

Abstract
Glia synthesize, package, and secrete several species of matrix proteases, including the gelatinases (pro-)MMP-2 and (pro-)MMP-9. In appropriate settings (e.g., experimental ischemia), these MMPs can be assayed from cerebral tissues or from astrocytes and microglia in culture by enzymatic substrate-dependent assays and by gelatin-based zymography. We describe the methodologies for the sensitive quantitative development of the inactive and active forms of both MMP-2 and MMP-9 from tissues and cells, by means of lysis of the collagen substrate in collagen-impregnated gel electropheresis by the zymogen and active gelatinases. These methodologies are a refinement of those used commonly, with instructions to increase sensitivity. Serious and often overlooked issues regarding sources of sample contamination and elements confounding the MMP band development and their interpretation are discussed.
Keywords: Metalloproteinases (-2 and -9), Gelatin zymography, Gelatinases, Extracellular matrix, Astrocytes
1. Introduction
This chapter describes the development of gel zymography for use in samples from astrocyte cultures for experiments with experimental ischemia (oxygen-glucose deprivation, OGD), or from cerebral tissues subject to focal ischemia.
Recent interest in the generation and fate of matrix metalloproteinases (MMPs), Zn+2 endopeptidases capable of activating specific zymogen precursors or degrading specific extracellular matrix (ECM) proteins, has made use of rather novel assay methods that can specifically identify two of these MMP species, the gelatinases. This assay, gel zymography, employs a sensitive quantifiable polyacrylamide gel-based electrophoretic approach that incorporates the ability of these two proteases to degrade one of their substrates, gelatin (1). A number of laboratories have used similar techniques to assay gelatinases in isolated cells or in central nervous system (CNS) tissues. Other MMP and matrix protease family members can be assayed by similar methods using specific substrates embedded in the gels (2–4).
Gelatinases in the CNS are associated with microvascular matrix degradation, intracerebral hemorrhage, angiogenesis and vascular remodeling, cellular trafficking and transmembrane migration, inflammatory responses to injury (e.g., leukocyte migration), and other processes.
With the exception of a few plasma membrane-bound members, the majority of MMPs are secreted as inactive proteases (5). While mainly involved in connective tissue turnover, MMPs display a variety of substrate specificities. The two gelatinases, MMP-2 and MMP-9, also known as gelatinase A and B, respectively, are secreted and are unique among MMPs for preferentially degrading type IV collagen, the major component of cerebrovascular basement membranes. Furthermore, while both MMP-2 and MMP-9 can proteolyze gelatin, collagen IV, laminin, and elastin in similar fashion, MMP-2 more efficiently degrades fibronectin than does MMP-9 (6–8). Other discrete differences in substrate specificity and degradation profile between the two gelatinases are now evident.
To assure temporal and spatial control of their activity in vivo, MMPs are synthesized as inactive proenzymes (pro-MMPs) or zymogens. pro-MMP-2 and pro-MMP-9 are composed of a signal peptide found in the N-terminal portion of the proteins, which is necessary for their secretion, followed by the propeptide domain, a catalytic domain that includes fibronectin type-II-related repeats necessary for substrate interaction, a hinge region, and a hemopexin-like C-terminal domain. The catalytic domains are already preformed in both zymogens; however, formation of a bond between a cysteine residue within the prodomain and Zn+2 found in the catalytic domain folds the prodomain in a way that shields the active site cleft and prevents substrate access (9, 10). Disruption of this bond, also called a “cysteine-switch,” allows the entry of a water molecule that binds Zn+2 and replaces the cysteine after dissociation. The presentation of the noncatalytic Zn+2 in this way results in an intermediate active enzyme. Removal of the prodomain by autocatalytic cleavage is necessary to fully activate both enzymes (11–13). This can be accomplished by the involvement of upstream MMPs (e.g., MMP-3 for pro-MMP-9 (14), in addition to activation of pro-MMP-9 by MMP-1 and MMP-7, and MMP-14 and -16 for pro-MMP-2 (15)), and other proteases (e.g., activation of pro-MMP-9 by plasmin). Exposure of the intrinsic activity by autocatalytic cleavage is believed to be responsible for the detection of pro-MMP-2 and pro-MMP-9, in addition to the active forms, by gel zymography.
The discussion of control of MMP activities must also acknowledge the involvement of the tissue inhibitors of metalloproteinases (TIMPs)-1, -2, and -3 in the regulation of the extracellular tissue MMPs. This zymographic method is not capable of detecting these inhibitors as the sample preparation methods dissociate the noncovalent MMP-TIMP complexes. The reader is referred to the excellent review by Stetler-Stevenson for further information on TIMPs (16).
Gel zymography with collagen as the substrate can only identify the latent and active gelatinases, (pro-)MMP-2 and (pro-)MMP-9 (1). Unfortunately, the methodology does not allow assay of other MMPs or TIMPs. While relatively straightforward, the use of gel zymography to identify the appearance of gelatinases in glial cells (and other cells), in their secretions, and in CNS tissues can, with some refinement, be used for quantitation of each gelatinase. Care must be taken in the preparation of the specimens to be assayed as the result interpretation is sensitive to the methodology.
To study the synthesis of MMP-2 and MMP-9 in brain tissue under normoxia and focal ischemia, or in mouse primary glial cells following in vitro ischemia, a well-defined method of zymography has been used by our group. The zymographic approach allows sensitive, quantifiable, and functional analysis of these (pro-)MMPs based on their molecular weight. The denaturing, nonreducing conditions under which the biological samples are separated by gel electrophoresis during zymography contribute to unfolding of all proteins, including the MMPs. Hence, the identification of the latent and active forms relies upon the opening/exposure of the active site in the proforms by partial denaturation of the molecule, separation by gel electrophoresis, and subsequent renaturation of the molecule, followed by cation exchange that allows both pro-MMP and MMP forms to interact with collagen. Among the advantages of gel zymography are the ability to distinguish the presence of both latent and active gelatinases simultaneously, acceptable quantitation and relative ease of performance, and (by altering the substrate) protease activity definition (see Note 1). Because the technique makes use of the enzymatic cleavage of the gelatin substrate, this provides a much more sensitive assay than Western blots to quantify these MMPs.
2. Materials
2.1. Gel Zymography Equipment
Gel casting stand and frame.
Glass plates with 1.5 mm spacers.
Short plates.
Combs.
Mini-Protein electrophoresis system.
Power supply.
Gel spatula.
15- and 50-mL polypropylene centrifuge tubes, sterile.
Plastic gel wash containers.
2.2. Gel Zymography Reagents and Solutions
2.2.1. Resolving Gel
1.5 M Tris-Cl, pH 8.8.
20% (w/v) sodium dodecyl sulfate (SDS).
30% polyacryamide (1:29).
10% (w/v) ammonium persulfate (APS).
TEMED.
Sterile deionised water (diH2O).
1% Gelatin, from bovine skin, type B (10 mg/mL).
2.2.2. Stacking Gel
0.5 M Tris-Cl with 0.4% SDS, pH 6.8.
30% polyacrylamide (1:29).
10% (w/v) ammonium persulfate (APS).
TEMED.
Sterile diH2O.
2.2.3. 10× Electrophoresis Running Buffer
30.3 g Tris-base.
14.4 g glycine.
10 g SDS.
1 L diH2O.
2.2.4. 5× Sample Buffer
0.313 M Tris–HCl, pH 6.8.
10% SDS.
50% glycerol.
0.05% bromphenol blue.
2.2.5. Gel Washing Buffer
25 mL Triton X-100.
975 mL sterile diH2O.
2.2.6. Gel Development Buffer
50 mM Tris, pH 7.4.
10 mM CaCl2.
0.02% NaN3.
Sterile diH2O.
2.2.7. Gel Staining Solution
Amido Black Staining Solution
0.1 g Amido black.
10 mL acetic acid.
30 mL methanol.
60 mL sterile diH2O.
Coomassie Blue Staining Solution
0.125 g Coomassie brilliant blue R-250.
1 mL acetic acid.
45 mL ethanol.
54 mL sterile diH2O.
2.2.8. Gel Destaining Solution
Solution I
125 mL ethanol.
50 mL acetic acid.
325 mL diH2O.
Solution II
25 mL ethanol.
37.5 mL acetic acid.
437.5 mL diH2O.
2.2.9. Gel Preservative Solution
30 mL methanol.
3 mL glycerol.
67 mL sterile diH2O.
2.3. MMP Positive Loading Control
Recombinant human MMP-2 (molecular mass 73 kDa).
Recombinant human MMP-9 (molecular mass 92 kDa).
2.4. Serum-Free Media for (In Vitro) Experimental Ischemia
10 mL Dulbeccos's Minimum Essential Medium (DMEM) containing either 1,000 mg/L (low) or 4,500 mg/L (high) glucose. DMEM media
0.2 mL of 200 mM L-glutamine.
0.1 mL of penicillin/streptomycin.
0.1 mL of N1 Medium Supplement.
2.5. Equipment for In Vitro Ischemia
Hypoxia incubator chamber (Billups-Rothenberg, Del Mar, CA).
Flow Meter (Billups-Rothenberg).
Oxygen meter (Microelectrodes Inc, OM-4 Oxygen Meter).
3. Methods
In this section, we describe how to analyze levels of MMP-2 and MMP-9, both in lysates prepared from brain samples, and from supernatants derived from primary glial cultures.
3.1. Experimental Ischemia in Astrocyte Cultures
To address the question of the origin of MMPs in the perivascular region following cerebral ischemia, we have examined the hypothesis that extracellular matrix (ECM) proteins present in the blood, such as vitronectin and fibronectin, might stimulate the secretion of MMPs from glial cells following blood extrusion during ischemia. Our data demonstrated that levels of secreted pro-MMP-2 from mouse primary astrocytes were down-regulated following in vitro ischemia independent of the ECM the cells were cultured on. In contrast, exposure of mouse primary microglia cells to vitronectin and fibronectin increased pro-MMP-9 levels significantly under ischemia when compared to poly-D-Lysine (PDL)-treated cells (del Zoppo et al. submitted, 2011).
To model the effects of experimental ischemia in vitro, we establish cultures of astrocytes or pure microglia on different substrates (poly-D-lysine, vitronectin, or fibronectin) and subject these cultures to oxygen-glucose deprivation (OGD), by switching the cultures from DMEM medium containing high glucose (4,500 mg/L) to low glucose (1,000 mg/L) and by reducing the ambient oxygen concentration to 1%. This can be performed as follows:
Coat each well in 6-well plates for 3 h at 37°C with 10 μg/mL vitronectin, fibronectin, or poly-D-lysine (control).
Wash the plates twice with 1× PBS prior to plating the cells.
Seed primary mouse astrocytes or microglia at a concentration of 4 × 105 cells per well and allow the cells to adhere overnight in a tissue culture incubator maintained at 5% CO2and 37°C (see Note 2).
The next day, 2 h prior to the induction of OGD, wash the cultures twice with 3 mL of 1× PBS (with Ca+2, Mg+2) or DMEM (see Notes 3–5).
Following the wash, add 1.5 mL of low glucose, serum-free media to cells undergoing OGD, and 1.5 mL of serum-free media containing high glucose to control cells (normoxia) (see Note 6).
Incubate the control plate (normoxia) in the tissue culture incubator at 5% CO2and 37°C for 18 h.
Enclose the experimental plate (hypoxia) in an incubator chamber and flush the chamber with 5% CO2-balanced N2at 10 L/min for 1 h.
After flushing, ensure that the chamber is well-sealed and place the chamber in a 37°C incubator for 18 h.
The next morning, following 18 h incubation, harvest 1 mL of the supernatants from normoxia- and hypoxia-treated wells (see Note 7).
To remove any contaminating floating cells, centrifuge the supernatants at 300 ×g for 3 min, and transfer only the top part of the supernatant to a fresh tube.
Store all samples at –80°C.
For samples of supernatants for gel zymography, we maximize the opportunity to detect (pro-)MMP-2 and (pro-)MMP-9 by loading 40 μL of each supernatant aliquot with 5× sample buffer into the 50 μL wells. Of course, smaller samples can be run with the appropriate adjustment in sample buffer. For cell lysates or fractions, smaller sample loads may be required so as not to overload the gel and cause bands to be exaggerated and hence out of the linear range for quantitation.
3.2. Zymographic Gel Preparation
The zymographic gel is made fresh by using a modified protein gel casting method.
Prepare the 1% solution of type B gelatin by dissolving 0.1 g gelatin in 10 mL water. To speed solubility, place the gelatin solution in a beaker of hot water (see Note 8).
Prepare 10 mL of the 8% resolving gel solution per gel by thoroughly mixing 3.26 mL sterile diH2O, 2.5 mL 1.5 M Tris-Cl, 0.2 mL 20% SDS, 2.7 mL 30% polyacrylamide, and 1.5 mL gelatin solution in a tube. The mixture is allowed to stand on a bench for 3 min before adding APS and TEMED (add 40 μL 10% APS and 8 μL TEMED per gel).
Pour the gel solution into the gel casting chamber (with a 1.5-mm spacer) and gently pour deionised water over the gel solution as an overlay (use a 1-mL pipette). Let the gel stand for 1 h until polymerized (see Note 9).
Prepare 2 mL of the 4% stacking gel (2 mL required for each gel) by combining 1.2 mL sterile diH2O, 500 μL 0.5 M Tris-Cl w/0.4% SDS, with 300 μL 30% polyacrylamide. Add 13 μL 10% APS and 7 μL TEMED per 2 mL of stacking gel solution.
Remove the water overlay solution from the polymerized resolving gel (by pouring off), and pour the stacking gel over the polymerized resolving gel and place combs into the stacking gel, let the assembly stand for 15~20 min until the stacking gel has polymerized.
3.3. Sample Preparation and Loading
Mix the glial cell sample (4 parts) with 1 part of 5× sample buffer. Incubate the mixture at room temperature for 10 min, then load the mixture into each well of the 10-well gel (see Notes 10 and 11).
Load 25 ng recombinant human MMP-2 and 25 ng recombinant human MMP-9 into one well of the same gel as positive standard controls.
3.4. Gel Electrophoresis
Run the gel at 110 V (e.g., apparatus from Bio-Rad, Inc; other instruments will vary), using 1× precooled electrophoresis running buffer at 4°C (see Note 12) for 60 min or until the dye front reaches the bottom of the gel.
3.5. Gel Activation
Carefully remove the gel from the electrophoretic plates with a gel spatula and place the gel in a washing container. Wash the gel 3 times with 60 mL gel washing buffer for 15 min each time, while agitating the container and gel on a rotary shaker (see Note 13).
Incubate the gel with zymographic development buffer in the gel container at 37°C for 42 h (see Note 13).
3.6. Gel Staining and Destaining
3.7. Gel Scan and Quantification
Wash the destained gel with sterile water for 30 min and incubate the gel in gel preservative solution for 15 min. Seal the gel in a plastic wrap with a sealer.
Immediately scan the gel with a digital scanner. An external gray scale (used for photography) must be scanned with the gel. Adjust all scans to the same gray scale.
Make the MMP-2 and MMP-9 density standard curves with NIH ImageJ 1.42q, or similar software.
Calibrate the MMP-2 or MMP-9 images to a proper set of density standards.
Quantify the integrative intensity of the gelatinase bands using NIH ImageJ 1.42q software.
Fig. 1.
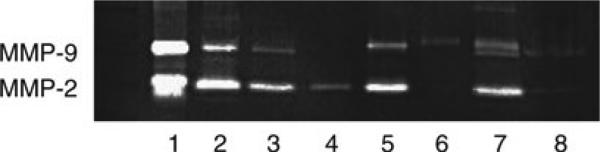
Gel zymogram depicting differences in (pro-)MMP-2 and (pro-)MMP-9 content among discrete samples of glial cells, serum supplements, and cerebral tissue. After activation, the gel was stained with Coomassie blue by the methods outlined here (note that a parallel gel stained with amido black was more sensitive with regard to the samples with larger contents of gelatinase, suggesting a different slope of linearity of that system.). The samples in the individual lanes are: Lane 1, human recombinant MMP-2 and MMP-9. Lane 2, fetal bovine serum (FBS, 2%). Lane 3, serum-free medium in the well that had previously been incubated with 10% FBS-containing medium. Lane 4, astrocytes (from C57 Bl/6 mice) followed by PBS wash prior to serum-free medium; note the single pro-MMP-2 band. Lane 5, astrocytes without PBS wash prior to serum-free medium; note the presence of both a pro-MMP-9 band (bovine) and a pro-MMP-2 band (murine). Lane 6, microglia (from C57 Bl/6 mice) followed by PBS wash prior to serum-free medium. Note the single pro-MMP-9 band. Lane 7, microglia without a PBS wash prior to serum-free medium. Note that the MMP-9 “doublet” represents pro-MMP-9 from both murine (cell) and bovine (FBS) sources. The lower band of the doublet is not active murine MMP-9, but derived from FBS. Lane 8, sample of cortical tissue (Papio anubis cynocephalus) taken 7 days after middle cerebral artery occlusion. Note the faint bands of pro-MMP-2 and pro-MMP-9.
Acknowlegments
This work was supported in part by NIH grants NS 053716, NS 038710, and NS 036945 to Dr. del Zoppo. Special thanks to Greta Berg for manuscript preparation.
Footnotes
In our laboratory, we prepare zymographic gels fresh. This has a number of advantages: (1) While precast gels are available from commercial sources that make the procedures convenient and relatively easier, they lack the flexibility, ability to troubleshoot, and personal oversight demanded by preparation of the gels just prior to assay. They may also make quantitation more difficult as a result. (2) Adjustments in gelatin concentration and source affect the quality and reproducibility of the lysis bands, and therefore study outcomes. (3) Careful management of gel preparation is required for quantitation. In our experience, reproducibility to a coefficient of variance (cv) of 6–8% is possible (1).
We usually harvest microglia and astrocytes from the mixed glial culture on day 12 and day 13 of growth, respectively. After isolation, microglial cells are plated overnight in the mixed glial culture-conditioned media. Astrocytes are further purified by shaking overnight, then harvested and plated the next day.
This wash is essential to remove all traces of FBS before adding serum-free media. MMPs are secreted by a variety of tissues. For this reason, it is not uncommon to detect (pro-)MMP-2 and (pro-)MMP-9 in biological samples of various origins, including serum (17). Gel zymography analysis of samples of FBS, used in isolated cell culture, revealed both gelatinases comigrating with the human recombinant pro-MMP-2 (72 kDa) and pro-MMP-9 (93 kDa) ( see Fig. 1). Simply replacing serum-containing media with serum-free media before the induction of in vitro ischemia does not totally prevent contamination of mouse experimental samples with bovine gelatinases. Furthermore, serum can bind to the walls and floor of the wells and can be eluted with the sample, thereby contaminating it (see lane 3, Fig. 1). However, we have found that contamination by serum-derived gelatinases can be significantly reduced to nondetectable levels by a thorough wash of cell cultures, removing all traces of serum or other purified biological factors that might be present. Thereupon, only a single band of pro-MMP-2 or pro-MMP-9 is usually detectable from cultures of mouse primary astrocytes or microglia cells, respectively. In contrast, in the absence of washing, while mouse microglial pro-MMP-9 can be readily distinguished from bovine gelatinases due to its different molecular weight, astrocyte pro-MMP-2 is indiscernible from the bovine MMP-2 band. Note that another clue to significant serum contamination is the detection of MMP bands of variable intensity between different replicates due to the uneven distribution of retained serum among samples.
To further improve analysis, control wells should be prepared which lack cells but receive the same treatment. These controls, run on the gel alongside the other samples, will help to detect potential contamination and further improve the technique of sample preparation.
Treat each well separately by adding wash solution or media back to the culture right away since cells are very sensitive and do not like to be left without media for an extended period of time.
OGD should be performed the day after plating, in order to maintain accurate cell counts. If the ratio between cell number and the amount of the media is not maintained, this can either result in cell death following ischemia, or incomplete ischemia due to increased oxygen levels, if the volume of the media has been increased.
O2levels in the media can be monitored using an oxygen meter. In our culture conditions, following a 1-h flush with 5% CO2/95% N2, O2levels usually decreased to 0.2–0.4% 4 h after flushing and remained at this level throughout the remainder of the experiment.
The gelatin is slow to dissolve at room temperature, so we recommend warming the solution by placing the tube in a beaker of hot water. Sometimes a concentrated pellet of gelatin may form at the bottom of the tube. This can be avoided by holding the tube containing gelatin powder at an oblique angle when adding water to the tube, then shaking the mixture before warming up.
To determine whether the gel has polymerized, we examine the unused polymerized gel solution left in the tube. In addition, one can see a line between the polymerized gel and the water overlay interface once the gel has set.
These methods can be used with cerebral tissues from a number of species. We recommend that tissues be completely perfused of blood under anesthesia via the transcardiac route with clamping of the jugular veins and the inferior vena cava and the descending aorta. Perfusion should be performed with a chilled (4°C) heparinized iso-osmotic fluid containing protein (e.g., bovine serum albumin) to mimic the characteristics of plasma, until red cell exclusion is complete (18).
The brain samples must be prepared immediately, and quick frozen, then stored at –80°C until used. Please see Heo et al. for the proper method of preparation (1).
We make up 1× electrophoresis running buffer and store at 4°C before casting the gels to ensure this is cool before running the gel.
Washing of the gel post-electrophoresis is a critical step in this procedure. During this time, the Triton-X100 in the washing buffer is exchanged for the SDS present within the gel. This is important because SDS, which denatures proteins, must be thoroughly removed to allow the temporarily denatured gelatinases to renature. Once renatured, the ability of gelatinases ability to cleave substrates is restored. Importantly, this rena-turation step activates not only the active form of the enzymes, but also the normally inactive proforms. Thus, this technique permits the detection of both the pro- and active forms of these enzymes. In our hands, often a weak or absent gelatinase signal can be traced back to inadequate washing of gels, so thorough washing is important. This step also affects quantitation. Although we often use 42 h for activation/development, it is advised to perform the proper controls with known quantities of the recombinant MMP controls to establish linearity of the gels, the development procedure, the scanning system, and data acquisition.
Note that together with the duration of activation, the durations of staining and destaining are variables that will also influence band quantitation. Typically, we stain for a fixed period of 1 h, and then adjust the time in destaining solution I until the lysis bands appear transparent, and the best contrast is observed. By not destaining long enough, it is hard to visualize the bands, but if destained for too long, then the blue background staining of the gelatin falls too low and the contrast is diminished.
Issues regarding timing or duration of development of the MMP activities in the gels are considered by Heo et al. (1). Here, the adherence to strict time and buffer concentrations as well as the use of longitudinal control samples to adjust for gelto-gel variability is extremely important.
Similar observations are possible with cell cultures from different origins. But, there are important species differences in the molecular weights of MMP-2 and MMP-9 to be aware of. According to the UniProt Knowledgebase (http://www.uniprot.org/), human, bovine, and mouse pro-MMP-2 have a molecular mass of 73.8, 73.8, and 74 kDa, respectively, while the masses for pro-MMP-9 are 78.5, 79.0, and 80.5 kDa, respectively. Posttranscriptionally, pro-MMP-2 is phosphorylated and its molecular weight remains unchanged. Thus, whether it is recombinant or not, pro-MMP-2 migrates around 72 kDa on an 8% SDS-PAGE gel. In contrast, pro-MMP-9 is variably glycosylated and sporadically associated with micro-globulin, a 25-kDa protein, which can increase its apparent molecular mass to 93 kDa or higher (19, 20). In our experience, however, bovine pro-MMP-9 comigrates with human recombinant MMP-9 at around 93 kDa on 8% SDS-PAGE, suggesting that the bovine gelatinase found in processed fetal bovine serum (FBS) may either be an active form of MMP-9 or is not fully glycosylated. In contrast, mouse pro-MMP-9 from glial cells is observed around 105 kDa.
References
- 1.Heo JH, Lucero J, Abumiya T, et al. Matrix metalloproteinases increase very early during experimental focal cerebral ischemia. J Cereb Blood Flow Metab. 1999;19:624–633. doi: 10.1097/00004647-199906000-00005. [DOI] [PubMed] [Google Scholar]
- 2.Fernandez-Resa P, Mira E, Quesada AR. Enhanced detection of casein zymography of matrix metalloproteinases. Anal Biochem. 1995;224:434–435. doi: 10.1006/abio.1995.1063. [DOI] [PubMed] [Google Scholar]
- 3.Yu WH, Woessner JF., Jr. Heparin-enhanced zymographic detection of matrilysin and collagenases. Anal Biochem. 2001;293:38–42. doi: 10.1006/abio.2001.5099. [DOI] [PubMed] [Google Scholar]
- 4.Hosomi N, Lucero J, Heo JH, et al. Rapid differential endogenous plasminogen activator expression after acute middle cerebral artery occlusion. Stroke. 2001;32:1341–1348. doi: 10.1161/01.str.32.6.1341. [DOI] [PubMed] [Google Scholar]
- 5.Massova I, Kotra LP, Fridman R, et al. Matrix metalloproteinases: structures, evolution, and diversification. FASEB J. 1998;12:1075–1095. [PubMed] [Google Scholar]
- 6.Morodomi T, Ogata Y, Sasaguri Y, et al. Purification and characterization of matrix metalloproteinase 9 from U937 monocytic leukaemia and HT1080 fibrosarcoma cells. Biochem J. 1992;285:603–611. doi: 10.1042/bj2850603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Senior RM, Griffin GL, Fliszar CJ, et al. Human 92- and 72-kilodalton type IV collagenases are elastases. J Biol Chem. 1991;266:7870–7875. [PubMed] [Google Scholar]
- 8.Prudova A, auf dem Keller U, Butler GS, et al. Multiplex N-terminome analysis of MMP-2 and MMP-9 substrate degradomes by iTRAQ-TAILS quantitative proteomics. Mol Cell Proteomics. 2010;9:894–911. doi: 10.1074/mcp.M000050-MCP201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tallant C, Marrero A, Gomis-Ruth FX. Matrix metalloproteinases: fold and function of their catalytic domains. Biochim Biophys Acta. 2010;1803:20–28. doi: 10.1016/j.bbamcr.2009.04.003. [DOI] [PubMed] [Google Scholar]
- 10.Morgunova E, Tuuttila A, Bergmann U, et al. Structure of human pro-matrix metal-loproteinase-2: activation mechanism revealed. Science. 1999;284:1667–1670. doi: 10.1126/science.284.5420.1667. [DOI] [PubMed] [Google Scholar]
- 11.Snoek-van Beurden PA, Von den Hoff JW. Zymographic techniques for the analysis of matrix metalloproteinases and their inhibitors. BioTechniques. 2005;38:73–83. doi: 10.2144/05381RV01. [DOI] [PubMed] [Google Scholar]
- 12.Springman EB, Angleton EL, Birkedal-Hansen H, et al. Multiple modes of activation of latent human fibroblast collagenase: evidence for the role of a Cys73 active-site zinc complex in latency and a “cysteine switch” mechanism for activation. Proc Natl Acad Sci USA. 1990;87:364–368. doi: 10.1073/pnas.87.1.364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Van Wart HE, Birkedal-Hansen H. The cysteine switch: a principle of regulation of metalloproteinase activity with potential applicability to the entire matrix metalloproteinase gene family. Proc Natl Acad Sci USA. 1990;87:5578–5582. doi: 10.1073/pnas.87.14.5578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ramos-DeSimone N, Hahn-Dantona E, Sipley J, et al. Activation of matrix metalloproteinase-9 (MMP-9) via a converging plasmin/stromelysin-1 cascade enhances tumor cell invasion. J Biol Chem. 1999;274:13066–13076. doi: 10.1074/jbc.274.19.13066. [DOI] [PubMed] [Google Scholar]
- 15.Morrison CJ, Butler GS, Bigg HF, et al. Cellular activation of MMP-2 (gelatinase A) by MT2-MMP occurs via a TIMP-2-independent pathway. J Biol Chem. 2001;276:47402–47410. doi: 10.1074/jbc.M108643200. [DOI] [PubMed] [Google Scholar]
- 16.Stetler-Stevenson WG. Tissue inhibitors of metalloproteinases in cell signaling: metalloproteinase-independent biological activities. Sci Signal. 2008;1:re6. doi: 10.1126/scisignal.127re6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Somiari SB, Somiari RI, Heckman CM, et al. Circulating MMP2 and MMP9 in breast cancer -- potential role in classification of patients into low risk, high risk, benign disease and breast cancer categories. Int J Cancer. 2006;119:1403–1411. doi: 10.1002/ijc.21989. [DOI] [PubMed] [Google Scholar]
- 18.Abumiya T, Lucero J, Heo JH, et al. Activated microvessels express vascular endothelial growth factor and integrin alpha(v) beta3 during focal cerebral ischemia. J Cereb Blood Flow Metab. 1999;19:1038–1050. doi: 10.1097/00004647-199909000-00012. [DOI] [PubMed] [Google Scholar]
- 19.Kotra LP, Zhang L, Fridman R, et al. N-Glycosylation pattern of the zymogenic form of human matrix metalloproteinase-9. Bioorg Chem. 2002;30:356–370. doi: 10.1016/s0045-2068(02)00501-1. [DOI] [PubMed] [Google Scholar]
- 20.Triebel S, Blaser J, Reinke H, et al. A 25 kDa alpha 2-microglobulin-related protein is a component of the 125 kDa form of human gelatinase. FEBS Lett. 1992;314:386–388. doi: 10.1016/0014-5793(92)81511-j. [DOI] [PubMed] [Google Scholar]