

Abstract
Megacystis microcolon intestinal hypoperistalsis syndrome (MMIHS) is a multisystemic disorder in which impaired intestinal motor activity causes recurrent symptoms of intestinal obstruction in the absence of mechanical occlusion, associated with bladder distention without distal obstruction of the urinary tract. MMIHS and prune belly syndrome may overlap in most of the clinical features and discrimination of these two entities is important because the prognosis, management and consulting with parents are completely different. MMIHS outcome is very poor and in this article we present two neonates with MMIHS that both died in a few days.
Background
Megacystis microcolon intestinal hypoperistalsis syndrome (MMIHS) is a rare congenital anomaly with decreased muscular tone in the urinary tract and intestine that was first described in 1976.1 Hypoperistalsis causes a pseudo-obstruction which antenatally leads to a shortened and malrotated microcolon. The bladder is distended, hypotonic and non-obstructed with variable upper urinary tract dilation.2 Patients with MMIHS present with a flaccid abdomen, massive abdominal distention and inability to spontaneous voiding in neonatal period.2 227 cases of MIHHS were reported in literature up to 2011 with a female predominance of 2–4:1.3
Evidence for autosomal recessive inheritance comes from several reports of MMIHS in sibling pairs although myogenic, neurogenic and hormonal causes are also discussed in pathogenesis of MMIHS.4
Therapeutic interventions include surgery, prokinetic agents, gastrointestinal (GI) hormones are of limited value in treatment of MMIHS.5–7 Pseudo-obstruction of both GI and urinary systems are common and final prognosis is always poor.8 Most MMIHS cases die because of severe sepsis or end-stage renal failure. Parentral nutrition is inevitable because of feeding intolerance. Prolonged total parenteral nutrition (TPN) also causes other morbidities such as infection complications and hepatotoxicity.2 3
Pathogenesis of MMIHS remains unclear but impaired peristalsis seems to be owing to abnormal ganglion cells pattern and absence of interstitial Cajal cells with marked reduction of contractile and cytoskeleton proteins in smooth muscle cells.4
MMIHS and prune belly syndrome (PBS) may overlap in most of the clinical features. Considering the poor prognosis of MMIHS compared to PBS, it would be very important to differentiate these two entities.9
We report two cases of MMIHS that referred to our paediatric surgery department with prune belly like syndrome and discuss their clinical course and outcome.
Case presentation
Case A
A 2-day-old girl with a history of abdominal distension and delayed meconium passage was referred to our centre with prune belly like syndrome. She was a term baby with APGAR 8 at the first minute and APGAR 9 at the fifth minute. Birth weight was 3850 g and parents were consanguinous. Head circumference was over 90th percentile and height was between 75th and 90th centile.
Prenatal ultrasound studies by gynaecologist revealed cephalic position and normal placenta and antenatal diagnosis of MMHIS was not obtained.
Physical examination revealed abdominal distention, but no mass was present. After insertion of a nasogastric tube and the Foley catheter, stomach and bladder were decompressed (400ml residual urine) and a flaccid abdomen was observed (figure 1A).
Figure 1.
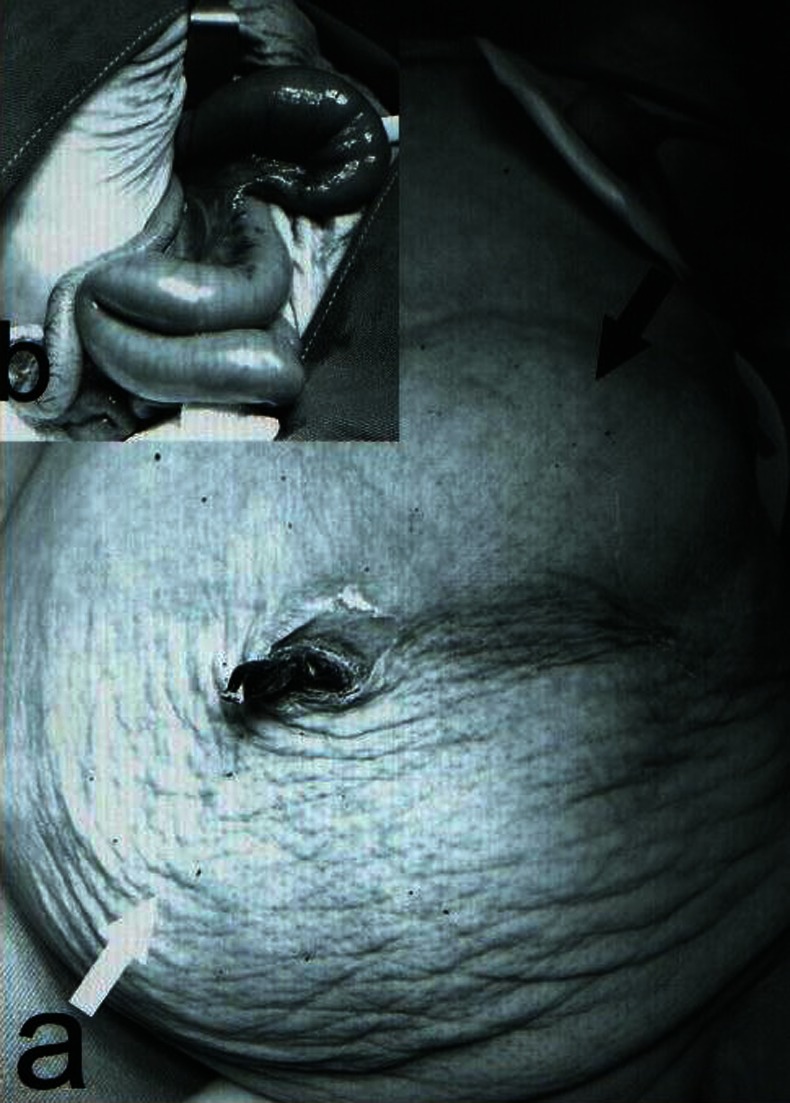
(A) Photography of the baby after insertion of the nasogastric tube and the Foley catheter. Stomach is seen in upper abdomen (black arrow) and abdominal wall became soft and prune shape (white arrow). (B) Intraoperative image: small intestine was showed that became dilated and short in diameter.
A mild leucocytosis was detected with normal haematocrit and platelet count. Plasma biochemistry studies were in normal range. A dilated stomach was seen in thoraco-abdominal radiography (figure 2A).
Figure 2.
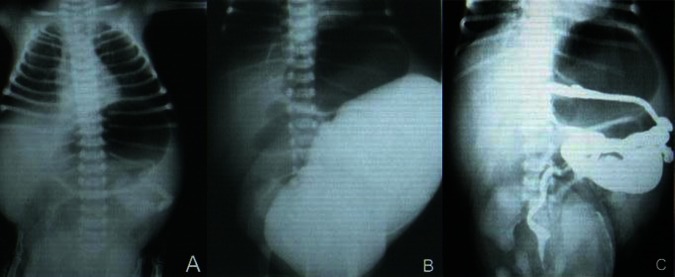
(A) Plain chest and abdominal x-ray showed a dilated stomach. (B) Voiding cystourethrogram (VCUG) shows very dilated bladder without vesico-ureteral reflux which occupied most of abdomen. (C) BE revealed malrotated microcolon.
Ultrasound studies showed bilateral dilation in upper urinary tracts, distended bladder which occupied most of the abdominal cavity with an irregular wall but normal thickness. Contrast study of upper GI tract showed a dilated oesophagus, stomach and small bowel. A dilated bladder without any evidence of vesicoureteral reflux was observed in voiding cystoureterogram (figure 2B). Barium enema revealed malrotated microcolon (figure 2C).
Conservative management and TPN were applied. Cisapride was also tried with cardiac monitoring for prolonged QTbut no therapeutic response was observed. She had persistent food intolerance without any mechanical obstruction. Finally, surgical intervention was decided. Intraoperative findings revealed a very short small bowel (total length was 75 cm) with dilation in proximal part (figure 1B) ileostomy and vesicostomy were performed and transverse colon biopsy was taken. Bowel transit did not observe despite the presence of G cells in bowel biopsies. The patient died after almost a month due to sepsis and respiratory failure.
Case B
A 3-day-old female neonate with the history of abdominal distension and delay in meconium passage and urination was referred to our centre with fare clinical condition due to sepsis. Prenatal ultrasound study was not performed and the baby was born by normal vaginal delivery in a rural place. First day weight was 2750 g and the initial symptom was feeding intolerance.
Initial physical examinations demonstrated a distended abdomen and after Foley catheter insertion, 50 ml of urine drained and the abdomen became flaccid but the patient was febrile and her clinical condition was poor. The patient was admitted in neonatal intensive care unit and sepsis workup and primary laboratory and imaging evaluations were requested.
Ultrasound study demonstrated bilateral hydrouretronephrosis with a very dilated bladder.
Contrast enema showed a microcolon and serum biochemistry studies revealed anaemia and hyperkalaemia and hyperbilirubinaemia and increased urea and creatinin.
Nephrologist consultation was requested according to hyperkalaemia and increased blood urine nitrogen. Clinical condition deteriorated gradually. The nasogasteric tube was inserted and bilious gastric contents were detected. TPN was started. Because of persistent hyperkalaemia, acute peritoneal dialysis was indicated and intraperitoneal catheter was inserted by a nephrology fellow. Although the catheter was inserted above the umbilicus, dialysis fluid was drained through the Foley catheter which demonstrated the insertion of dialysis catheter into the dilated urinary system. Dialysis was stopped and the catheter withdrawn but foley catheter maintained. A permanent peritoneal dialysis catheter was inserted then by open fashion in the operative room.
Finally the patient died due to renal failure and severe electrolytes imbalance, fever and clinical signs of sepsis. Autopsy demonstrated the diagnosis of MMIHS.
Outcome and follow-up
Both cases died in hospital after fruitless palliative and therapeutic interventions.
Discussion
MMIHS is a rare autosomal recessive condition which is almost lethal in the first year of life. Although pro-kinetic agents and alimentation have prolonged life in some cases, but the long-term outcome remains poor.9
The aetiology of MMIHS is unknown. Several reports have mentioned defective autonomic innervations but variety of genetic, myogenic and hormonal factors are also discussed.2 3
It is suggested that the initial event in the pathogenesis is an intramural inflammatory process that affects the GI and urinary tract, leading to extensive fibrosis that destroys the intestinal neural network and produces hypoperistalsis.10
MMIHS usually affects women and clinical hallmarks of MMIHS include a flaccid and distended abdomen with hypoplastic abdominal wall that resembles PBS.11
Clinical features in PBS and MMIHS may overlap and a distended abdomen and bladder and abdominal wall laxity are frequently seen in both entities so many patients may be labelled as PBS on the first visit. This may lead to underdiagnosis of MMIHS and missing the other gastro-intestinal abnormalities that are expected in MMIHS. Dilated small bowel loops may be reported as a dilated ureter in ultrasound.1 Bowel dysfunction and microcolon in MMIHS cause obstipation and food intolerance.
MMIHS cases mostly die because of renal failure, sepsis or other comorbidities due to prolonged TPN. Despite recent improvements in parenteral nutrition and neonatal critical care, death rate still remains as high as 80% so it is critical to differentiate these two entities for predicting the prognosis, therapeutic plan and consulting with parents.
It seems that gender predominance and GI symptoms such as bilious vomiting are the main diagnostic keys.
The other clinical point in these patients is considering the dilated urinary tract that may not collapse even with catheter drainage. As many of these cases may be complicated with renal failure during their admission course, we suggest to insert the peritoneal dialysis catheter just in open method and under direct vision because iatrogenic urinary and GI tract trauma may be encountered.
Learning points.
Megacystis microcolon intestinal hypoperistalsis syndrome (MMIHS) may be difficult to differentiate from prune belly syndrome according to overlapping presentations.
Any female infant with pseudo-PBS should be considered as MMIHS, unless GI involvement is ruled out.
Peritoneal catheter insertion should be carried out under direct vision and in the upper abdomen due to dilated urinary system.
Footnotes
Competing interests: None.
Patient consent: Obtained.
Provenance and peer review: Not commissioned; externally peer reviewed.
References
- 1.Berdon WE, Baker DH, Blanc WA, et al. Megacystis-microcolon-intestinal hypoperistalsis syndrome: a new cause of intestinal obstruction in the newborn. Report of radiologic findings in five newborn girls. Am J Roentgenol 1976;2013:957–64 [DOI] [PubMed] [Google Scholar]
- 2.Puri P, Shinkai M. Megacystis microcolon intestinal hypoperistalsis syndrome. Semin Pediatr Surg 2005;2013:58–63 [DOI] [PubMed] [Google Scholar]
- 3.Gosemann JH, Puri P. Megacystis microcolon intestinal hypoperistalsis syndrome: systematic review of outcome. Pediatr Surg Int 2011;2013:1041–6 [DOI] [PubMed] [Google Scholar]
- 4.Piotrowska AP, Rolle U, Chertin B, et al. Alterations in smooth muscle contractile and cytoskeleton proteins and interstitial cells of Cajal in megacystis microcolon intestinal hypoperistalsis syndrome. J Pediatr Surg 2003;2013:749–55 [DOI] [PubMed] [Google Scholar]
- 5.Raofi V, Beatty E, Testa G, et al. Combined living-related segmental liver and bowel transplantation for megacystis-microcolon-intestinal hypoperistalsis syndrome. J Pediatr Surg 2008;2013:e9–11 [DOI] [PubMed] [Google Scholar]
- 6.Lorenzo AJ, Twickler DM, Baker LA. Megacystis microcolon intestinal hypoperistalsis syndrome with bilateral duplicated systems. Urology 2003;2013:144. [DOI] [PubMed] [Google Scholar]
- 7.Masetti M, Rodriguez MM, Thompson JF, et al. Multivisceral transplantation for megacystis microcolon intestinal hypoperistalsis syndrome. Transplantation 1999;2013:228–32 [DOI] [PubMed] [Google Scholar]
- 8.Loinaz C, Rodriguez MM, Kato T, et al. Intestinal and multivisceral transplantation in children with severe gastrointestinal dysmotility. J Pediatr Surg 2005;2013:1598–604 [DOI] [PubMed] [Google Scholar]
- 9.Giuliani S, Vendryes CH, Malhotra A, et al. Prune belly syndrome associated with cloacal anomaly, patent urachal remnant, and omphalocele in a female infant. J Ped Surg 2010;2013:P39–42 [DOI] [PubMed] [Google Scholar]
- 10.Rolle U, O'Briain S, Pearl RH. Megacystis-microcolon-intestinal hypoperistalsis syndrome: evidence of intestinal myopatty. Pediatr surg Int 2002;2013:2–5 [DOI] [PubMed] [Google Scholar]
- 11.Terry L, Lamia S, Netta MB, et al. Megacystis-microcolon-intestinal hypoperistalsis and prune belly: overlapping syndromes. Pediatr Radiol 2004;2013:995–8 [DOI] [PubMed] [Google Scholar]