

Abstract
A middle aged female patient presented with generalised palpable purpura associated with intense pruritus along with subconjunctival haemorrhage and orbital inflammation. There was extensive dermographism. Other systemic examinations were within normal limits. Haematological profile was normal except raised D-dimer. Skin biopsy revealed the presence of leucocytoclastic vasculitis. Antinuclear antibody was positive in a titre of 1 : 160, but antidouble-stranded DNA was negative. Urine examination revealed haematuria and proteinuria. Complement C3, C4 and C1q levels were decreased with the presence of anti-C1q antibody. There was a diagnostic dilemma between systemic lupus erythematosus and hypocomplementaemic urticarial vasculitis syndrome. However, as the patient did not fulfil the American College of Rheumatology criteria for systemic lupus erythematosus, but fulfilled all the criteria for hypocomplementaemic urticarial vasculitis syndrome, the case was finally diagnosed as hypocomplementaemic urticarial vasculitis syndrome and treated accordingly with favourable outcome.
Background
Chronic urticaria and urticarial vasculitis often points the diagnosis of a systemic disease. However, hypocomplementaemic urticarial vasculitis syndrome (HUVS) is by itself an independent autoimmune disease that may present with only cutaneous manifestations or may present with florid systemic features. HUVS often mimics other autoimmune diseases particularly systemic lupus erythematosus (SLE). Prompt diagnosis of this disease entity helps in its better management and reduces unnecessary investigations in the patients suffering from this disease.
Case presentation
A 36-year-old female patient was admitted under our care with purpuric spots distributed all over the body and redness of eyes. There was an associated history of low grade fever and polyarthralgia involving the large joints in the preceding 4 months. However, on admission, there was no fever, retro-orbital pain, symptomatic anaemia, bruise on minor trauma, abnormal bleeding from any natural orifices or nodular swelling anywhere in the body. The patient also gave a history of severe generalised pruritus and the appearance of purpuric lesions at the site of scratching for last 6 months. These purpuric lesions disappeared on their own within 48 h. On examination, there was a presence of subconjunctival haemorrhage in both eyes. Vision in right eye was 6/18 and left eye was 6/12. On ophthalmoscopic examination, vitreous haemorrhage was found in both the eyes and fundus could not be visualised clearly. There was presence of palpable purpuric spots over lower extremities, back and front of chest, superior extremities and face. There was presence of purpuric spots on hard palate as well. There was no sternal tenderness, no lymphadenopathy and no hepatosplenomegaly. Dermographism (figure 1) was present. All other systemic examinations were within normal limits.
Figure 1.
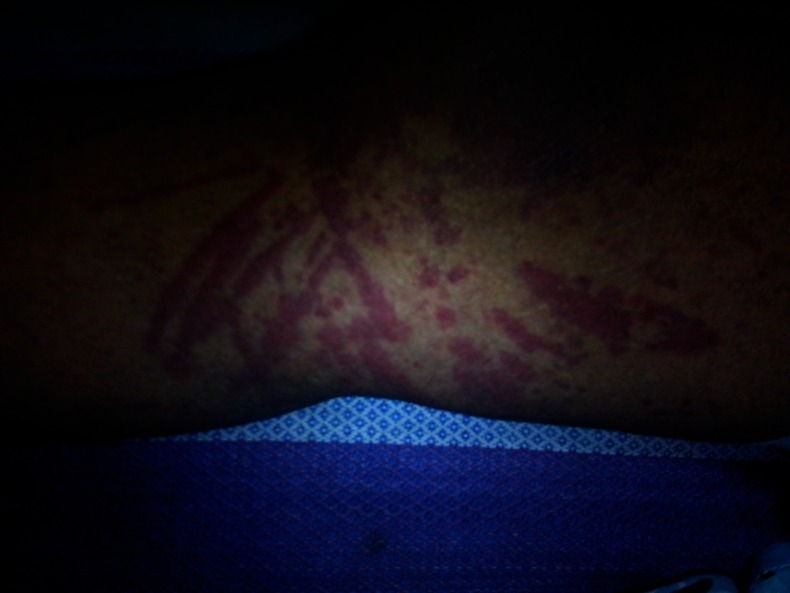
Purpuric skin lesions of the patient showing dermographism.
Investigations
Investigations revealed a haemoglobin level of 8 g/dl, normal total and differential count, platelet count of 150 000/ml and an erythrocyte sedimentation rate of 25 mm in first hour. Packed cell volume and RBC indices were within normal limits. Liver function test was normal except for a reduced serum albumin of 2.6 g/dl. Serum globulin was 2.3 g/dl. Random blood sugar, serum urea and creatine were within normal limits. Bleeding time, clotting time, activated partial thromboplastin time (APTT), prothrombin time (PT) and fibrinogen were within normal limits. However, D-dimer level was 8026 ng/ml D-dimer units which is much above the normal reference range (Normal reference range is <250 ng/ml D-Dimer units). Dengue serology, HIV serology, hepatitis B virus surface antigen and anti-hepatitis C virus antibody were negative. A skin biopsy was done from the palpable purpura present over the lower extremities and was sent for histopathological examination. Meanwhile, the purpuric spots disappeared over a period of 2–3 days with residual hyperpigmentation. Subconjunctival haemorrhage also got slowly absorbed. The skin biopsy sent earlier revealed the presence of leucocytoclastic vasculitis (figure 2) with occasional eosinophil within the vessel wall. A number of tests were performed for the evaluation of leucocytoclastic vasculitis. Antinuclear antibody test was positive in a titre of 1 : 160. Anti-SSA and anti-SSB was positive in high titre; however, antidouble-stranded DNA (dsDNA) was negative. An antineutrophil cytoplasmic antibody (ANCA) was negative. A Schirmer test was performed which was 12 mm in right eye and 15 mm in left eye. Ultrasonogram (USG) of the whole abdomen revealed the presence of minimal bilateral pleural effusion and mild ascites. Chest radiograph was suggestive of minimal bilateral pleural effusion. Routine and microscopic examination of urine revealed the presence of trace albumin with plenty of RBC per high power field. Twenty-four hour urinary protein excretion was 370 mg. Complement C3, C4 and C1q levels were decreased. Anti C1qantibody was also present in the serum. Our patient did not fulfil the American College of Rheumatology (ACR) criteria for SLE, but fulfilled all the criteria for HUVS. So, the case was finally diagnosed as HUVS.
Figure 2.
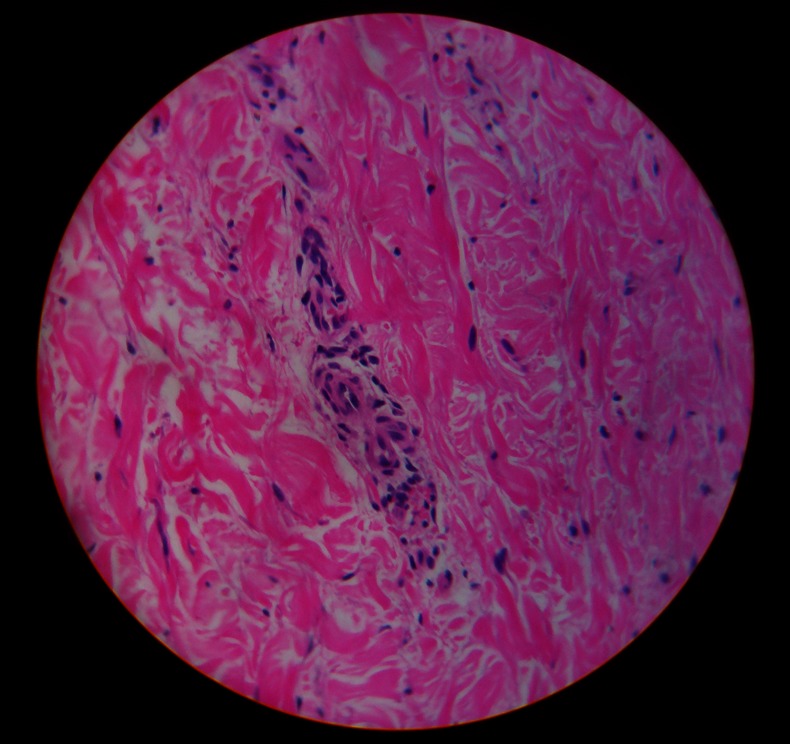
Skin biopsy showing leucocytoclastic vasculitis.
Differential diagnosis
Systemic lupus erythematosus.
Treatment
The patient was put on oral steroids at a dose of 1 mg/kg/day and discharged in stable condition.
Outcome and follow-up
However, the patient returned a week later with a sudden onset swelling of right eye, recurrence of purpuric spots on skin and swelling over left side of neck. There was associated intermittent abdominal pain. On examination, there was presence of oedematous eye lids of right eye (figure 3) and presence of significantly enlarged anterior cervical group of lymph nodes. An ophthalmological opinion was sought who were of the opinion of a non-specific orbital inflammatory syndrome. A fine needle aspiration cytology of the cervical lymph node was performed which came out to be reactive hyperplasia. An USG of the orbit was done which showed the presence of subretinal fluid collection. The patient was put on pulse methyl prednisolone at a dose of 1 gm by intravenous route daily for 5 days and was continued on oral steroids thereafter. The purpuric spots disappeared within 2–3 days and the orbital swelling subsided (figure 4) within 3–4 days. The patient stayed in the hospital for another 1 week, during which there were no further exacerbations of the disease. She was discharged in stable condition. She is presently on regular follow-up and is asymptomatic after 1 year.
Figure 3.
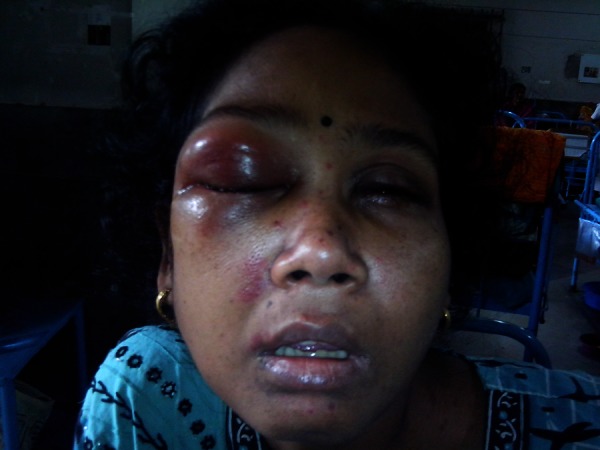
Non-specific orbital inflammatory syndrome presenting as swelling of right eye.
Figure 4.
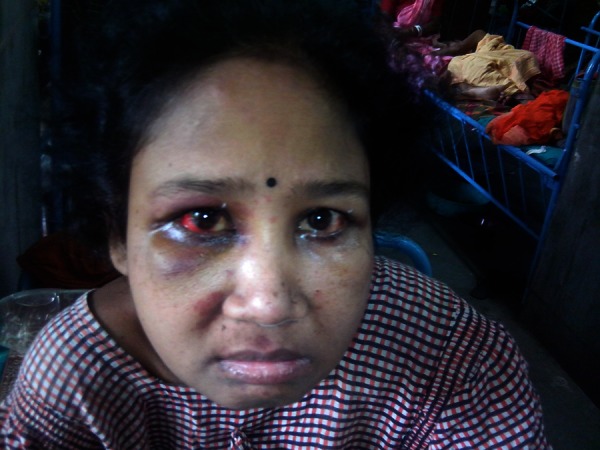
Swelling of right eye subsiding after corticosteroid treatment.
Discussion
Urticaria is a cutaneous vascular reaction characterised by locally increased vascular permeability. Clinically, urticaria manifests as erythema and wheals often accompanied by itching sensation. Urticarial vasculitis (UV) is a discrete disease entity, distinct from urticaria, in which the urticarial lesions persist for more than 24 h and heal leaving brownish residues.1 UV is characterised histopathologically by leucocytoclastic vasculitis. In leucocytoclastic vasculitis, there is a leakage of erythrocytes from blood vessels. So, hyperpigmentation remains after the resolution of the urticarial lesion. In contrast, the exanthema seen in acute or chronic urticaria resolves completely within 24 h.2 HUVS is a specific autoimmune disease with involvement of the skin, joints, kidneys and gastrointestinal tract manifested by vasculitis, arthritis, polyserositis and glomerulonephritis. Sometimes, other organ systems are also involved. HUVS affects women more frequently than men (ratio 2 : 1). The peak incidence is seen in the fifth decade of life, but HUVS has also been observed in children.3 The pathophysiology of HUVS has not been fully clarified, the following mechanism has been proposed. IgG antibodies to the collagen-similar regions of C1q are formed that initiate the cascade. These C1q antibodies and immune complexes are also detected in the plasma of patients with HUVS.4 These immune complexes activate the complement cascade that results in the formation of a variety of vasoactive substances, thereby resulting in an increased vascular permeability with urticaria and sometimes even angioedema.5 The criteria for diagnosis of HUVS were established by Schwartz in 1982.6 These include two major criteria—chronic urticarial exanthema for more than 6 months and hypocomplementaemia, and six minor criteria—leucocytoclastic vasculitis, arthralgia or arthritis, orbital inflammation, glomerulonephritis, abdominal pain, positive C1q antibody. Both the major criteria and two out of six minor criteria are required for the diagnosis of HUVS. Our patient had a history suggestive of chronic urticaria for more than 6 months and hypocomplementaemia in serum. She also had leucocytoclastic vasculitis on skin biopsy, arthralgia, non-specific orbital inflammation, abdominal pain, haematuria and proteinuria and positive C1q antibody. So, she fulfilled all the major and minor criteria for the diagnosis of HUVS. The most common differential diagnosis of HUVS is SLE. Differentiation from SLE is sometimes difficult because 50% of patients with HUVS show positive antinuclear antibody and often also fulfil the ACR criteria of SLE. The patient whose case is presented above, however, fulfilled only two of the 11 SLE criteria. Inversely, in 50% of patients with confirmed UV, the cause is SLE.7 The C1q antibodies are also not specific for HUVS and may be found in one-third of patients with SLE.4 In addition to the ACR critera, HUVS and SLE can often be differentiated on the basis of dsDNA and the typical clinical presentations. High titres of dsDNA or the demonstration of SLE-typical extractable nuclear antigen speaks against HUVS. So, our final diagnosis was hypocomplementemic urticarial vasculitis syndrome. The patient responded well with corticosteroids and is on regular follow-up for last 1 year. We the authors want to stress here that HUVS is not merely a cutaneous manifestation of an autoimmune disease, rather it is an autoimmune disease itself with multisystemic involvement and leucocytoclastic vasculitis.
Learning points.
Hypocomplementemic urticarial vasculitis syndrome (HUVS) is an autoimmune disease itself with leucocytoclastic vasculitis and multisystemic involvement.
HUVS may present with distinct urticarial exanthema without any systemic manifestations or may present with life-threatening systemic vasculitis in the form of ocular, rheumatological, renal, gastrointestinal, neurological and cardiopulmonary involvement.
Diagnosis of HUVS requires the presence of chronic arterial exanthema, leucocytoclastic vasculitis on dermal biopsy samples and hypocomplementaemia in blood.
The closest differential diagnosis is systemic lupus erythematosus (SLE). In fact, many patients of HUVS may fulfil the ACR criteria for SLE.
Early diagnosis and recognition of HUVS as a discrete clinical entity prevents superfluous diagnostic investigations in patients suffering from it.
Footnotes
Contributors: KR and SS were the treating physicians. BK coordinated all necessary investigation and follow-up. AT and BK did literature search and manuscript write up.
Competing interests: None.
Patient consent: Obtained.
Provenance and peer review: Not commissioned; externally peer reviewed.
References
- 1.Maurer M, Grabbe J. Urticaria: its history-based diagnosis and etiologically oriented treatment. Dtsch Arztebl Int 2008;2013:458–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Guha B, Youngberg G, Krishnaswamy G. Urticaria and urticarial vasculitis. Comp Ther 2003;2013:146–56 [DOI] [PubMed] [Google Scholar]
- 3.Mehregan DR, Hall MJ, Gibson LE. Urticarial vasculitis: a histopathologic and clinical review of 72 cases. J Am Acad Dematol 1992;2013:441–8 [DOI] [PubMed] [Google Scholar]
- 4.Wisnieski JJ. Urticarial vasculitis. Curr Opin Rheumatol 2000;2013:24–31 [DOI] [PubMed] [Google Scholar]
- 5.Wisnieski JJ, Jones SM. IgG autoantibody to collagen like region of C1q in hypocomplementemic urticarial vasculitic syndrome, systemic lupus erythematosus and 6 other musculoskeletal or rheumatic diseases. J Rheumatol 1992;2013:884–8 [PubMed] [Google Scholar]
- 6.Schwartz HR, Mc Duffie FC, Black LF, et al. Hypocomplementemic urticarial vasculitis: association with chronic obstructive pulmonary disease. Mayo Clinic Proc 1982;2013:231–8 [PubMed] [Google Scholar]
- 7.Davies MD, Daoud MS, Kirby B, et al. Clinicopathologic corelation of hypocomplementemic and normocomplementemic. Urticarial vasculitis. J Am Acad Dematol 1998;2013:899–905 [PubMed] [Google Scholar]