

Abstract
A middle-aged female patient, previously diagnosed with asthma, presented with a large spontaneous left pneumothorax. She had a history of nephrectomy for a ruptured renal angiomyolipoma (AML) with a postoperative spontaneous pneumothorax when she was an adolescent. High-resolution CT chest revealed multiple scattered thin-walled lung parenchyma cysts consistent with lymphangioleiomyomatosis (LAM). Hepatic AMLs and adenoma sebaceum skin lesions were also noted, consistent with an overall diagnosis of tuberous sclerosis. Her acute management included lung re-expansion via chest tube insertion, antibiotics for concurrent chest infection, nebulisation and chest physiotherapy. Since discharge, the patient had only occasional shortness of breath, relieved by bronchodilators. She is considering expanded immunisation as well as enrolment in a clinical trial. Her hepatic AMLs will be monitored via ultrasound for growth. LAM treatment is generally aimed at its complications with lung transplantation reserved for severe disease; however, hormonal therapy and the mTOR inhibitor aim at targeting systemic disease.
Background
This is a rare case of tuberous sclerosis complex associated lymphangioleiomyomatosis (TSC-LAM), a cystic interstitial lung disease. Tuberous sclerosis is a rare autosomal dominant disorder with a minimum prevalence of 1 case in 15 000–30 000 persons.1 Although 30–40% of adult women with TSC are found to have TSC-LAM,1 the pulmonary disease is usually less severe than in women with sporadic LAM (S-LAM).2 3 This patient, however, suffered with persistent pulmonary symptoms including pneumothoraces, which was thought to be secondary to asthma. Her presentation, however, began decades earlier with a ruptured renal angiomyolipoma (AML). Taking this history into consideration, the differential diagnoses of LAM and TSC should have been considered, given the association between AMLs and LAM.
Case presentation
A woman in her early 40s presented with worsening shortness of breath along with wheezing and a productive cough for 1 week. Significant medical history included a left nephrectomy as an adolescent for a ruptured AML, with a spontaneous right-sided pneumothorax occurring postoperatively. She was a non-smoker and had no history of seizures. On examination air entry was significantly decreased on the left side. Multiple small red-brown papules were noted over the central part of the face consistent with adenoma sebaceum. General examination was otherwise unremarkable. In particular, she was of normal intelligence.
Investigations
Chest x-ray on admission revealed a large left pneumothorax (figure 1). The right lung appeared normal in volume with no radiographic evidence of cysts. Biochemical and haematological investigations were normal apart from an elevated white cell count of 14.3×103/µl. Connective tissue screen was negative. High-resolution CT (HRCT) of the chest post re-expansion of the left lung via chest tube revealed multiple well-defined thin-walled cysts scattered throughout the parenchyma (figure 2). No pleural effusion was present. Abdominal imaging revealed postsurgical absence of left kidney and a normal right kidney; however, multiple AMLs were seen in the liver (figure 3). CT and MRI of the brain as well as echocardiogram were normal.
Figure 1.
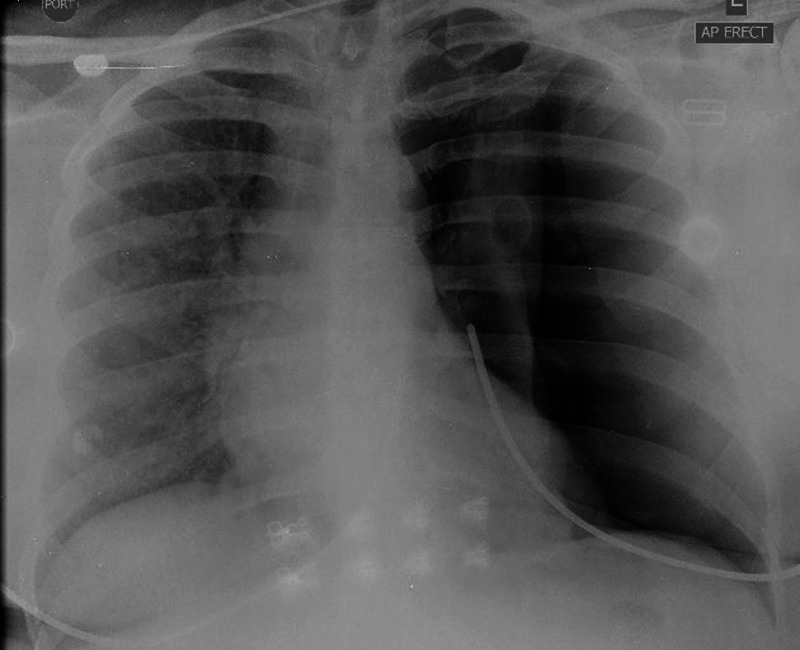
Chest radiograph showing large left spontaneous pneumothorax with associated mediastinal shift. A chest tube was promptly inserted.
Figure 2.
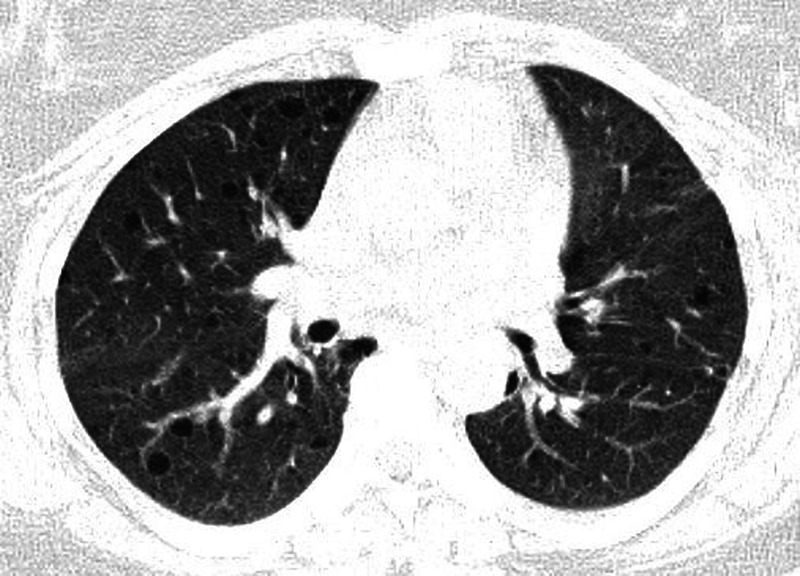
High-resolution CT scan of the chest revealed multiple well-defined, thin-walled cysts scattered throughout both lungs, consistent with lymphangioleiomyomatosis. The left lung is re-expanded postchest tube drainage.
Figure 3.
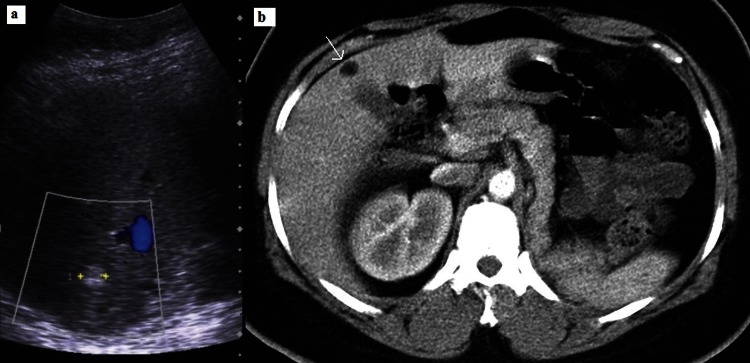
(A) Ultrasound of the liver showed well-circumscribed hyperechoic lesions (callipers). (B) which were of fat density (≤10 Hounsfield units; arrow) on CT, consistent with angiomyolipomas. Surgical absence of left kidney noted (owing to previous ruptured angiomyolipoma). Normal right kidney.
Differential diagnosis
LAM diagnosis is typically delayed by 3–5 years as more common dyspnoea causes such as asthma or chronic obstructive pulmonary disease are often diagnosed initially.4 The two most common presenting symptoms of LAM are dyspnoea on exertion and spontaneous pneumothorax.5 Pneumothoraces have been reported in 40–80% of patients with LAM and are often recurrent,6 more commonly occurring in S-LAM than in TSC-LAM.3 Less common manifestations which develop with disease progression include haemoptysis, non-productive cough, chylous pleural effusions and chylous ascites.5 Abnormalities related to the lymphatics, such as chylous effusions and ascites, dilation of the thoracic duct and lymphatic masses or lymphangioleiomyomas, are more common in S-LAM than in TSC-LAM.2
The finding of lung cysts on imaging also leads to a radiological differential diagnosis, including centrilobular emphysema and langerhans cell histiocytosis (LCH).3 Emphysematous spaces lack a perceptible wall with a central location of the centrilobular artery7; the ‘central dot sign’. LAM and LCH; however, show the vascular structures along the periphery of the cyst walls.8 LCH cysts are thicker walled, sometimes irregular in shape, predominantly in mid and upper zones with costophrenic angle sparing.3 7 Cysts in LAM typically exhibit thin regular walls and are distributed throughout the lungs with no zone predilection.7 9 Additionally, LCH typically occurs in tobacco smokers unlike LAM with no smoking association.7 8
S-LAM and TSC-LAM can be identical radiologically and pathologically.10 Characteristic microscopic features are cysts and proliferation of smooth muscle cells or LAM cells.10 LAM cells infiltrate distal airways leading to airway narrowing, air trapping, bulla formation and pneumothoraces.10 The presence of LAM cells can be confirmed with HMB-45 staining in available settings.10 The only distinguishing histological feature between these two entities is multifocal micronodular pneumocyte hyperplasia, present only in TSC-LAM, which may appear as lung nodules on imaging.10
Renal AMLs are present in up to 80% of patients with TSC-LAM and up to 54% of patients with S-LAM.2 Renal AMLs in TSC-LAM tend to be multiple and bilateral whereas unilateral and solitary AMLs are more common with S-LAM.2 3 Frequency of haemorrhage from renal AML is also higher in TSC-LAM.2 Hepatic AMLs are also more common in TSC-LAM, occurring in 13–45% of these patients.2 Hence the history of intra-abdominal AMLs with chest symptoms should prompt clinicians to consider LAM. Other manifestations of tuberous sclerosis should also be sought whenever LAM is suspected.
The classic clinical triad for tuberous sclerosis consists of seizures, intellectual disability and cutaneous angiofibromas; however, this occurs in less than 30% of affected patients.1 Interestingly, patients with TSC with pulmonary involvement tend to have a lower incidence of epilepsy and mental retardation.6 Common imaging central nervous system abnormalities in TSC include cortical tubers, subependymal nodules, subependymal giant cell astrocytomas and white matter lesions,11 however 5–10% of patients may not show any of these changes on MRI.12 Hence the absence of this clinical triad or normal brain imaging should not dissuade physicians from considering tuberous sclerosis.
Treatment
The acute management of the patient included prompt chest tube insertion for lung re-expansion as well as antibiotics for concurrent chest infection. She was also nebulised with salbutamol and ipratropium and administered corticosteroids and oxygen therapy. She underwent chest physiotherapy.
Outcome and follow-up
The patient's symptoms fully resolved following lung re-expansion, antibiotics and chest physiotherapy. She was discharged and only has occasional shortness of breath, relieved by inhaled bronchodilators. She was referred to another hospital for future management in a thoracic specialist clinic. She will undergo interval monitoring of her hepatic AMLs via ultrasound for growth. She is considering expanded immunisation and enrolment in a clinical trial.
Discussion
TSC is autosomal dominant while S-LAM is sporadic.4 S-LAM and TSC-LAM, however, share a genetic relationship, the tumour suppressor gene TSC2.4 Whereas TSC is caused by a germline mutation of TSC1 or TSC2 on chromosomes 9q34 and 16p13, respectively, mutations and loss of heterozygosity in TSC2 have been detected in LAM cells.4 Interestingly, although TSC affects both sexes equally, TSC-LAM, like S-LAM, occurs almost exclusively in women of childbearing age.6
When a diagnosis of TSC is made, a baseline abdominal ultrasound should be performed and repeated every 3–5 years if normal.13 Renal AMLs can be monitored every 1–2 years via ultrasound when asymptomatic and less than 4 cm.14 When greater than 4 cm or symptomatic, risk of spontaneous life-threatening haemorrhage increases hence arterial embolisation or nephron sparing surgery can be performed, to conserve renal tissue.14 No clear guidelines are yet available, however, for monitoring of hepatic AMLs. HRCT of the chest should be performed in women at age 18 and again at 30–40 years, or more frequently in patients with respiratory symptoms.1 When HRCT appearance is characteristic of LAM and other typical manifestations such as AMLs are present, tissue biopsy is not necessary for definitive diagnosis.14
Bronchodilators, supplemental oxygen, pulmonary rehabilitation and smoking cessation can be useful for airway disease and hypoxaemia.3 6 Prophylactic pneumococcal and influenza vaccines are also important.14 An initial pneumothorax or effusion is treated the same as with other lung diseases14; however, thoracic surgery referral is indicated for recurrent pneumothorax or persistent symptomatic effusions after aspiration (small chylous effusions often require no intervention).3 14 In these patients pleurodesis may prevent or decrease recurrence but can make future lung transplantation more difficult.10 Lung transplantation is usually reserved for progressive disease and severe disability, in patients who are oxygen-dependent or where FEV1 is less than 30% predicted.14
As LAM is seen almost exclusively in premenopausal women and exacerbated by oestrogen exposure, hormonal therapy has been used in its treatment including oophorectomy, tamoxifen, GnRH agonists and progesterone therapy.14 There has not been definitive evidence, however, showing their effectiveness at this point.14 Currently, progesterone therapy is the most common hormone therapy in use, where some studies show slightly better lung function in these patients.14 Cessation of the oral contraceptive pill is advised and pregnancy risk including LAM exacerbation and pneumothorax must be explained to patients.14
A recent and promising systemic therapy is a mTOR (mammalian target of rapamycin) inhibitor called sirolimus.4 15 TSC genes are believed to be regulated by the mTOR protein, hence its inhibition prevents proliferation of LAM cells.3 4 15 Clinical trials have shown reduction in AML size and slowing of lung function decline in patients with TSC and LAM.4 Renal and pulmonary improvements, however, tend to reverse after withdrawal of drug, hence long-term treatment would likely be necessary.4 15 Sirolimus has also been reported to cause regression of astrocytomas in TSC.16
In conclusion LAM is a rare lung disease that is often delayed in its diagnosis because of its many clinical and imaging mimickers. When encountered, associated systemic manifestations and signs of TSC should be sought. The clinical course of TSC is variable with a multidisciplinary approach required for management; depending on the patient's manifestations.
Learning points.
Consider lymphangioleiomyomatosis (LAM) in female patients of childbearing age with dyspnoea, particularly with a history of recurrent pneumothoraces or angiomyolipomas (AML).
When high-resolution CT is characteristic for LAM in a woman and typical associated manifestations such as AMLs are present, lung biopsy is not necessary for definitive diagnosis.
Patients with AMLs should be monitored for interval growth, which increases the risk for spontaneous rupture and life-threatening haemorrhage.
Not all patients with tuberous sclerosis suffer with seizures or intellectual disability, and MRI brain may show no abnormality in a small percentage.
Tuberous sclerosis complex-LAM has a greater incidence of renal and hepatic AMLs and lung nodules than sporadic-LAM (S-LAM); however, S-LAM more commonly has lymphatic involvement, that is, thoracic duct dilation, chylous pleural effusions or ascites and lymphangioleiomyomas.
Footnotes
Contributors: All authors contributed to the conception, drafting, revision and final approval of the manuscript.
Competing interests: None.
Patient consent: Obtained.
Provenance and peer review: Not commissioned; externally peer reviewed.
References
- 1.Seibert D, Hong CH, Takeuchi F, et al. Recognition of tuberous sclerosis in adult women: delayed presentation with life-threatening consequences. Ann Intern Med 2011;2013:806–13,W-294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Avila NA, Dwyer AJ, Rabel A, et al. Sporadic lymphangioleiomyomatosis and tuberous sclerosis complex with lymphangioleiomyomatosis: comparison of CT features. Radiology 2007;2013:277–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.The LAM foundation http://www.thelamfoundation.org (accessed Apr 2013).
- 4.Henske EP, McCormack FX. Lymphangioleiomyomatosis—a wolf in sheep's clothing. J Clin Invest 2012;2013:3807–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Taylor JR, Ryu J, Colby TV, et al. Lymphangioleiomyomatosis. Clinical course in 32 patients. N Engl J Med 1990;2013:1254–60 [DOI] [PubMed] [Google Scholar]
- 6.Hohman DW, Noghrehkar D, Ratnayake S. Lymphangioleiomyomatosis: a review. Eur J Intern Med 2008;2013:319–24 [DOI] [PubMed] [Google Scholar]
- 7.Seaman DM, Meyer CA, Gilman MD, et al. Diffuse cystic lung disease at high-resolution CT. AJR Am J Roentgenol 2011;2013:1305–11 [DOI] [PubMed] [Google Scholar]
- 8.Cosgrove GP, Frankel SK, Brown KK. Challenges in pulmonary fibrosis. 3: cystic lung disease. Thorax 2007;2013:820–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Islam A. Lymphangioleiomyomatosis imaging. http://emedicine.medscape.com/article/358917 (accessed Apr 2013).
- 10.Abbott GF, Rosado-de-Christenson ML, Frazier AA, et al. From the archives of the AFIP: lymphangioleiomyomatosis: radiologic-pathologic correlation. Radiographics 2005;2013:803–28 [DOI] [PubMed] [Google Scholar]
- 11.Umeoka S, Koyama T, Miki Y, et al. Pictorial review of tuberous sclerosis in various organs. Radiographics 2008;2013:e32. [DOI] [PubMed] [Google Scholar]
- 12.Wu WE, Kirov II, Tal A, et al. Brain MR spectroscopic abnormalities in ‘MRI-negative’ tuberous sclerosis complex patients. Epilepsy Behav 2013;2013:319–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rouvière O, Nivet H, Grenier N, et al. Guidelines for the management of tuberous sclerosis complex renal disease. Prog Urol 2012;2013:367–79 [DOI] [PubMed] [Google Scholar]
- 14.Johnson SR. Lymphangioleiomyomatosis. Eur Respir J 2006;2013:1056–65 [DOI] [PubMed] [Google Scholar]
- 15.Bissler JJ, McCormack FX, Young LR, et al. Sirolimus for angiomyolipoma in tuberous sclerosis complex or lymphangioleiomyomatosis. N Engl J Med 2008;2013:140–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Franz DN, Leonard J, Tudor C, et al. Rapamycin causes regression of astrocytomas in tuberous sclerosis complex. Ann Neurol 2006;2013:490–8 [DOI] [PubMed] [Google Scholar]