry sorting still holds that resident microglial cells are CD45lowCD11b+, while infiltrating hematogenous myeloid cells are CD45highCD11b+ [24]. Experimental stroke studies with two photon imaging [25] and IHC [26] also demonstrated that infiltrating leukocytes in the ischemic hemisphere showed a higher expression of CD45, whereas microglia had a fainter CD45 immunostaining, which is consistent with our flow cytometry data (Figure 2). Although CD11chigh and CD14– have sometimes been used to label microglia in flow cytometry, their similarity in expression on other peripheral cell types makes it difficult to justify resident microglial specificity [24,27].
Figure 2.
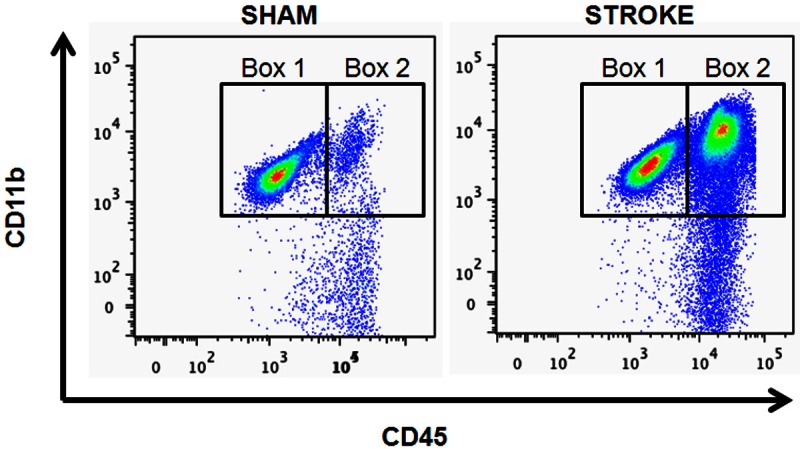
Representative flow cytometry plots of resident microglia and peripheral leukocytes in the mouse brain. In the brains of sham mice (left), CD45lowCD11b+ (box 1) and CD45highCD11b+ (box 2) cells were identified as resident microglia and peripheral leukocytes respectively. 72 hours after 90 min MCAO (right), both CD45lowCD11b+ and CD45highCD11b+ cells were increased in the brain.
There are, however, limitations to using variable levels of CD45 expression to distinguish between microglia and other monocyte/macrophage populations. CD45 on microglia can be upregulated in mouse models of EAE in spinal cord towards a CD45 high phenotype [28]. Inflammation in the periphery of humans may also upregulate CD45 in CNS resident microglia with no changes in CD11b expression [29]. The possible instability of this marker’s levels during pathological states reiterates the need for a better method of tracking resident microglia and peripheral monocyte/macrophages.
Transgenic mice
A multitude of transgenic mice can also be used to label cell populations by attaching a fluorescent reporter to a myeloid promoter. For example, CD11b-GFP can label all cells of myeloid origin including microglia in the mouse brain [30]. Similarly, Iba-1-EGFP transgenic mice have shown successful fluorescent labeling of CNS microglia/macrophages in tissue samples [31]. More recently, a fluorescent knock-in transgenic mice line created using CX3CR1-GFP and CCR2-RFP has shown to be the most useful to the field to differentiate resident microglia from peripheral monocytes. It is now known that CCR2, though differentially expressed, is specific to the periphery in naïve and EAE mice [32], and unlike CD45, is not upregulated significantly in pathological states. In addition, fractalkine receptor (CX3CR1) is present on microglial progenitors in the yolk sac and, in the CNS, remains specific to microglia into adulthood [13].
Studies using systemic and region-specific depletion of myeloid cells or macrophages can be done using DTR-CD11b transgenic mice. These mice have diphtheria toxin receptors (DTR) linked to the CD11b promoter. Administration of diphtheria toxin will deplete CD11b+ (myeloid) cells [30,33]. Another commonly used technique to selectively ablate CD11b+ cells involves treating CD11b-HSV TK transgenic mice with injection of ganciclovir (GCV) [34]. Inducible death of CD11b+ cells has more recently become a useful technique in the field.
Chimeras
Irradiation chimera models are a popular technique to differentiate microglia from peripheral myeloid cells since there is no exclusive antigenic marker [35]. To generate a BM chimera (Figure 1), BM cells of a mouse with ubiquitously expressing fluorescent (GFP+, YFP+, RFP+, etc) protein is extracted and injected into a mouse of interest whose own marrow is first irradiated and eliminated. After weeks to months of reconstitution of the new cell population, it is possible to track the origins of cells in CNS tissue, particularly after neurological diseases, where cells from the periphery that cross the BBB have been labeled with fluorescence. BM chimeras can be performed with transgenic knockout or knockin mice to selectively target cells of the CNS or of the periphery. Unfortunately, the effects of irradiation are confounding and have been implicated in physiological alterations such as weakening of the BBB, potential cell death and activation of microglia [36-40]. Though microglia are mostly radio-resistant, their numbers in the CNS may also be altered in uninjured and ischemic chimeric mice compared to nonchimeric mice in the hippocampus and the cortex [41]. Changes in gene regulation with higher expression of proinflammatory cytokine and CCL2 can also accompany irradiation [38,42]. These undesireable effects of irradiation pose a caveat to the technique. However, preventive measures can be taken to study the brain using chimeras. For example, targeted, instead of whole body irradiation using lead shielding of mouse heads is now often used in an effort to prevent damage or brain environmental changes due to radiation exposure [36,43].
Resting microglia
Resting phenotype
Resting microglia in a healthy brain, more representatively known as “surveying microglia”, are constantly extending and retracting their thin ramified processes in an effort to inspect the CNS microenvironment [44,45] (Table 1). As the central macrophages of the brain, microglia are implicated in controlling synapse number and remodeling in the developing brain, and function to prevent accumulation of debris in the healthy adult CNS. They are not present uniformly in the adult brain and express differences in location, protein expression, and morphology [46]. Heterogeneity of morphology and location of microglia translates to differences in microglial responses to injuries and in activation states [47]. The majority of microglia are found in the gray matter and express more ramified structure with radial branches than those found in the white matter with longitudinal processes [46]. In the ischemic lesion induced by MCAO modeling, the penumbral area ( the border zone of dead and living tissue) showed highly ramified cells (resting), while the ischemic core housed amoeboid bodies with thick ramifications (activated) [25]. To conclude, resting microglia are not “sleeping”; instead they are poised to respond to stimuli resulting from disturbances in the CNS environment by drastically altering their phenotypes and functions.
Table 1.
Phenotypes of microglia
| Phenotype | Identification Markers | References |
|---|---|---|
| Resting State | Iba-1 | [21,143] |
| CD45int CD11b+ | [23,24] | |
| F4/80 | [46,144] | |
| Isolectin (IB4) | [145] | |
| Classical Activation (M1) | MHCII | [28,45,146] |
| CD16 (FcγR III) | [147,148] | |
| CD 32 (FcγR II) | [147] | |
| [149,150] | ||
| CD80 (B7-1) | [149,150] | |
| CD86 (B7-2) | [151 | |
| CD40 (TNFR) | ||
| Alternative Activation (M2) | Arg-1 | [152] |
| CD68 (ED1) | [153] | |
| Fizz1 (Relmα) | [152,154] | |
| Ym-1 | [152,154] | |
| CD206 (MR) | [155,156] | |
| Dectin-1 | [155] |
MHC, major histocompatibility complex; Arg, arginase; TNFR, tumor necrosis factor receptor; Fizz1, resting-like molecule alpha; MR, mannose receptor.
Inhibitory signaling of microglia
In the healthy brain, microglia express numerous inhibitory proteins, and interact with neurons to form a “brake” on inflammation [48]. The endogenous mechanisms in the form of neuronal-glial interactions may prevent excessive microglial activation in the CNS. Following injury, these interactions may be disrupted due to neuronal cell death and structural tissue damage. Regulating these inhibitions to keep microglia from acquiring a proinflammatory phenotype has been shown to prevent uncontrolled damage in models of Alzheimers Disease (AD), multiple sclerosis (MS), and neurodegeneration [49-53]. Similarly, regulation of these inhibitory proteins may also have beneficial effects on ischemic stroke.
CD200/CD200R1
CD200 is a transmembrane glycoprotein mainly expressed on neurons [54,55]. The cell-cell contact between CD200 and its receptor (CD200R1), expressed on the surface of all myeloid origin cells, provides microglia with stability to remain in a resting state under normal CNS conditions [56,57]. Cleavage of this interaction and subsequent microglial activation leads to the upregulation of proinflammatory cytokines and an inflammatory profile [54]. Increased tissue damage was related to a decrease of CD200 level in mouse models of EAE, suggesting a detrimental effect of the activated microglia unbound from CD200/CD200R1 interaction [58]. Aging is also associated with a reduced level of CD200 and long-term potentiation (LTP), as well as an increased microglial activation in the brain; however, treatment of CD200R1 agonist (CD200Fc) can attenuate the LTP deficit and ameliorate microglial activation, even after Lipopolysaccharide (LPS) stimulation [59]. Little is known about the role of CD200/CD200R1 signaling in ischemic stroke, but a primarily descriptive study of CD200 on Iba+ cells showed a decrease in gene transcripts of CD200 in the ischemic hemisphere [60].
Fractalkine (CX3CL1)/CX3CR1
Much like the immunoglobulin superfamily member CD200R1, fractalkine receptor (CX3CR1) on microglia can bind the soluble and membrane bound forms of CX3CL1, to keep microglia quiescent [61]. However, under injury, neurons significantly decrease CX3CL1 release thereby enabling microglial activation [62]. Loss of this contact has been shown to be neurotoxic in many disease models including Parkinson’s disease and ALS by exacerbating neuronal loss [50]. Similarly, LPS stimulation of microglia on CX3CR1(-/-) mice leads to greater IL1-β secretion compared to CX3CR1(+/-) mice [50].
However, the effect of CX3CL1/CX3CR1 signaling in neuroinflammation is controversial as deleterious roles for the CX3CL1/CX3CR1 pair were reported in rodent models of AD and cerebral ischemia [51,52]. Twenty four hours after a transient MCAO, CX3CR1(gfp/gfp) mice (GFP is inserted into both alleles of the CX3CR1 locus) were noted to have less severe cerebral infarct volumes than WT mice, possibly associated with a coinciding decrease in IL-1β and TNF-α gene transcripts [63], as in the case of fractalkine knockout mice [64]. Similar results were seen in a recent study by Ciprani et al with a pMCAO model in CX3CL1-/- and CX3CR1(gfp/gfp) rodents, both of which showed less severe ischemic damage than WT mice [52]. Furthermore, CX3CL1-/- animals have increased damage after ischemia with exogenous intracerebroventricular CX3CL1 administration. Interestingly, WT rodents show less severe infarct volumes and better functional outcomes with the addition of CX3CL1 in a dose dependent manner [52]. The disparity in the effect of exogenous CX3CL1 between KO and WT animals suggests that CX3CL1 may be protective only when microglia exhibited a normal constitutive CX3CR1-mediated signaling throughout development in the WT brain [52].
SIRPα/CD47
Signal-regulatory protein alpha (SIRPα), expressed on myeloid cells including microglia, binds integrin associated protein CD47 on neurons to activate an intracellular immunotyrosine inhibitory motif [65,66], keeping microglia silenced and thereby suppressing phagocytosis [67,68]. Human MS lesions have shown a decrease in CD47 expression [53], though little is known to date about the role of CD47 and SIRPα in ischemia specifically. A reduction of infarct was seen 24 and 72 hours after 90 min MCAO in CD47 knockout mice, potentially due to a decrease in peripheral inflammatory cell infiltration [69]. Wang et al recently also reported a reduction of infarct and improvement of behavior deficit after transient MCAO in SIRPα mutant mice [70].
Triggering receptor expressed on myeloid cells 2 (TREM2)
The neuronal-microglial connection of heat shock protein 60 (HSP60)-TREM2 is important for the clearance of apoptotic neurons by microglial phagocytosis [71]. Microglia, via its intracellular adaptor protein DAP12, prevent the release of proinflammatory cytokines and maintain an anti-inflammatory microglial profile [72,73]. In EAE, mice lacking TREM-2 by antibody blockage suffered worse pathology, but are rescued with greater recovery and tissue repair by injection of myeloid cells expressing TREM-2 [73]. Contrarily, TREM-2 KO mice showed a decrease in amoeboid Iba1+ and CD68+ microglial activation and an overall decreased inflammatory response 7 days post-reperfusion after a 30 minute MCAO model that did not translate into a decreased infarct volume [74]. The inhibitory effect of TREM-2 on microglial activation following stroke is still uncertain at this time.
Activation of microglia
A commonly held assertion is that distinct activation states (based on protein expression signatures) impart defined functional roles of microglia and may account for heterogenic responses to CNS injury. These activation states are generally described in terms of the class of activating signals and selectively induced expression of unique markers. Microglia can change patterns of migration, cell surface protein expression, and functions in response to tissue damage or dysfunction (Figure 3). The rapid responses to altered homeostasis of the brain parenchyma can be visualized in two photon imaging to show immediate (within minutes) increase in the motility of microglial processes towards area of injury in a chemical gradient dependent manner, while the cell bodies remain stationary [44,45]. In vivo data suggest that ATP signaling of P2Y12 receptors on the cell surface is responsible for such extensions and directional guidance [44,75,76].
Figure 3.
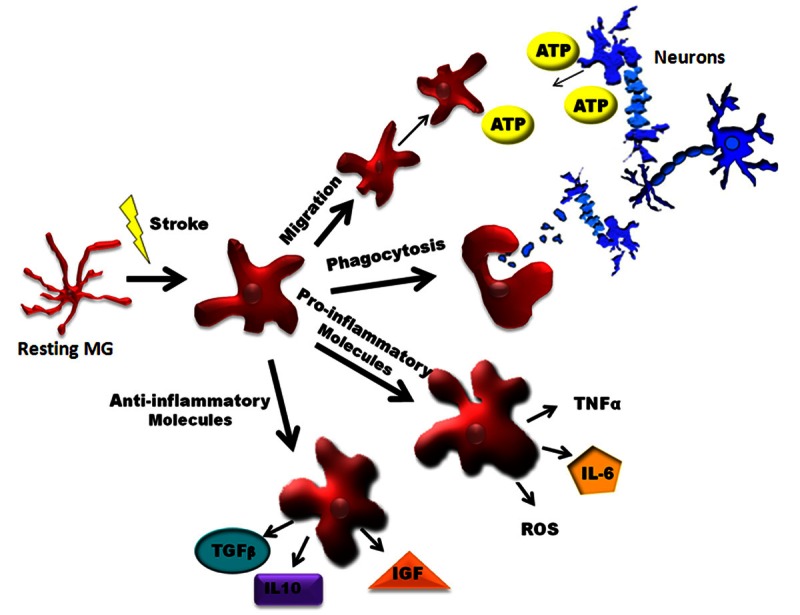
Schematic of Microglia Activation after Ischemic Brain Injury. In the ischemic brain, microglia display prominent changes in morphology associated with various functional states. Activation results in upregulation of transcriptional machinery which serves to increase the production of inflammatory mediators. In response to chemotaxic factors (e.g. ATP, etc.), microglia can migrate to sites of ischemic tissue injury to increase phagocytic uptake of cellular debris and cytotoxic substances. Depending on their activation state, microglia may promote a proinflammatory environment (via M1 activation) or regenerative milieu (via M2 activation). IGF-1: Insulin like growth factor 1; IL-1β: interleukin 1 beta; IL-6: interleukin 6; IL-10: interleukin 10; MG, microglia; ROS: reactive oxidative species; TGFβ: Transforming growth factor beta; TNFα: Tumor necrosis factor alpha.
Microglial activation phenotypes: M1 and M2
Changes in microglial phenotype during activation may be analogous to that of peripheral macrophages, as the two cell types are indistinguishable without definitive surface markers for either. Microglial responses to stimuli from a changing brain environment are characterized as either M1, classical activation, or M2, alternative activation (Table 1). M1 is a proinflammatory cellular state associated with an increase in protein synthesis of pro-inflammatory mediators (IFNγ, Il-1β, TNFα, Il-6, CXCL10, etc.), ROS and NO production, and proteolytic enzymes (MMP 9, MMP3) that act on the extracellular matrix leading to BBB breakdown [77,78]. M1 phenotype can lead to increased neuronal death compared to alternatively activated M2 microglia [79]; therefore, there is a growing interest to pharmacologically interfere with the signaling mechanisms that give rise to the classical activation phenotype of microglia. M2 microglia release anti-inflammatory mediators (IL-10, TGF-β, IL-4, IL-13, IGF-1, etc.) [80], leading to enhanced expression of genes associated with inflammation resolution, scavenging, and homeostasis [77,81-83]. Levels of IL-10, TGF-β and CD206 mRNA increased as early as day 1 after ischemic injury and peaked at 4-6 days. In addition, TGF-β released by microglia promotes an anti-inflammatory profile associated with increased proliferation and neuroprotection in the ischemic brain [84]. This may be therapeutically relevant because TGF-β1 is specifically found in the salvageable peri-infarcted region of the cortex 24 hours after a 60 min MCAO and involved in distinct spatiotemporally regulated inflammatory and neuroprotective processes [85].
During disease progression and in normal ageing, microglial activation phenotypes can switch from M2 to M1 [86]. One recent study suggests that microglia are activated early after MCAO and morph into a reactive M1 phenotype by 7 days [79]. The balance between the M1 and M2 states is dynamic in inflammatory responses and may be offset in chronic disease states such as stroke, representing a novel mechanistic target for therapy [77]. Several proteins have been identified as markers for M1 or M2. MHC II, implicated in antigen presentation as an immune reaction, is upregulated on classically activated microglia, and is commonly used as a marker for M1 [28]. The alternatively activated M2 cells up-regulate presentation of several antigens. Ym-1 (Chitinase 3-like 3), for example, has been found to be associated with the protective, pre-phagocytic state of macrophages after ischemia [25]. Some of Ym-1+ cells co-express CD206 (mannose receptor), which is another marker for M2 activation known to be involved in antigen internalization and processing [25]. CD68 (macrosialin) glycoprotein is another accepted marker for phagocytotic cells and is often used to distinguish the M2 debris clearing state of microglia [25]. Stroke-induced changes in the expression of specific cell surface proteins probably reflect a continuum of the microglial activation spectrum. There is a growing need to identify subpopulations of M1/M2 microglia versus those of peripheral myeloid cells and the relative percentages of each over time to determine the overall functional contribution to stroke injury.
Migration
Microglia, as the first immune responders in the CNS, migrate to areas of injury through detection of chemoattractant gradients to subsequently phagocytize debris in damaged tissue, neutrophils, and apoptotic cells that have the potential to release damaging molecules [44,87,88]. CX3CL1 and ATP released from dying neurons can act on microglial receptors to induce chemotaxis. In addition, monocyte chemotactic protein-1 (MCP-1, CCL2) is a chemokine expressed both in the brain and in some peripheral organs that can induce migration of leukocytes and macrophages/microglia to the ischemic area [89,90]. After MCAO, MCP-1 expression increases at injured region and peaks at 2-3 days [91] when monocytes/macrophages also start to peak in the ischemic brain [92]. Transgenic CCL2 knockout mice show decreased ischemic injury [93], suggesting that the recruitment of monocytes/macrophages to the injured area may be detrimental and that therapies designed to block the migration of leukocytes may have translational value. Microglia at the border region of the infarct, “penumbral” microglia, may be the major target of therapy given their large numbers, and activated status after stroke [94]. Unlike microglia of the ischemic core, which appear to be dying with increased disease severity, penumbral microglia are presumably living and highly activated [47]. This suggests a regional target of interest to deliver treatments.
Phagocytosis by microglia and macrophages
As the primary phagocytes in the brain, activated microglia phagocytize and uptake damaging components in an effort to reestablish homeostasis after insults [5]. Microglial phagocytosis of neuronal cells begins early even before peripheral macrophages infiltrate into the brain after stroke [95]. These phagocytotic cells were seen interacting with neurons and show neuron engulfment in the ischemic brain. CD68 is a commonly used marker of macrophage phagocytosis and is expressed as early as 6h after pMCAO on ramified CD11b+ cells in the penumbra, and continues to increase later in the hypertrophic ameboid cells of the ischemic core [25]. However, this phagocytosis marker can be expressed on both the resident and infiltrating phagocytes, and therefore may not be exclusive to microglia [25].
Phagocytosis is able to attenuate inflammation but can also be implicated in neuronal damage. A recent study of primary microglial cultures assessed the sequence of events from microglial activation to the phagocytosis of neurons [96]. Production of peroxynitrite during a microglial immune response to TLR4 and TLR2 activation leads neurons to externalize phosphatidylserine on the outer membrane to act as an “eat me” signal to elicit phagocytosis. Interestingly, inhibition of this process at any stage not only leads to the decrease in phagocytosis of the neurons, as would be suspected, but also prevents 90% of neuronal cell death [96]. The potential for microglia to phagocytize viable neurons illustrates the importance of microglial modulation in ischemic stroke [96].
Purinergic receptors
Purinergic receptors (P2X7, etc.) are upregulated on microglia in the periinfarct region of injury [97] and have been shown to interact with ATP that is accumulated extracellularly in the ischemic brain [98], subsequently leading to microglial activation [99]. Activation of these receptors leads to cell death [100] and the prominent release of proinflammatory cytokine IL-1β through caspase-1 pathway [101]. ATP leaked from dying neuronal cells and released by astrocytes [44,102] can act as a chemoattractant on microglia by interacting with P2Y12 receptor [103]; the microglia attracted to sites of ischemic injury can further amplify activation by autocrine signaling of ATP [104]. This positive feedback loop leads to increased proliferation and secretion of IL-1β, TNF-α, and ROS [99], and exacerbates inflammatory responses. Blocking P2X7 receptors was shown to improve behavior deficits in a model of transient global ischemia where decreases in microglial activation and proinflammatory cytokines (TNF-α, IL1-β, and IL-6) were noted [105]. A nonselective P2 blocker, Reactive Blue 2, was shown to reduce infarction in a focal pMCAO model. However, P2X7 receptors are also expressed on neurons and astrocytes. Therefore, the function may not be solely attributed to microglia [106].
Toll like receptor (TLR)
The stroke-induced innate immune response is also associated with the release of various damage associated molecular patterns (DAMPs), which can further activate pattern recognition receptors (PRRs), including members of the TLR family on microglia [107,108]. TLRs are important in innate immunity in both the central and peripheral systems. TLR4 is the prominent LPS receptor that can lead to activation of LPS-responsive cells, such as monocytes and macrophages, and subsequently causes upregulation of proinflammatory genes via NFκB signaling [109]. The most prominent TLRs on microglia are TLR4 and TLR2, both of which are upregulated after ischemia [108,110-113]. TLR4 deficient mice have smaller infarct sizes, better neurological scores and decreased downstream NFκB signaling in experimental stroke studies [112,114]. Recent studies have shown that CNS preconditioning with TLR4 agonist can lead to tolerance, eliciting a diminished proinflammatory response with subsequent less ischemic injury [115,116]. Although there has been controversy as to the role of TLR2 deficiency in cerebral ischemia, recent studies have shown an exacerbation of injury in TLR2 KO mice [108,111,117]. Some endogenous ligands of TLRs have recently been identified. Purines and peroxiredoxin (prx) released to the extracellular space from dying cells can bind TLRs on macrophages and produce proinflammatory cytokines IL-23 and IL-17 [118]. Heat shock proteins (HSP) and High mobility group box 1 (HMGB1) are other endogenous ligands for TLRs that result in upregulation of NOS and proinflammatory cytokines (TNF-α, IL-6, and IL-1β) [119,120].
Functional insights of resident microglia and peripheral macrophages
Microglial proliferation and renewal
There is evidence that activated microglia are proliferative in the ischemic brain. Using carboxyfluorescein diacetate succinimidyl ester (CSFE) to intracellularly label peripheral cells before a 30-60 min MCAO, one study found higher amounts of BrdU+Iba1+CSFE- colabeling microglia after stroke indicating increased proliferation [88]. Interestingly, accumulating data show microglial proliferation is beneficial after ischemia. In vitro studies have shown that increased microglial proliferation by stimulation of CSF-1R, which is upregulated in ischemia, is neuroprotective [28,121]. An in vivo study also showed that defective microglial proliferation was associated with significant increase in the size of ischemic lesion and a 2-fold increase in the number of apoptotic neurons [122].
In addition to microglia, choroid plexus macrophages, perivascular macrophages, and meningeal macrophages inhabit various regions around the CNS [123]. This heterogeneous population of tissue macrophages is continuously replenished by circulating and peripheral monocytes, unlike microglia that are largely thought to be resident in the adult CNS from early development [38,124,125]. Theories of another wave of microglial establishment postembryonic from peripheral monocytic precursers that last into adulthood are part of an ongoing debate [38,126]. It is uncertain whether these monocytic precursors become integrated into the microglial population or remain bone marrow-derived monocytes. Nevertheless, Varvel et al. [127] suggest yet another mode of microglial replacement apart from proliferation exists in the adult brain. They ablated microglia using intracerebroventricular ganciclovir treatment in Tk+/Ccr2+/rfp mice and found abundant levels of CD45high monocytes in neocortical areas of microglial depletion before engraftment. In addition, there were increased levels of Ccr2 RFP expression which returned to normal levels after engraftment. These infiltrating cells of monocytic origin are morphologically similar to microglia and functionally active in surveying the microenvironment of the brain, without changes in number over time [127]. It’s possible that a population of dying microglia in the ischemic brain could be replenished by peripheral monocyte/macrophages infiltrating into the injured region and downregulating CCR2 and CD45 to morph into microglia. The topic of microglial renewal and proliferation, however, is still controversial.
MMPS
Macrophages, including microglia, are major contributors to the release of MMPs (mainly MMP3 and MMP9) which are responsible for the breakdown of the extracellular matrix and the BBB after ischemia [88,128]. MMP-3 and MMP-9 knockout mice were shown to suffer less neuronal injury after an ischemic episode [129,130]. Since MMP-9 can be derived from both brain and peripheral immune cells, its origin after ischemia has been debated. Recently, chimeric studies showed that the increased damage from MMP-9 toxicity in the ischemic brain are attributed to bone marrow cells as opposed to resident microglia [130,131]. This suggests that infiltrating monocytes/macrophages can also release MMPs and modulate their own entry into the CNS by weakening BBB.
TNF
TNF-α can be released from both microglia and peripheral leukocytes. TNF-α has been largely regarded as neurotoxic [132]; however, chimeric studies have found that TNF-α produced locally by resident brain microglia (but not by peripheral macrophages and leukocytes) exerted neuroprotection in pMCAO model via TNF-p55 signaling [133]. The disparity in implications may be attributed to different signaling mechanisms between the neuroprotective TNF-p55 and neurotoxic TNF-p75 pathways [134].
iNOS
Local accumulation of nitric oxide (NO) is also involved in the inflammatory cascade after cerebral ischemia [135]. Equipped with inducible nitric oxide synthase (iNOS), M1 microglia are partially responsible for the changes in expression level of NO. The role of iNOS in ischemia is inconsistent in literature as both protective and deleterious roles have been reported [136-138]. Pruss et al [139] found that in chimeric iNOS-deficient mice transplanted with WT bone marrow (BM) cells and wild-type mice transplanted with iNOS-deficient BM cells, no difference in cerebral iNOS expression or in infarct volumes can be seen between the chimeric animals after MCAO, suggesting iNOS from microglia and peripheral myeloid cells may not be a significant regulator of ischemic injury.
Peroxiredoxin
The peroxiredoxin family of proteins (Prxs) are intracellular antioxidant enzymes that are needed for cell survival in the brain [140,141] . However, once released from necrotic brain cells, extracellular Prxs promote neural cell death in ischemia by inducing expression of inflammatory cytokines including IL-23 in macrophages [118]. This study also utilized chimeric mice models and further showed that the infiltrating bone marrow-derived macrophages but not the resident microglia, are responsible for increased ischemic volume and inflammatory response triggered by Prxs release [118]. In addition, they found that neutralization of the released extracellular Prxs with a prx antibody is protective.
Summary
Despite more than one century of research since they were first investigated by Rio Del Hortega [142], “the father of microglia”, the precise role of microglia is still shrouded in mystery due to a current lack of tools and fundamental understanding of the heterogeneity of their observed activation spectrum. The nature of the microglial response to stroke is multi-faceted and complicated by the aggregate immune response. The significance of the local and systemic inflammatory response as well as the interplay between the two is widely argued. While many studies suggested that anti-inflammatory agents confer neuroprotection following ischemic brain injury, others pointed to a requirement for pro-inflammatory cytokines and leukocyte activation in orchestrating repair. Enhancing repair may be possible by targeting distinct populations of microglia with special attention to temporal and spatial specific therapeutic intervention in ischemic stroke and other neurological disorders. There is room for improvements of methods for better identification and manipulation of microglia. The roles of microglia in stroke-induced inflammatory responses merit further investigation, in hope that fine-tuning immunomodulatory therapies could be available to avoid the deleterious effects of total immunosuppression and the possible detrimental effects of chronic microglial inhibition.
Acknowledgement
This work was supported by the NIH/NINDS (grants NS050505 and NS055215 to LDM), and by AHA (grant 12SDG9030000 to FL).
Conflict of interest
None.
References
- 1.Wang Q, Tang XN, Yenari MA. The inflammatory response in stroke. J Neuroimmunol. 2007;184:53–68. doi: 10.1016/j.jneuroim.2006.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Iadecola C, Anrather J. The immunology of stroke: from mechanisms to translation. Nat Med. 2011;17:796–808. doi: 10.1038/nm.2399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wang X, Feuerstein GZ. The Janus face of inflammation in ischemic brain injury. Acta Neurochir Suppl. 2004;89:49–54. doi: 10.1007/978-3-7091-0603-7_6. [DOI] [PubMed] [Google Scholar]
- 4.Wood PL. Microglia as a unique cellular target in the treatment of stroke: potential neurotoxic mediators produced by activated microglia. Neurol Res. 1995;17:242–248. doi: 10.1080/01616412.1995.11740321. [DOI] [PubMed] [Google Scholar]
- 5.Neumann H, Kotter MR, Franklin RJ. Debris clearance by microglia: an essential link between degeneration and regeneration. Brain. 2009;132:288–295. doi: 10.1093/brain/awn109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jin R, Yang G, Li G. Inflammatory mechanisms in ischemic stroke: role of inflammatory cells. J Leukoc Biol. 2010;87:779–789. doi: 10.1189/jlb.1109766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chan WY, Kohsaka S, Rezaie P. The origin and cell lineage of microglia: new concepts. Brain Res Rev. 2007;53:344–354. doi: 10.1016/j.brainresrev.2006.11.002. [DOI] [PubMed] [Google Scholar]
- 8.Gomez Perdiguero E, Schulz C, Geissmann F. Development and homeostasis of “resident” myeloid cells: The case of the microglia. Glia. 2013;61:112–120. doi: 10.1002/glia.22393. [DOI] [PubMed] [Google Scholar]
- 9.Ginhoux F, Greter M, Leboeuf M, Nandi S, See P, Gokhan S, Mehler MF, Conway SJ, Ng LG, Stanley ER, Samokhvalov IM, Merad M. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science. 2010;330:841–845. doi: 10.1126/science.1194637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kissa K, Herbomel P. Blood stem cells emerge from aortic endothelium by a novel type of cell transition. Nature. 2010;464:112–115. doi: 10.1038/nature08761. [DOI] [PubMed] [Google Scholar]
- 11.Lichanska AM, Browne CM, Henkel GW, Murphy KM, Ostrowski MC, McKercher SR, Maki RA, Hume DA. Differentiation of the mononuclear phagocyte system during mouse embryogenesis: the role of transcription factor PU. 1. Blood. 1999;94:127–138. [PubMed] [Google Scholar]
- 12.Kierdorf K, Erny D, Goldmann T, Sander V, Schulz C, Perdiguero EG, Wieghofer P, Heinrich A, Riemke P, Holscher C, Muller DN, Luckow B, Brocker T, Debowski K, Fritz G, Opdenakker G, Diefenbach A, Biber K, Heikenwalder M, Geissmann F, Rosenbauer F, Prinz M. Microglia emerge from erythromyeloid precursors via Pu. 1- and Irf8-dependent pathways. Nat Neurosci. 2013;46:273–80. doi: 10.1038/nn.3318. [DOI] [PubMed] [Google Scholar]
- 13.Mizutani M, Pino PA, Saederup N, Charo IF, Ransohoff RM, Cardona AE. The fractalkine receptor but not CCR2 is present on microglia from embryonic development throughout adulthood. J Immunol. 2012;188:29–36. doi: 10.4049/jimmunol.1100421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mukouyama Y, Chiba N, Mucenski ML, Satake M, Miyajima A, Hara T, Watanabe T. Hematopoietic cells in cultures of the murine embryonic aorta-gonad-mesonephros region are induced by c-Myb. Curr Biol. 1999;9:833–836. doi: 10.1016/s0960-9822(99)80368-6. [DOI] [PubMed] [Google Scholar]
- 15.Schulz C, Gomez Perdiguero E, Chorro L, Szabo-Rogers H, Cagnard N, Kierdorf K, Prinz M, Wu B, Jacobsen SE, Pollard JW, Frampton J, Liu KJ, Geissmann F. A lineage of myeloid cells independent of Myb and hematopoietic stem cells. Science. 2012;336:86–90. doi: 10.1126/science.1219179. [DOI] [PubMed] [Google Scholar]
- 16.Labastie MC, Cortes F, Romeo PH, Dulac C, Peault B. Molecular identity of hematopoietic precursor cells emerging in the human embryo. Blood. 1998;92:3624–3635. [PubMed] [Google Scholar]
- 17.Erblich B, Zhu L, Etgen AM, Dobrenis K, Pollard JW. Absence of colony stimulation factor-1 receptor results in loss of microglia, disrupted brain development and olfactory deficits. PLoS One. 2011;6:e26317. doi: 10.1371/journal.pone.0026317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ovchinnikov DA. Macrophages in the embryo and beyond: much more than just giant phagocytes. Genesis. 2008;46:447–462. doi: 10.1002/dvg.20417. [DOI] [PubMed] [Google Scholar]
- 19.Tambuyzer BR, Ponsaerts P, Nouwen EJ. Microglia: gatekeepers of central nervous system immunology. J Leukoc Biol. 2009;85:352–370. doi: 10.1189/jlb.0608385. [DOI] [PubMed] [Google Scholar]
- 20.Imai Y, Ibata I, Ito D, Ohsawa K, Kohsaka S. A novel gene iba1 in the major histocompatibility complex class III region encoding an EF hand protein expressed in a monocytic lineage. Biochem Biophys Res Commun. 1996;224:855–862. doi: 10.1006/bbrc.1996.1112. [DOI] [PubMed] [Google Scholar]
- 21.Ito D, Imai Y, Ohsawa K, Nakajima K, Fukuuchi Y, Kohsaka S. Microglia-specific localisation of a novel calcium binding protein, Iba1. Brain Res Mol Brain Res. 1998;57:1–9. doi: 10.1016/s0169-328x(98)00040-0. [DOI] [PubMed] [Google Scholar]
- 22.Ito D, Tanaka K, Suzuki S, Dembo T, Fukuuchi Y. Enhanced expression of Iba1, ionized calcium-binding adapter molecule 1, after transient focal cerebral ischemia in rat brain. Stroke. 2001;32:1208–1215. doi: 10.1161/01.str.32.5.1208. [DOI] [PubMed] [Google Scholar]
- 23.Sedgwick JD, Schwender S, Imrich H, Dorries R, Butcher GW, ter Meulen V. Isolation and direct characterization of resident microglial cells from the normal and inflamed central nervous system. Proc Natl Acad Sci U S A. 1991;88:7438–7442. doi: 10.1073/pnas.88.16.7438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ford AL, Goodsall AL, Hickey WF, Sedgwick JD. Normal adult ramified microglia separated from other central nervous system macrophages by flow cytometric sorting. Phenotypic differences defined and direct ex vivo antigen presentation to myelin basic protein-reactive CD4+ T cells compared. J Immunol. 1995;154:4309–4321. [PubMed] [Google Scholar]
- 25.Perego C, Fumagalli S, De Simoni MG. Temporal pattern of expression and colocalization of microglia/macrophage phenotype markers following brain ischemic injury in mice. J Neuroinflammation. 2011;8:174. doi: 10.1186/1742-2094-8-174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gesuete R, Storini C, Fantin A, Stravalaci M, Zanier ER, Orsini F, Vietsch H, Mannesse ML, Ziere B, Gobbi M, De Simoni MG. Recombinant C1 inhibitor in brain ischemic injury. Ann Neurol. 2009;66:332–342. doi: 10.1002/ana.21740. [DOI] [PubMed] [Google Scholar]
- 27.Guillemin GJ, Brew BJ. Microglia, macrophages, perivascular macrophages, and pericytes: a review of function and identification. J Leukoc Biol. 2004;75:388–397. doi: 10.1189/jlb.0303114. [DOI] [PubMed] [Google Scholar]
- 28.Ponomarev ED, Shriver LP, Maresz K, Dittel BN. Microglial cell activation and proliferation precedes the onset of CNS autoimmunity. J Neurosci Res. 2005;81:374–389. doi: 10.1002/jnr.20488. [DOI] [PubMed] [Google Scholar]
- 29.Melief J, Koning N, Schuurman KG, Van De Garde MD, Smolders J, Hoek RM, Van Eijk M, Hamann J, Huitinga I. Phenotyping primary human microglia: tight regulation of LPS responsiveness. Glia. 2012;60:1506–1517. doi: 10.1002/glia.22370. [DOI] [PubMed] [Google Scholar]
- 30.Hume DA. Applications of myeloid-specific promoters in transgenic mice support in vivo imaging and functional genomics but do not support the concept of distinct macrophage and dendritic cell lineages or roles in immunity. J Leukoc Biol. 2011;89:525–538. doi: 10.1189/jlb.0810472. [DOI] [PubMed] [Google Scholar]
- 31.Hirasawa T, Ohsawa K, Imai Y, Ondo Y, Akazawa C, Uchino S, Kohsaka S. Visualization of microglia in living tissues using Iba1-EGFP transgenic mice. J Neurosci Res. 2005;81:357–362. doi: 10.1002/jnr.20480. [DOI] [PubMed] [Google Scholar]
- 32.Saederup N, Cardona AE, Croft K, Mizutani M, Cotleur AC, Tsou CL, Ransohoff RM, Charo IF. Selective chemokine receptor usage by central nervous system myeloid cells in CCR2-red fluorescent protein knock-in mice. PLoS One. 2010;5:e13693. doi: 10.1371/journal.pone.0013693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Goren I, Allmann N, Yogev N, Schurmann C, Linke A, Holdener M, Waisman A, Pfeilschifter J, Frank S. A transgenic mouse model of inducible macrophage depletion: effects of diphtheria toxin-driven lysozyme M-specific cell lineage ablation on wound inflammatory, angiogenic, and contractive processes. Am J Pathol. 2009;175:132–147. doi: 10.2353/ajpath.2009.081002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gowing G, Vallieres L, Julien JP. Mouse model for ablation of proliferating microglia in acute CNS injuries. Glia. 2006;53:331–337. doi: 10.1002/glia.20288. [DOI] [PubMed] [Google Scholar]
- 35.Tang X, Zheng Z, Yenari MA. Bone Marrow Chimeras in the Study of Experimental Stroke. Transl. Stroke Res. 2012;3:341–347. doi: 10.1007/s12975-012-0169-6. [DOI] [PubMed] [Google Scholar]
- 36.Monje ML, Mizumatsu S, Fike JR, Palmer TD. Irradiation induces neural precursor-cell dysfunction. Nat Med. 2002;8:955–962. doi: 10.1038/nm749. [DOI] [PubMed] [Google Scholar]
- 37.Davoust N, Vuaillat C, Androdias G, Nataf S. From bone marrow to microglia: barriers and avenues. Trends Immunol. 2008;29:227–234. doi: 10.1016/j.it.2008.01.010. [DOI] [PubMed] [Google Scholar]
- 38.Mildner A, Schmidt H, Nitsche M, Merkler D, Hanisch UK, Mack M, Heikenwalder M, Bruck W, Priller J, Prinz M. Microglia in the adult brain arise from Ly-6ChiCCR2+ monocytes only under defined host conditions. Nat Neurosci. 2007;10:1544–1553. doi: 10.1038/nn2015. [DOI] [PubMed] [Google Scholar]
- 39.Wirenfeldt M, Dissing-Olesen L, Anne Babcock A, Nielsen M, Meldgaard M, Zimmer J, Azcoitia I, Leslie RG, Dagnaes-Hansen F, Finsen B. Population control of resident and immigrant microglia by mitosis and apoptosis. Am J Pathol. 2007;171:617–631. doi: 10.2353/ajpath.2007.061044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li YQ, Chen P, Jain V, Reilly RM, Wong CS. Early radiation-induced endothelial cell loss and blood-spinal cord barrier breakdown in the rat spinal cord. Radiat Res. 2004;161:143–152. doi: 10.1667/rr3117. [DOI] [PubMed] [Google Scholar]
- 41.Lambertsen KL, Clausen BH, Babcock AA, Gregersen R, Fenger C, Nielsen HH, Haugaard LS, Wirenfeldt M, Nielsen M, Dagnaes-Hansen F, Bluethmann H, Faergeman NJ, Meldgaard M, Deierborg T, Finsen B. Microglia protect neurons against ischemia by synthesis of tumor necrosis factor. J Neurosci. 2009;29:1319–1330. doi: 10.1523/JNEUROSCI.5505-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Linard C, Marquette C, Mathieu J, Pennequin A, Clarencon D, Mathe D. Acute induction of inflammatory cytokine expression after gamma-irradiation in the rat: effect of an NF-kappaB inhibitor. Int J Radiat Oncol Biol Phys. 2004;58:427–434. doi: 10.1016/j.ijrobp.2003.09.039. [DOI] [PubMed] [Google Scholar]
- 43.Mizumatsu S, Monje ML, Morhardt DR, Rola R, Palmer TD, Fike JR. Extreme sensitivity of adult neurogenesis to low doses of X-irradiation. Cancer Res. 2003;63:4021–4027. [PubMed] [Google Scholar]
- 44.Davalos D, Grutzendler J, Yang G, Kim JV, Zuo Y, Jung S, Littman DR, Dustin ML, Gan WB. ATP mediates rapid microglial response to local brain injury in vivo. Nat Neurosci. 2005;8:752–758. doi: 10.1038/nn1472. [DOI] [PubMed] [Google Scholar]
- 45.Nimmerjahn A, Kirchhoff F, Helmchen F. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science. 2005;308:1314–1318. doi: 10.1126/science.1110647. [DOI] [PubMed] [Google Scholar]
- 46.Lawson LJ, Perry VH, Dri P, Gordon S. Heterogeneity in the distribution and morphology of microglia in the normal adult mouse brain. Neuroscience. 1990;39:151–170. doi: 10.1016/0306-4522(90)90229-w. [DOI] [PubMed] [Google Scholar]
- 47.Carson MJ, Bilousova TV, Puntambekar SS, Melchior B, Doose JM, Ethell IM. A rose by any other name? The potential consequences of microglial heterogeneity during CNS health and disease. Neurotherapeutics. 2007;4:571–579. doi: 10.1016/j.nurt.2007.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Biber K, Neumann H, Inoue K, Boddeke HW. Neuronal ‘On’ and ‘Off’ signals control microglia. Trends Neurosci. 2007;30:596–602. doi: 10.1016/j.tins.2007.08.007. [DOI] [PubMed] [Google Scholar]
- 49.Hoek RM, Ruuls SR, Murphy CA, Wright GJ, Goddard R, Zurawski SM, Blom B, Homola ME, Streit WJ, Brown MH, Barclay AN, Sedgwick JD. Down-regulation of the macrophage lineage through interaction with OX2 (CD200) Science. 2000;290:1768–1771. doi: 10.1126/science.290.5497.1768. [DOI] [PubMed] [Google Scholar]
- 50.Cardona AE, Pioro EP, Sasse ME, Kostenko V, Cardona SM, Dijkstra IM, Huang D, Kidd G, Dombrowski S, Dutta R, Lee JC, Cook DN, Jung S, Lira SA, Littman DR, Ransohoff RM. Control of microglial neurotoxicity by the fractalkine receptor. Nat Neurosci. 2006;9:917–924. doi: 10.1038/nn1715. [DOI] [PubMed] [Google Scholar]
- 51.Fuhrmann M, Bittner T, Jung CK, Burgold S, Page RM, Mitteregger G, Haass C, LaFerla FM, Kretzschmar H, Herms J. Microglial Cx3cr1 knockout prevents neuron loss in a mouse model of Alzheimer’s disease. Nat Neurosci. 2010;13:411–413. doi: 10.1038/nn.2511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cipriani R, Villa P, Chece G, Lauro C, Paladini A, Micotti E, Perego C, De Simoni MG, Fredholm BB, Eusebi F, Limatola C. CX3CL1 is neuroprotective in permanent focal cerebral ischemia in rodents. J Neurosci. 2011;31:16327–16335. doi: 10.1523/JNEUROSCI.3611-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Koning N, Bo L, Hoek RM, Huitinga I. Downregulation of macrophage inhibitory molecules in multiple sclerosis lesions. Ann Neurol. 2007;62:504–514. doi: 10.1002/ana.21220. [DOI] [PubMed] [Google Scholar]
- 54.Barclay AN, Wright GJ, Brooke G, Brown MH. CD200 and membrane protein interactions in the control of myeloid cells. Trends Immunol. 2002;23:285–290. doi: 10.1016/s1471-4906(02)02223-8. [DOI] [PubMed] [Google Scholar]
- 55.Koning N, Swaab DF, Hoek RM, Huitinga I. Distribution of the immune inhibitory molecules CD200 and CD200R in the normal central nervous system and multiple sclerosis lesions suggests neuron-glia and glia-glia interactions. J Neuropathol Exp Neurol. 2009;68:159–167. doi: 10.1097/NEN.0b013e3181964113. [DOI] [PubMed] [Google Scholar]
- 56.Webb M, Barclay AN. Localisation of the MRC OX-2 glycoprotein on the surfaces of neurones. J Neurochem. 1984;43:1061–1067. doi: 10.1111/j.1471-4159.1984.tb12844.x. [DOI] [PubMed] [Google Scholar]
- 57.Barclay AN. The localization of populations of lymphocytes defined by monoclonal antibodies in rat lymphoid tissues. Immunology. 1981;42:593–600. [PMC free article] [PubMed] [Google Scholar]
- 58.Chitnis T, Imitola J, Wang Y, Elyaman W, Chawla P, Sharuk M, Raddassi K, Bronson RT, Khoury SJ. Elevated neuronal expression of CD200 protects Wlds mice from inflammation-mediated neurodegeneration. Am J Pathol. 2007;170:1695–1712. doi: 10.2353/ajpath.2007.060677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Cox FF, Carney D, Miller AM, Lynch MA. CD200 fusion protein decreases microglial activation in the hippocampus of aged rats. Brain Behav Immun. 2012;26:789–796. doi: 10.1016/j.bbi.2011.10.004. [DOI] [PubMed] [Google Scholar]
- 60.Matsumoto H, Kumon Y, Watanabe H, Ohnishi T, Takahashi H, Imai Y, Tanaka J. Expression of CD200 by macrophage-like cells in ischemic core of rat brain after transient middle cerebral artery occlusion. Neurosci Lett. 2007;418:44–48. doi: 10.1016/j.neulet.2007.03.027. [DOI] [PubMed] [Google Scholar]
- 61.Ludwig A, Weber C. Transmembrane chemokines: versatile ‘special agents’ in vascular inflammation. Thromb Haemost. 2007;97:694–703. [PubMed] [Google Scholar]
- 62.Fumagalli S, Perego C, Ortolano F, De Simoni MG. CX3CR1 deficiency induces an early protective inflammatory environment in ischemic mice. Glia. 2013;61:827–42. doi: 10.1002/glia.22474. [DOI] [PubMed] [Google Scholar]
- 63.Denes A, Ferenczi S, Halasz J, Kornyei Z, Kovacs KJ. Role of CX3CR1 (fractalkine receptor) in brain damage and inflammation induced by focal cerebral ischemia in mouse. J Cereb Blood Flow Metab. 2008;28:1707–1721. doi: 10.1038/jcbfm.2008.64. [DOI] [PubMed] [Google Scholar]
- 64.Soriano SG, Amaravadi LS, Wang YF, Zhou H, Yu GX, Tonra JR, Fairchild-Huntress V, Fang Q, Dunmore JH, Huszar D, Pan Y. Mice deficient in fractalkine are less susceptible to cerebral ischemia-reperfusion injury. J Neuroimmunol. 2002;125:59–65. doi: 10.1016/s0165-5728(02)00033-4. [DOI] [PubMed] [Google Scholar]
- 65.Jiang P, Lagenaur CF, Narayanan V. Integrin-associated protein is a ligand for the P84 neural adhesion molecule. J Biol Chem. 1999;274:559–562. doi: 10.1074/jbc.274.2.559. [DOI] [PubMed] [Google Scholar]
- 66.Fujioka Y, Matozaki T, Noguchi T, Iwamatsu A, Yamao T, Takahashi N, Tsuda M, Takada T, Kasuga M. A novel membrane glycoprotein, SHPS-1, that binds the SH2-domain-containing protein tyrosine phosphatase SHP-2 in response to mitogens and cell adhesion. Mol Cell Biol. 1996;16:6887–6899. doi: 10.1128/mcb.16.12.6887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Okazawa H, Motegi S, Ohyama N, Ohnishi H, Tomizawa T, Kaneko Y, Oldenborg PA, Ishikawa O, Matozaki T. Negative regulation of phagocytosis in macrophages by the CD47-SHPS-1 system. J Immunol. 2005;174:2004–2011. doi: 10.4049/jimmunol.174.4.2004. [DOI] [PubMed] [Google Scholar]
- 68.Yamao T, Noguchi T, Takeuchi O, Nishiyama U, Morita H, Hagiwara T, Akahori H, Kato T, Inagaki K, Okazawa H, Hayashi Y, Matozaki T, Takeda K, Akira S, Kasuga M. Negative regulation of platelet clearance and of the macrophage phagocytic response by the transmembrane glycoprotein SHPS-1. J Biol Chem. 2002;277:39833–39839. doi: 10.1074/jbc.M203287200. [DOI] [PubMed] [Google Scholar]
- 69.Jin G, Tsuji K, Xing C, Yang YG, Wang X, Lo EH. CD47 gene knockout protects against transient focal cerebral ischemia in mice. Exp Neurol. 2009;217:165–170. doi: 10.1016/j.expneurol.2009.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wang L, Lu Y, Deng S, Zhang Y, Yang L, Guan Y, Matozaki T, Ohnishi H, Jiang H, Li H. SHPS-1 deficiency induces robust neuroprotection against experimental stroke by attenuating oxidative stress. J Neurochem. 2012;122:834–843. doi: 10.1111/j.1471-4159.2012.07818.x. [DOI] [PubMed] [Google Scholar]
- 71.Stefano L, Racchetti G, Bianco F, Passini N, Gupta RS, Panina Bordignon P, Meldolesi J. The surface-exposed chaperone, Hsp60, is an agonist of the microglial TREM2 receptor. J Neurochem. 2009;110:284–294. doi: 10.1111/j.1471-4159.2009.06130.x. [DOI] [PubMed] [Google Scholar]
- 72.Piccio L, Buonsanti C, Mariani M, Cella M, Gilfillan S, Cross AH, Colonna M, Panina-Bordignon P. Blockade of TREM-2 exacerbates experimental autoimmune encephalomyelitis. Eur J Immunol. 2007;37:1290–1301. doi: 10.1002/eji.200636837. [DOI] [PubMed] [Google Scholar]
- 73.Takahashi K, Rochford CD, Neumann H. Clearance of apoptotic neurons without inflammation by microglial triggering receptor expressed on myeloid cells-2. J Exp Med. 2005;201:647–657. doi: 10.1084/jem.20041611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Sieber MW, Jaenisch N, Brehm M, Guenther M, Linnartz-Gerlach B, Neumann H, Witte OW, Frahm C. Attenuated inflammatory response in triggering receptor expressed on myeloid cells 2 (TREM2) knock-out mice following stroke. PLoS One. 2013;8:e52982. doi: 10.1371/journal.pone.0052982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ohsawa K, Irino Y, Nakamura Y, Akazawa C, Inoue K, Kohsaka S. Involvement of P2X4 and P2Y12 receptors in ATP-induced microglial chemotaxis. Glia. 2007;55:604–616. doi: 10.1002/glia.20489. [DOI] [PubMed] [Google Scholar]
- 76.Haynes SE, Hollopeter G, Yang G, Kurpius D, Dailey ME, Gan WB, Julius D. The P2Y12 receptor regulates microglial activation by extracellular nucleotides. Nat Neurosci. 2006;9:1512–1519. doi: 10.1038/nn1805. [DOI] [PubMed] [Google Scholar]
- 77.Varnum MM, Ikezu T. The classification of microglial activation phenotypes on neurodegeneration and regeneration in Alzheimer’s disease brain. Arch Immunol Ther Exp (Warsz) 2012;60:251–266. doi: 10.1007/s00005-012-0181-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Yenari MA, Kauppinen TM, Swanson RA. Microglial activation in stroke: therapeutic targets. Neurotherapeutics. 2010;7:378–391. doi: 10.1016/j.nurt.2010.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Hu X, Li P, Guo Y, Wang H, Leak RK, Chen S, Gao Y, Chen J. Microglia/macrophage polarization dynamics reveal novel mechanism of injury expansion after focal cerebral ischemia. Stroke. 2012;43:3063–3070. doi: 10.1161/STROKEAHA.112.659656. [DOI] [PubMed] [Google Scholar]
- 80.Ponomarev ED, Veremeyko T, Weiner HL. MicroRNAs are universal regulators of differentiation, activation, and polarization of microglia and macrophages in normal and diseased CNS. Glia. 2013;61:91–103. doi: 10.1002/glia.22363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Liu HC, Zheng MH, Du YL, Wang L, Kuang F, Qin HY, Zhang BF, Han H. N9 microglial cells polarized by LPS and IL4 show differential responses to secondary environmental stimuli. Cell Immunol. 2012;278:84–90. doi: 10.1016/j.cellimm.2012.06.001. [DOI] [PubMed] [Google Scholar]
- 82.Shin WH, Lee DY, Park KW, Kim SU, Yang MS, Joe EH, Jin BK. Microglia expressing interleukin-13 undergo cell death and contribute to neuronal survival in vivo. Glia. 2004;46:142–152. doi: 10.1002/glia.10357. [DOI] [PubMed] [Google Scholar]
- 83.Zhou X, Spittau B, Krieglstein K. TGFbeta signalling plays an important role in IL4-induced alternative activation of microglia. J Neuroinflammation. 2012;9:210. doi: 10.1186/1742-2094-9-210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Hanisch UK, Kettenmann H. Microglia: active sensor and versatile effector cells in the normal and pathologic brain. Nat Neurosci. 2007;10:1387–1394. doi: 10.1038/nn1997. [DOI] [PubMed] [Google Scholar]
- 85.Pal G, Vincze C, Renner E, Wappler EA, Nagy Z, Lovas G, Dobolyi A. Time course, distribution and cell types of induction of transforming growth factor betas following middle cerebral artery occlusion in the rat brain. PLoS One. 2012;7:e46731. doi: 10.1371/journal.pone.0046731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Kanazawa H, Ohsawa K, Sasaki Y, Kohsaka S, Imai Y. Macrophage/microglia-specific protein Iba1 enhances membrane ruffling and Rac activation via phospholipase C-gamma -dependent pathway. J Biol Chem. 2002;277:20026–20032. doi: 10.1074/jbc.M109218200. [DOI] [PubMed] [Google Scholar]
- 87.Colton CA, Abel C, Patchett J, Keri J, Yao J. Lectin staining of cultured CNS microglia. J Histochem Cytochem. 1992;40:505–512. doi: 10.1177/40.4.1372634. [DOI] [PubMed] [Google Scholar]
- 88.Boscia F, Esposito CL, Casamassa A, de Franciscis V, Annunziato L, Cerchia L. The isolectin IB4 binds RET receptor tyrosine kinase in microglia. J Neurochem. 2013 doi: 10.1111/jnc.12209. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 89.Perry VH. A revised view of the central nervous system microenvironment and major histocompatibility complex class II antigen presentation. J Neuroimmunol. 1998;90:113–121. doi: 10.1016/s0165-5728(98)00145-3. [DOI] [PubMed] [Google Scholar]
- 90.Ulvestad E, Williams K, Vedeler C, Antel J, Nyland H, Mork S, Matre R. Reactive microglia in multiple sclerosis lesions have an increased expression of receptors for the Fc part of IgG. J Neurol Sci. 1994;121:125–131. doi: 10.1016/0022-510x(94)90340-9. [DOI] [PubMed] [Google Scholar]
- 91.Vedeler C, Ulvestad E, Grundt I, Conti G, Nyland H, Matre R, Pleasure D. Fc receptor for IgG (FcR) on rat microglia. J Neuroimmunol. 1994;49:19–24. doi: 10.1016/0165-5728(94)90176-7. [DOI] [PubMed] [Google Scholar]
- 92.Wolf SA, Gimsa U, Bechmann I, Nitsch R. Differential expression of costimulatory molecules B7-1 and B7-2 on microglial cells induced by Th1 and Th2 cells in organotypic brain tissue. Glia. 2001;36:414–420. doi: 10.1002/glia.1127. [DOI] [PubMed] [Google Scholar]
- 93.Becher B, Antel JP. Comparison of phenotypic and functional properties of immediately ex vivo and cultured human adult microglia. Glia. 1996;18:1–10. doi: 10.1002/(SICI)1098-1136(199609)18:1<1::AID-GLIA1>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- 94.Aloisi F, De Simone R, Columba-Cabezas S, Penna G, Adorini L. Functional maturation of adult mouse resting microglia into an APC is promoted by granulocyte-macrophage colony-stimulating factor and interaction with Th1 cells. J Immunol. 2000;164:1705–1712. doi: 10.4049/jimmunol.164.4.1705. [DOI] [PubMed] [Google Scholar]
- 95.Colton CA, Mott RT, Sharpe H, Xu Q, Van Nostrand WE, Vitek MP. Expression profiles for macrophage alternative activation genes in AD and in mouse models of AD. J Neuroinflammation. 2006;3:27. doi: 10.1186/1742-2094-3-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Ramprasad MP, Terpstra V, Kondratenko N, Quehenberger O, Steinberg D. Cell surface expression of mouse macrosialin and human CD68 and their role as macrophage receptors for oxidized low density lipoprotein. Proc Natl Acad Sci U S A. 1996;93:14833–14838. doi: 10.1073/pnas.93.25.14833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Raes G, De Baetselier P, Noel W, Beschin A, Brombacher F, Hassanzadeh Gh G. Differential expression of FIZZ1 and Ym1 in alternatively versus classically activated macrophages. J Leukoc Biol. 2002;71:597–602. [PubMed] [Google Scholar]
- 98.Shah VB, Huang Y, Keshwara R, Ozment-Skelton T, Williams DL, Keshvara L. Beta-glucan activates microglia without inducing cytokine production in Dectin-1-dependent manner. J Immunol. 2008;180:2777–2785. doi: 10.4049/jimmunol.180.5.2777. [DOI] [PubMed] [Google Scholar]
- 99.Giraldi-Guimaraes A, de Freitas HT, Coelho Bde P, Macedo-Ramos H, Mendez-Otero R, Cavalcante LA, Baetas-da-Cruz W. Bone marrow mononuclear cells and mannose receptor expression in focal cortical ischemia. Brain Res. 2012;1452:173–184. doi: 10.1016/j.brainres.2012.03.002. [DOI] [PubMed] [Google Scholar]
- 100.Jimenez S, Baglietto-Vargas D, Caballero C, Moreno-Gonzalez I, Torres M, Sanchez-Varo R, Ruano D, Vizuete M, Gutierrez A, Vitorica J. Inflammatory response in the hippocampus of PS1M146L/APP751SL mouse model of Alzheimer’s disease: age-dependent switch in the microglial phenotype from alternative to classic. J Neurosci. 2008;28:11650–11661. doi: 10.1523/JNEUROSCI.3024-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Katayama T, Kobayashi H, Okamura T, Yamasaki-Katayama Y, Kibayashi T, Kimura H, Ohsawa K, Kohsaka S, Minami M. Accumulating microglia phagocytose injured neurons in hippocampal slice cultures: involvement of p38 MAP kinase. PLoS One. 2012;7:e40813. doi: 10.1371/journal.pone.0040813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Denes A, Vidyasagar R, Feng J, Narvainen J, McColl BW, Kauppinen RA, Allan SM. Proliferating resident microglia after focal cerebral ischaemia in mice. J Cereb Blood Flow Metab. 2007;27:1941–1953. doi: 10.1038/sj.jcbfm.9600495. [DOI] [PubMed] [Google Scholar]
- 103.Sakanashi Y, Takeya M, Yoshimura T, Feng L, Morioka T, Takahashi K. Kinetics of macrophage subpopulations and expression of monocyte chemoattractant protein-1 (MCP-1) in bleomycin-induced lung injury of rats studied by a novel monoclonal antibody against rat MCP-1. J Leukoc Biol. 1994;56:741–750. doi: 10.1002/jlb.56.6.741. [DOI] [PubMed] [Google Scholar]
- 104.Kumar AG, Ballantyne CM, Michael LH, Kukielka GL, Youker KA, Lindsey ML, Hawkins HK, Birdsall HH, MacKay CR, LaRosa GJ, Rossen RD, Smith CW, Entman ML. Induction of monocyte chemoattractant protein-1 in the small veins of the ischemic and reperfused canine myocardium. Circulation. 1997;95:693–700. doi: 10.1161/01.cir.95.3.693. [DOI] [PubMed] [Google Scholar]
- 105.Che X, Ye W, Panga L, Wu DC, Yang GY. Monocyte chemoattractant protein-1 expressed in neurons and astrocytes during focal ischemia in mice. Brain Res. 2001;902:171–177. doi: 10.1016/s0006-8993(01)02328-9. [DOI] [PubMed] [Google Scholar]
- 106.Ceulemans AG, Zgavc T, Kooijman R, Hachimi-Idrissi S, Sarre S, Michotte Y. The dual role of the neuroinflammatory response after ischemic stroke: modulatory effects of hypothermia. J Neuroinflammation. 2010;7:74. doi: 10.1186/1742-2094-7-74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Dimitrijevic J, Dzirlo K, Bratic M, Hrnjica M, Hebib L, Alajbegovic A, Heco S, Bulic G. [10-year analysis of cerebrovascular accidents at the Neurology Clinic in Sarajevo (before, during and after the war)] . Med Arh. 2002;56:151–153. [PubMed] [Google Scholar]
- 108.del Zoppo GJ, Sharp FR, Heiss WD, Albers GW. Heterogeneity in the penumbra. J Cereb Blood Flow Metab. 2011;31:1836–1851. doi: 10.1038/jcbfm.2011.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Schilling M, Besselmann M, Muller M, Strecker JK, Ringelstein EB, Kiefer R. Predominant phagocytic activity of resident microglia over hematogenous macrophages following transient focal cerebral ischemia: an investigation using green fluorescent protein transgenic bone marrow chimeric mice. Exp Neurol. 2005;196:290–297. doi: 10.1016/j.expneurol.2005.08.004. [DOI] [PubMed] [Google Scholar]
- 110.Neher JJ, Neniskyte U, Zhao JW, Bal-Price A, Tolkovsky AM, Brown GC. Inhibition of microglial phagocytosis is sufficient to prevent inflammatory neuronal death. J Immunol. 2011;186:4973–4983. doi: 10.4049/jimmunol.1003600. [DOI] [PubMed] [Google Scholar]
- 111.Franke H, Gunther A, Grosche J, Schmidt R, Rossner S, Reinhardt R, Faber-Zuschratter H, Schneider D, Illes P. P2X7 receptor expression after ischemia in the cerebral cortex of rats. J Neuropathol Exp Neurol. 2004;63:686–699. doi: 10.1093/jnen/63.7.686. [DOI] [PubMed] [Google Scholar]
- 112.Melani A, Turchi D, Vannucchi MG, Cipriani S, Gianfriddo M, Pedata F. ATP extracellular concentrations are increased in the rat striatum during in vivo ischemia. Neurochem Int. 2005;47:442–448. doi: 10.1016/j.neuint.2005.05.014. [DOI] [PubMed] [Google Scholar]
- 113.Monif M, Reid CA, Powell KL, Smart ML, Williams DA. The P2X7 receptor drives microglial activation and proliferation: a trophic role for P2X7R pore. J Neurosci. 2009;29:3781–3791. doi: 10.1523/JNEUROSCI.5512-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Brough D, Le Feuvre RA, Iwakura Y, Rothwell NJ. Purinergic (P2X7) receptor activation of microglia induces cell death via an interleukin-1-independent mechanism. Mol Cell Neurosci. 2002;19:272–280. doi: 10.1006/mcne.2001.1054. [DOI] [PubMed] [Google Scholar]
- 115.Ferrari D, Pizzirani C, Adinolfi E, Lemoli RM, Curti A, Idzko M, Panther E, Di Virgilio F. The P2X7 receptor: a key player in IL-1 processing and release. J Immunol. 2006;176:3877–3883. doi: 10.4049/jimmunol.176.7.3877. [DOI] [PubMed] [Google Scholar]
- 116.Verderio C, Matteoli M. ATP mediates calcium signaling between astrocytes and microglial cells: modulation by IFN-gamma. J Immunol. 2001;166:6383–6391. doi: 10.4049/jimmunol.166.10.6383. [DOI] [PubMed] [Google Scholar]
- 117.Honda S, Sasaki Y, Ohsawa K, Imai Y, Nakamura Y, Inoue K, Kohsaka S. Extracellular ATP or ADP induce chemotaxis of cultured microglia through Gi/o-coupled P2Y receptors. J Neurosci. 2001;21:1975–1982. doi: 10.1523/JNEUROSCI.21-06-01975.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Pellegatti P, Falzoni S, Pinton P, Rizzuto R, Di Virgilio F. A novel recombinant plasma membrane-targeted luciferase reveals a new pathway for ATP secretion. Mol Biol Cell. 2005;16:3659–3665. doi: 10.1091/mbc.E05-03-0222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Chu K, Yin B, Wang J, Peng G, Liang H, Xu Z, Du Y, Fang M, Xia Q, Luo B. Inhibition of P2X7 receptor ameliorates transient global cerebral ischemia/reperfusion injury via modulating inflammatory responses in the rat hippocampus. J Neuroinflammation. 2012;9:69. doi: 10.1186/1742-2094-9-69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Melani A, Amadio S, Gianfriddo M, Vannucchi MG, Volonte C, Bernardi G, Pedata F, Sancesario G. P2X7 receptor modulation on microglial cells and reduction of brain infarct caused by middle cerebral artery occlusion in rat. J Cereb Blood Flow Metab. 2006;26:974–982. doi: 10.1038/sj.jcbfm.9600250. [DOI] [PubMed] [Google Scholar]
- 121.Piccinini AM, Midwood KS. DAMPening inflammation by modulating TLR signalling. Mediators Inflamm. 2010:2010. doi: 10.1155/2010/672395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Ziegler G, Prinz V, Albrecht MW, Harhausen D, Khojasteh U, Nacken W, Endres M, Dirnagl U, Nietfeld W, Trendelenburg G. Mrp-8 and -14 mediate CNS injury in focal cerebral ischemia. Biochim Biophys Acta. 2009;1792:1198–1204. doi: 10.1016/j.bbadis.2009.10.003. [DOI] [PubMed] [Google Scholar]
- 123.Chow JC, Young DW, Golenbock DT, Christ WJ, Gusovsky F. Toll-like receptor-4 mediates lipopolysaccharide-induced signal transduction. J Biol Chem. 1999;274:10689–10692. doi: 10.1074/jbc.274.16.10689. [DOI] [PubMed] [Google Scholar]
- 124.Lehnardt S, Lehmann S, Kaul D, Tschimmel K, Hoffmann O, Cho S, Krueger C, Nitsch R, Meisel A, Weber JR. Toll-like receptor 2 mediates CNS injury in focal cerebral ischemia. J Neuroimmunol. 2007;190:28–33. doi: 10.1016/j.jneuroim.2007.07.023. [DOI] [PubMed] [Google Scholar]
- 125.Bohacek I, Cordeau P, Lalancette-Hebert M, Gorup D, Weng YC, Gajovic S, Kriz J. Toll-like receptor 2 deficiency leads to delayed exacerbation of ischemic injury. J Neuroinflammation. 2012;9:191. doi: 10.1186/1742-2094-9-191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Hyakkoku K, Hamanaka J, Tsuruma K, Shimazawa M, Tanaka H, Uematsu S, Akira S, Inagaki N, Nagai H, Hara H. Toll-like receptor 4 (TLR4), but not TLR3 or TLR9, knock-out mice have neuroprotective effects against focal cerebral ischemia. Neuroscience. 2010;171:258–267. doi: 10.1016/j.neuroscience.2010.08.054. [DOI] [PubMed] [Google Scholar]
- 127.Lalancette-Hebert M, Phaneuf D, Soucy G, Weng YC, Kriz J. Live imaging of Toll-like receptor 2 response in cerebral ischaemia reveals a role of olfactory bulb microglia as modulators of inflammation. Brain. 2009;132:940–954. doi: 10.1093/brain/awn345. [DOI] [PubMed] [Google Scholar]
- 128.Suzuki Y, Hattori K, Hamanaka J, Murase T, Egashira Y, Mishiro K, Ishiguro M, Tsuruma K, Hirose Y, Tanaka H, Yoshimura S, Shimazawa M, Inagaki N, Nagasawa H, Iwama T, Hara H. Pharmacological inhibition of TLR4-NOX4 signal protects against neuronal death in transient focal ischemia. Sci Rep. 2012;2:896. doi: 10.1038/srep00896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Chen Z, Jalabi W, Shpargel KB, Farabaugh KT, Dutta R, Yin X, Kidd GJ, Bergmann CC, Stohlman SA, Trapp BD. Lipopolysaccharide-induced microglial activation and neuroprotection against experimental brain injury is independent of hematogenous TLR4. J Neurosci. 2012;32:11706–11715. doi: 10.1523/JNEUROSCI.0730-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Vartanian KB, Stevens SL, Marsh BJ, Williams-Karnesky R, Lessov NS, Stenzel-Poore MP. LPS preconditioning redirects TLR signaling following stroke: TRIF-IRF3 plays a seminal role in mediating tolerance to ischemic injury. J Neuroinflammation. 2011;8:140. doi: 10.1186/1742-2094-8-140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Lu C, Liu L, Chen Y, Ha T, Kelley J, Schweitzer J, Kalbfleisch JH, Kao RL, Williams DL, Li C. TLR2 ligand induces protection against cerebral ischemia/reperfusion injury via activation of phosphoinositide 3-kinase/Akt signaling. J Immunol. 2011;187:1458–1466. doi: 10.4049/jimmunol.1003428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Shichita T, Hasegawa E, Kimura A, Morita R, Sakaguchi R, Takada I, Sekiya T, Ooboshi H, Kitazono T, Yanagawa T, Ishii T, Takahashi H, Mori S, Nishibori M, Kuroda K, Akira S, Miyake K, Yoshimura A. Peroxiredoxin family proteins are key initiators of post-ischemic inflammation in the brain. Nat Med. 2012;18:911–917. doi: 10.1038/nm.2749. [DOI] [PubMed] [Google Scholar]
- 133.Li G, Liang X, Lotze MT. HMGB1: The Central Cytokine for All Lymphoid Cells. Front Immunol. 2013;4:68. doi: 10.3389/fimmu.2013.00068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Vabulas RM, Ahmad-Nejad P, Ghose S, Kirschning CJ, Issels RD, Wagner H. HSP70 as endogenous stimulus of the Toll/interleukin-1 receptor signal pathway. J Biol Chem. 2002;277:15107–15112. doi: 10.1074/jbc.M111204200. [DOI] [PubMed] [Google Scholar]
- 135.Mitrasinovic OM, Grattan A, Robinson CC, Lapustea NB, Poon C, Ryan H, Phong C, Murphy GM Jr. Microglia overexpressing the macrophage colony-stimulating factor receptor are neuroprotective in a microglial-hippocampal organotypic coculture system. J Neurosci. 2005;25:4442–4451. doi: 10.1523/JNEUROSCI.0514-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Lalancette-Hebert M, Swarup V, Beaulieu JM, Bohacek I, Abdelhamid E, Weng YC, Sato S, Kriz J. Galectin-3 is required for resident microglia activation and proliferation in response to ischemic injury. J Neurosci. 2012;32:10383–10395. doi: 10.1523/JNEUROSCI.1498-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.McMenamin PG. Distribution and phenotype of dendritic cells and resident tissue macrophages in the dura mater, leptomeninges, and choroid plexus of the rat brain as demonstrated in wholemount preparations. J Comp Neurol. 1999;405:553–562. [PubMed] [Google Scholar]
- 138.Bechmann I, Priller J, Kovac A, Bontert M, Wehner T, Klett FF, Bohsung J, Stuschke M, Dirnagl U, Nitsch R. Immune surveillance of mouse brain perivascular spaces by blood-borne macrophages. Eur J Neurosci. 2001;14:1651–1658. doi: 10.1046/j.0953-816x.2001.01793.x. [DOI] [PubMed] [Google Scholar]
- 139.Lassmann H, Schmied M, Vass K, Hickey WF. Bone marrow derived elements and resident microglia in brain inflammation. Glia. 1993;7:19–24. doi: 10.1002/glia.440070106. [DOI] [PubMed] [Google Scholar]
- 140.Geissmann F, Auffray C, Palframan R, Wirrig C, Ciocca A, Campisi L, Narni-Mancinelli E, Lauvau G. Blood monocytes: distinct subsets, how they relate to dendritic cells, and their possible roles in the regulation of T-cell responses. Immunol Cell Biol. 2008;86:398–408. doi: 10.1038/icb.2008.19. [DOI] [PubMed] [Google Scholar]
- 141.Varvel NH, Grathwohl SA, Baumann F, Liebig C, Bosch A, Brawek B, Thal DR, Charo IF, Heppner FL, Aguzzi A, Garaschuk O, Ransohoff RM, Jucker M. Microglial repopulation model reveals a robust homeostatic process for replacing CNS myeloid cells. Proc Natl Acad Sci U S A. 2012;109:18150–18155. doi: 10.1073/pnas.1210150109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Candelario-Jalil E, Yang Y, Rosenberg GA. Diverse roles of matrix metalloproteinases and tissue inhibitors of metalloproteinases in neuroinflammation and cerebral ischemia. Neuroscience. 2009;158:983–994. doi: 10.1016/j.neuroscience.2008.06.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Walker EJ, Rosenberg GA. TIMP-3 and MMP-3 contribute to delayed inflammation and hippocampal neuronal death following global ischemia. Exp Neurol. 2009;216:122–131. doi: 10.1016/j.expneurol.2008.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Gidday JM, Gasche YG, Copin JC, Shah AR, Perez RS, Shapiro SD, Chan PH, Park TS. Leukocyte-derived matrix metalloproteinase-9 mediates blood-brain barrier breakdown and is proinflammatory after transient focal cerebral ischemia. Am J Physiol Heart Circ Physiol. 2005;289:H558–568. doi: 10.1152/ajpheart.01275.2004. [DOI] [PubMed] [Google Scholar]
- 145.Wang G, Guo Q, Hossain M, Fazio V, Zeynalov E, Janigro D, Mayberg MR, Namura S. Bone marrow-derived cells are the major source of MMP-9 contributing to blood-brain barrier dysfunction and infarct formation after ischemic stroke in mice. Brain Res. 2009;1294:183–192. doi: 10.1016/j.brainres.2009.07.070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Dawson DA, Martin D, Hallenbeck JM. Inhibition of tumor necrosis factor-alpha reduces focal cerebral ischemic injury in the spontaneously hypertensive rat. Neurosci Lett. 1996;218:41–44. doi: 10.1016/0304-3940(96)13116-5. [DOI] [PubMed] [Google Scholar]
- 147.Lambertsen KL, Biber K, Finsen B. Inflammatory cytokines in experimental and human stroke. J Cereb Blood Flow Metab. 2012;32:1677–1698. doi: 10.1038/jcbfm.2012.88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Fontaine V, Mohand-Said S, Hanoteau N, Fuchs C, Pfizenmaier K, Eisel U. Neurodegenerative and neuroprotective effects of tumor Necrosis factor (TNF) in retinal ischemia: opposite roles of TNF receptor 1 and TNF receptor 2. J Neurosci. 2002;22:RC216. doi: 10.1523/JNEUROSCI.22-07-j0001.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Iadecola C, Zhang F, Xu S, Casey R, Ross ME. Inducible nitric oxide synthase gene expression in brain following cerebral ischemia. J Cereb Blood Flow Metab. 1995;15:378–384. doi: 10.1038/jcbfm.1995.47. [DOI] [PubMed] [Google Scholar]
- 150.Loihl AK, Asensio V, Campbell IL, Murphy S. Expression of nitric oxide synthase (NOS)-2 following permanent focal ischemia and the role of nitric oxide in infarct generation in male, female and NOS-2 gene-deficient mice. Brain Res. 1999;830:155–164. doi: 10.1016/s0006-8993(99)01388-8. [DOI] [PubMed] [Google Scholar]
- 151.Iadecola C, Zhang F, Casey R, Nagayama M, Ross ME. Delayed reduction of ischemic brain injury and neurological deficits in mice lacking the inducible nitric oxide synthase gene. J Neurosci. 1997;17:9157–9164. doi: 10.1523/JNEUROSCI.17-23-09157.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Iadecola C, Zhang F, Xu X. Inhibition of inducible nitric oxide synthase ameliorates cerebral ischemic damage. Am J Physiol. 1995;268:R286–292. doi: 10.1152/ajpregu.1995.268.1.R286. [DOI] [PubMed] [Google Scholar]
- 153.Pruss H, Prass K, Ghaeni L, Milosevic M, Muselmann C, Freyer D, Royl G, Reuter U, Baeva N, Dirnagl U, Meisel A, Priller J. Inducible nitric oxide synthase does not mediate brain damage after transient focal cerebral ischemia in mice. J Cereb Blood Flow Metab. 2008;28:526–539. doi: 10.1038/sj.jcbfm.9600550. [DOI] [PubMed] [Google Scholar]
- 154.Hu X, Weng Z, Chu CT, Zhang L, Cao G, Gao Y, Signore A, Zhu J, Hastings T, Greenamyre JT, Chen J. Peroxiredoxin-2 protects against 6-hydroxydopamine-induced dopaminergic neurodegeneration via attenuation of the apoptosis signal-regulating kinase (ASK1) signaling cascade. J Neurosci. 2011;31:247–261. doi: 10.1523/JNEUROSCI.4589-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Rashidian J, Rousseaux MW, Venderova K, Qu D, Callaghan SM, Phillips M, Bland RJ, During MJ, Mao Z, Slack RS, Park DS. Essential role of cytoplasmic cdk5 and Prx2 in multiple ischemic injury models, in vivo. J Neurosci. 2009;29:12497–12505. doi: 10.1523/JNEUROSCI.3892-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Del Rio Hortega P, Penfield W. Cerebral Cicatrix: the reaction of neuroglia and microglia to brain wounds. Bulletin of the Johns Hopkins Hospital. 1892;41:278–303. [Google Scholar]