

Abstract
Both the core JAK-STAT pathway components and their in vivo roles have been widely conserved between vertebrates and invertebrate models such as Drosophila melanogaster. Misregulation of JAK-STAT pathway activity has also been identified as a key factor in the development of multiple human malignancies. Recently, whole genome RNA interference (RNAi) screens in cultured Drosophila cells have identified both positively and negatively acting JAK-STAT pathway regulators. Here, we describe the analysis of 73 human genes representing homologs of 56 Drosophila genes originally identified by genome-wide RNAi screening as regulators of JAK-STAT signaling. Using assays for human STAT1 and STAT3 protein levels and phosphorylation status, as well as assays measuring the expression of endogenous STAT1 and STAT3 transcriptional targets, we have tested siRNAs targeting these 73 human genes and have identified potential JAK-STAT pathway regulatory roles in 69 (95%) of these. The genes identified represent a wide range of human JAK-STAT pathway regulators and include genes not previously known to modulate this signaling cascade. These results underline the value of model system based approaches for the identification of pathway regulators and have led to the identification of loci whose misregulation may ultimately be implicated in JAK-STAT pathway-mediated human disease.
Keywords: assay, Drosophila, GBP1, HeLa, phosphorylation, RNAi, screening, SOCS3
Introduction
The accurate, timely and proportionate regulation of a small number of evolutionarily conserved signal transduction cascades is essential for embryonic development and adult life.1 However, while the components central to most signaling pathways have been identified, the regulators of pathway activity required in vivo are less well known. One example of such a signal transduction pathway is the JAK-STAT signaling cascade, named after its two central components, the Janus kinase (JAK) and the Signal Transducer and Activator of Transcription (STAT).2,3 In vertebrates, four JAK-like genes (JAK1–3 and Tyk2) and seven STAT transcription factors (STAT1–4, 5a, 5b and 6) have been identified. These have been shown to function in a range of processes including development, cellular proliferation and response to infection.2 In the canonical model of pathway activation, binding of an extracellular ligand to a trans-membrane receptor activates a receptor-associated JAK tyrosine kinase. Following activation, JAK phosphorylation of the receptor complex provides docking sites for STAT transcription factors, which are themselves phosphorylated on a conserved C-terminal tyrosine residue. STAT molecules activated in this manner translocate to the nucleus where they bind to DNA within the promoters of pathway target genes to activate transcription.4
In vertebrates, JAK-STAT pathway signaling can be modulated by many different ligands leading to the specific activation of STATs with distinct biological consequences. For example, the interferon family of ligands activates STAT1 and STAT2 via JAK1 and JAK2. This in turn induces the expression of target genes including GBP1 and other cytokines.5 The cellular functions mediated by STAT1 include potent anti-proliferative and pro-apoptotic responses, tumor immuno-surveillance6 and responses to viral infection.7 By contrast, constitutive activation of STAT3, as well as STAT5A and 5B, can result in oncogenic cellular responses with multiple tumors and tumor-derived cell lines displaying high levels of phosphorylated-STAT3 activity.8 During normal cellular processes, ligands such as Interleukin 6 (IL-6) and Oncostatin M (OSM), also acting via JAK1, lead to STAT3 phosphorylation and the expression of specific target genes including SOCS3.9 Interestingly, the proliferative and anti-proliferative functions performed by different STATs in vertebrates can be exerted by the single STAT protein present in Drosophila melanogaster.10
In addition to the core pathway components, ligands and receptors a number of ‘non-core’ pathway regulators have also been identified. These include the SOCS proteins, such as the pathway target gene SOCS3, which act to negatively regulate the stimulated receptor/JAK complex forming negative feedback loops that reduce the duration and intensity of pathway activation.11 In addition, the PIAS proteins and the SHP1/2 tyrosine phosphatases also act as negative regulators of pathway activity.12 However, although understanding of these factors has advanced significantly in recent years, a comprehensive search for novel modulators of vertebrate pathway activity has not been undertaken and it remains likely that a number of regulatory mechanisms are yet to be identified.
To circumvent the difficulties inherent in screening the vertebrate genome for regulators of the high complexity and semi-redundant JAK-STAT pathway, we have previously used Drosophila melanogaster to undertake a whole-genome cell culture based RNAi screen. This approach led to the identification and validation of 90 Drosophila regulators of JAK-STAT pathway signaling including 66 positive and 24 putative negative pathway regulators. Many of these show in vivo, genetic and molecular interactions consistent with their proposed role in pathway signaling.13 One of the central tenets of this approach was the anticipation that low levels of genetic redundancy within the Drosophila genome would allow the identification of factors that might not otherwise be detected in similar vertebrate screens. At the same time, it was anticipated that the regulatory activities identified in Drosophila would have been evolutionary conserved with homologous gene-products exerting specific effects on the JAK-STAT pathways of vertebrate systems.
In this report we ask whether factors important for JAK-STAT signal transduction in Drosophila are required for the activity of one or more of the STATs that make up the human pathway. We identified 73 human genes, which represent putative homologs of 56 Drosophila genes previously identified as pathway modulators.13 Using siRNA approaches in human HeLa cells, we knocked down the activity of these genes and, using phosphorylation and transcriptional assays for STAT1 and STAT3, have identified 67 human pathway regulators. The loci identified include genes encoding components of the endocytic machinery, chromatin remodeling enzymes and protein-modifying enzymes, which may provide post-translational modifications important for pathway activity.
This study highlights the strength of systematic cross-species approaches for the identification of cancer-pathway regulators and serves as a starting point for future analysis of potential disease-related molecules.
Results
STAT phosphorylation assays
One essential pre-requisite for canonical JAK-STAT pathway activity is the phosphorylation of a conserved tyrosine (Y) residue present in the C-terminal region of all STAT transcription factors. This post-translational modification is both essential for, and indicative of, pathway activation.14 Using HeLa cells as a tractable and representative human cancer-derived cell line, we therefore set out to assess the phosphorylation state of endogenous STAT1 and STAT3 as stimulated by upstream pathway components and receptors endogenously expressed in these cells. Both STAT1 and STAT3 are expressed in unstimulated cells with STAT3 S726 phosphorylation15 and low levels of STAT3 Y705 phosphorylation also detected in the absence of exogenous ligand (Fig. 1A). In order to determine the most appropriate pathway ligands we treated cells with IL-2, IL-3, IL-6, IL-6 with soluble IL-6 receptor (IL-6R), Interferon gamma (IFN-γ) and OSM for 15 min (Fig. 1A). While stimulation with IL-2 and IL-3 have no effect on either STAT, IL-6 + IL-6R, IFN-γ and OSM all result in a strong increase in the relative level of STAT1 phospho-Y701 (pSTAT1). Similarly, stimulation with IL6, IL-6 + IL-6R and OSM causes the phosphorylation of Y705 of STAT3 (pSTAT3). Based on these results we therefore focused on IFN-γ as a mediator of STAT1 stimulation and OSM as a mediator of STAT3.

Figure 1. STAT phosphorylation as a reporter for regulatory factors. (A) Antibodies detecting pSTAT1 and STAT1 (top two panels) as well as pSTAT3 and STAT3 (bottom 3 panels) were used to assess the effect of stimulation with the cytokines shown. (B) The effect of the indicated siRNAs on the levels of pSTAT1 and total STAT1 following IFN-γ stimulation as indicated. Note the loss of STAT1 following treatment with STAT1 siRNA and the reduction in pSTAT1 levels following knockdown with JAK1 siRNA. (C) The effect of the indicated siRNAs on the levels of pSTAT3 and total STAT3 following OSM stimulation as indicated. (D and E) The effect of the indicated siRNAs on the levels of pSTAT1 and total STAT1 following IFN-γ stimulation (D) and pSTAT3 and STAT3 following OSM stimulation (E).
In order to check the feasibility of using siRNA-mediated knockdown of JAK-STAT pathway regulators in conjunction with pSTAT1 and pSTAT3 assays we also set up experiments using either control siRNAs or siRNA pools knocking down known pathway components. Allowing 3 d for protein depletion, JAK1 knockdown reduces the intensity of both pSTAT1 and pSTAT3 detectable after ligand stimulation while siRNAs targeting the individual STAT transcripts specifically reduce both phosphorylated and non-phosphorylated forms (Fig. 1B and C) indicating that knockdown of genes known to modulate STAT phosphorylation can be identified by this approach.
It should however be noted that although tyrosine phosphorylation of STATs is required, it is not necessarily sufficient for transcriptional activity. Other post-translational modifications have been identified that modulate the transcriptional potential of activated STAT molecules.14 Conversely, constitutively phosphorylated dominant-negative mutations of Drosophila STAT92E have also been identified that are incapable of stimulating target gene transcription.16
Transcriptional assays
Although tyrosine phosphorylation of vertebrate STATs is essential for their activity, the principal biological consequence of JAK-STAT pathway stimulation is a change in pathway target gene expression.5,17 We therefore set out to measure the expression of endogenous target genes driven by native promoters in their normal chromatin context, thereby avoiding the limitations of transiently transfected reporters.13 We first tested nine endogenous genes previously reported to be STAT transcriptional targets5 for their potential suitability as pathway activity reporters. We stimulated with IL-6 and OSM to activate STAT3 and IFN-γ to activate STAT1 target genes and measured mRNA levels expressed relative to β-ACTIN (Fig. 2A). Of the target genes tested, IFN-γ-induced GBP1 and OSM-induced SOCS3 expression were most suitable as reporters for STAT1 and STAT3 activity respectively. However, while large increases in GBP1 expression are elicited by IFN-γ stimulation, the fold increase in SOCS3 expression elicited by OSM is less, with IFN-γ also leading to increased SOCS3 mRNA levels (Fig. 2A). The increase in the signal: noise ratio resulting from lower levels of SOCS3 expression, and as well as potential inter-pathway cross-talk must therefore be taken into account when analyzing results derived from this assay.
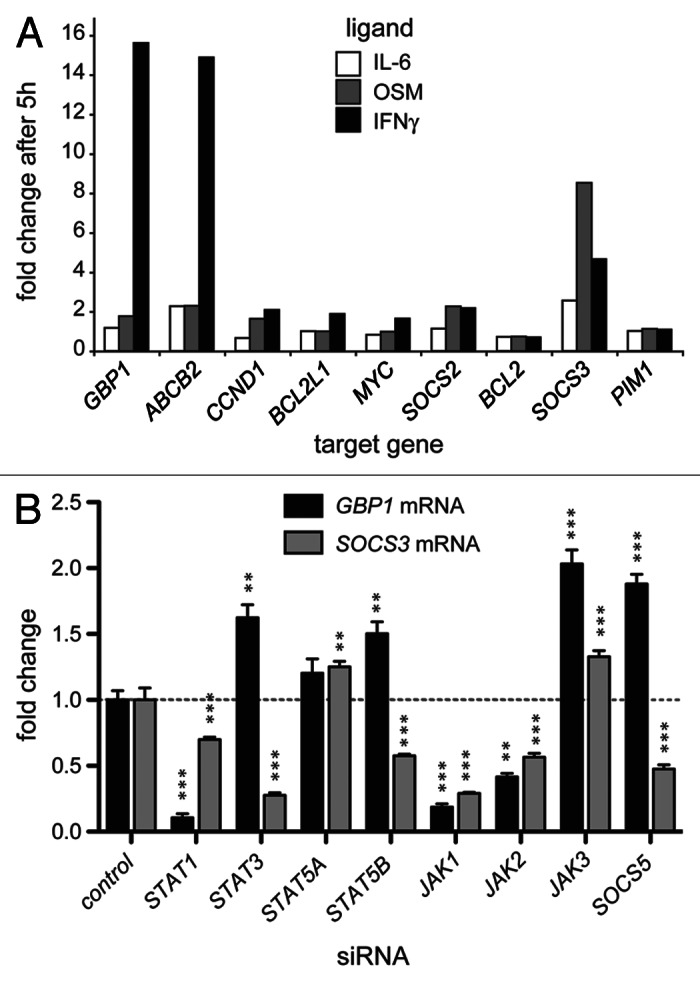
Figure 2. Assays for STAT-mediated transcriptional regulation. (A) Fold increase in the mRNA levels of the indicated putative STAT target genes following stimulation with the indicated ligands. (B) Fold increase in the mRNA levels of GBP1 (black bars) and SOCS3 (gray bars) following stimulation with IFN-γ and OSM respectively. Cells were previously treated with the indicated siRNAs targeting known JAK-STAT pathway components and demonstrate the specificity of these target genes for STAT1 and STAT3 as well as the compensatory effects that result from the knockdown of other related factors. Statistical significance is indicated by **p < 0.01, ***p < 0.001. Error bars show standard error.
We then set out to test the efficacy of siRNA-induced knockdown on GBP1 and SOSC3 transcription. As expected, knockdown of JAK1 and JAK2 significantly reduces expression of both target genes (Fig. 2B). Similarly, as would be expected of a bona fide target gene, knockdown of STAT1 strongly reduces expression of GBP1 while knockdown of STAT3 reduces the levels of OSM induced SOCS3 expression. However, a degree of crosstalk/redundancy is evident with the levels of OSM-induced SOCS3 mRNA falling following STAT1 knockdown while the level of IFN-γ-induced GBP1 increases following a reduction in STAT3 levels.
Intriguingly, compensatory mechanisms and crosstalk between JAK-STAT pathway components is also demonstrated by the knockdown of STAT5A and STAT5B as well as JAK3 which all result in statistically significant (p < 0.05) increases in IFN-γ-induced GBP1 expression. Consistent with these findings, it has been reported that activated STAT5 can protect cells from IFN-γ-induced apoptosis18 and that overexpression of STAT5 can counteract interferon signaling.19 However, the molecular basis of this interaction remains to be established.
Interestingly, while STAT5A and STAT5B are highly homologous at the protein level, OSM-induced SOCS3 mRNA is modulated in opposite directions following knockdown of these two closely related molecules (Fig. 2B).
Identification of human homologs
We recently identified 90 JAK-STAT pathway-regulating genes in a whole-genome RNAi based screen in Drosophila Kc167 cells.13 In order to identify potential human homologs we used HomoloGene, Inparanoid and best reciprocal BLAST searches as parsed from the Flight database (http://flight.licr.org)20 and identified 73 human candidate genes representing homologs of 56 interacting Drosophila genes (Table 1). This collection includes controls such as STAT1, STAT3 and JAK1 as well as previously uncharacterized loci. In order to address the potential role of these genes, siRNA pools targeting each transcript with four independent 21-mers were used to maximize the chance of effective knockdown while minimizing potential off-target effects.21
Table 1. Homolog selection.

Screening for human JAK-STAT pathway regulators
Having developed assays and identified the human homologs of interacting Drosophila genes we then tested all 73 siRNA-pools for their influence on STAT1 and pSTAT1 as well as STAT3, pSTAT3 and β-ACTIN levels following a 3 d knockdown protocol (Table 2 and Fig. 1D and E). Using the levels of β-ACTIN as a reference, the levels of total and phosphorylated STATs were measured (see Materials and Methods) and expressed as fold changes relative to controls run in parallel on the same protein gel blot (Table 2). Changes in the overall levels of unphosphorylated STAT1 and STAT3 as well as the level of pSTAT1 and pSTAT3 are indicated with increases highlighted in green and decreased levels in red. A subset of tested loci is shown in Figure 1D and E. Genes were identified that affected the overall levels of STAT1 and/or STAT3, the level of pSTAT1/pSTAT3 or the levels of both overall and phosphorylated STATs.
Table 2.
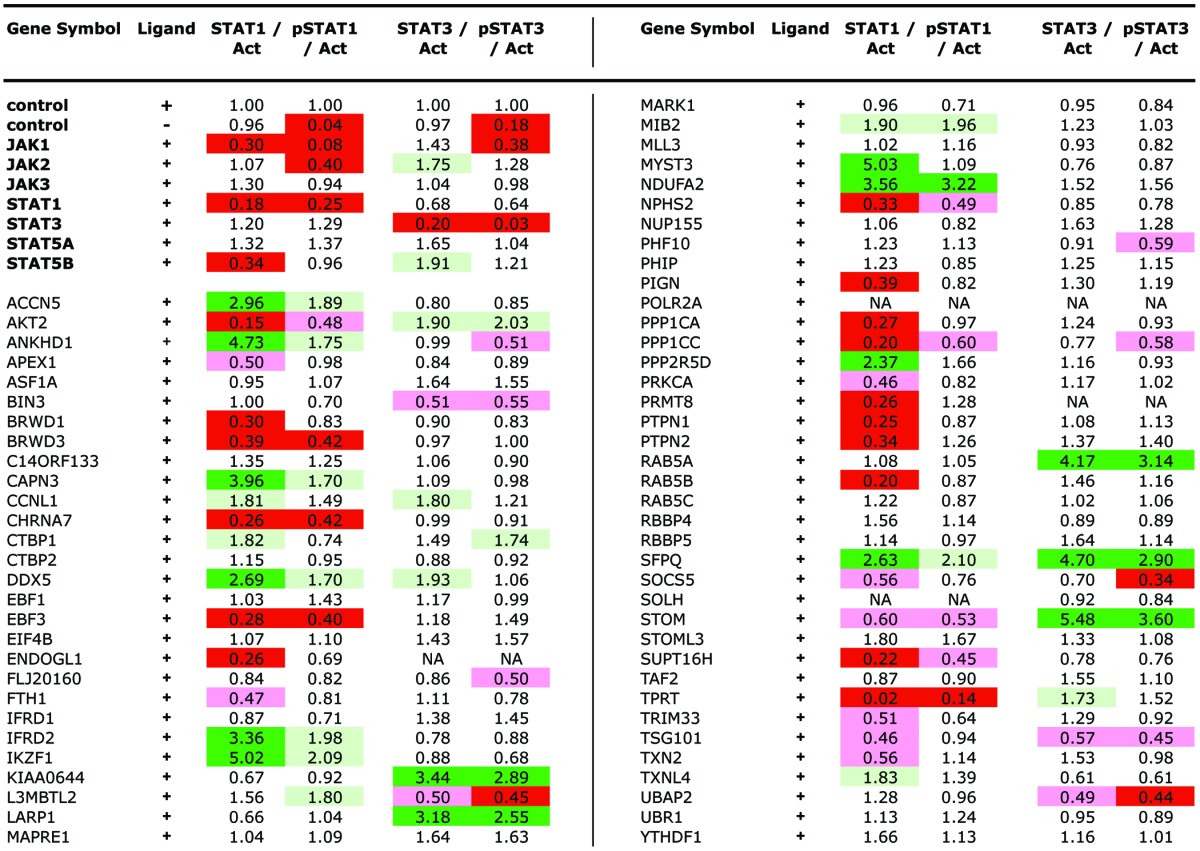
NA, not available due to low β-actin levels; light green, > 1.67-fold higher; green, > 2.2-fold higher; pink, < 0.6-fold higher; red, < 0.45-fold higher.
We next assayed the effects of the 73 siRNA pools on IFN-γ and OSM-stimulated target gene expression. Following treatment with siRNA for 3 d and stimulation with either IFN-γ or OSM for 6 h, cells were lysed and RNA prepared. The level of GBP1 and SOCS3 mRNAs expressed from their endogenous loci were detected by qPCR and normalized to β-actin mRNA levels. This was expressed as a fold change relative to cells treated with a control siRNA (Fig. 3A). Triplicate experimental replicates were used to calculate the mean change in gene expression (Fig. 3A). Overall 57 genes (excluding the known pathway components shown in Fig. 2B) produced significant changes in either GBP1 or SOCS3 expression. As shown in Figure 3A, genes were clustered into groups on the basis of their differential gene regulation representing loci that upregulate GBP1 (group a, c and d), or SOCS3 (group a and b), regulate only one target gene (group b, c, e and g) or differentially regulate expression in opposite directions (group d). The identity and potential roles of some of these STAT-regulating loci are discussed in greater detail below. Strikingly however, a greater number of STAT1-interacting v. STAT3-interacting loc were identified by both transcriptional and phosphorylation assays. While the reagents/assays for STAT1 activation are better than for STAT3 (note the comparatively lower background following STAT1 knockdown in Figs. 1D and 2B) it is unlikely that this is the only explanation and further investigation into the mechanisms underlying this observation will be required.

Figure 3. Screening for JAK-STAT modulating genes by qPCR. (A) Heat map showing the level of IFN-γ induced GBP1 mRNA and OSM induced SOCS3 mRNAs expressed following knock down of the indicated genes. Numbers represent the fold change relative to controls. Colors represent statistically significant increases (reds) or decreases (blues) in expression with dark red/blue p < 0.001 and light red/blue p < 0.05. Genes have been grouped according to phenotype and are discussed in the main text. (B and C) Graphs representing the interactions of genes falling into predicted endocytosis and protein stability (B) or chromatin modifier (C) ontologies. Error bars show standard error. *p < 0.05, **p < 0.01, ***p < 0.001.
Discussion
We have identified 73 putative human homologs (Table 1) of 53 Drosophila JAK-STAT pathway modulators originally identified by whole-genome RNAi screening.13 Using a combination of protein phosphorylation (Fig. 1) and transcriptional assays (Fig. 2) we examined STAT1 and STAT3 activity following knockdown of these 73 human homologs in HeLa cells to assess potential evolutionary conservation of their pathway modulating function. Overall, phosphorylation assays identified 39 modifiers of STAT1 and 19 modifiers of STAT3 (Table 2) while transcriptional assays identified 57 loci that significantly modulate the expression of endogenous STAT1 and/or STAT3 target genes (Fig. 3A). In total, 69 of the 73 human genes (95%) showed an interaction indicating that the majority of genes whose primary sequence has been conserved have also retained their functional biological roles during the evolutionary divergence that separates Drosophila and humans. It should however be noted that more detailed study in multiple human cell lines will ultimately be needed to confirm the role of these interacting genes in the wider human context.
The conservation of gene function between related STAT-regulators present in both humans and Drosophila demonstrates that it is feasible to use Drosophila as a relevant and tractable system for gene discovery approaches that can be applied to humans. This utility is especially striking given the significant differences in genomic and proteomic complexity between the two organisms. Indeed, well-characterized cellular processes such as endocytosis are frequently regulated by multiple closely related semi-redundant factors. For example, while Drosophila contains a single Rab5 protein,22 vertebrate trafficking utilizes three homologous Rab5-like proteins, Rab5A, Rab5B and Rab5C. Furthermore, removal/knockdown of all three is required to block endocytosis in cultured cells23 suggesting a significant level of redundancy. Despite this apparent redundancy, individual knockdown of each Rab5-like gene produces distinct transcriptional JAK-STAT phenotypes with loss of Rab5A leading to an increase in STAT1, loss of Rab5B an increase in STAT1 and a decrease in STAT3, and Rab5C producing no effect (Fig. 3B). Consistent with a link between endocytosis and STAT activity, STAT1-mediated signaling induced by IFN-γ has recently been shown to be differentially regulated depending on the Clathrin-dependent or Clathrin-independent route via which receptor endocytosis occurs.24 Our results suggest that the situation may be more complex and suggests that the Rab5 family proteins required for Clathrin dependent endocytosis perform distinct roles and that disruption of these roles is sufficient to modulate the activity of at least two independent STAT molecules.
Another striking phenotype observed is the strong upregulation of both GBP1 and SOCS3 mRNA following knockdown of UBR1 (Fig. 3B). UBR1 is a key component of the N-end rule machinery, a conserved cellular process that affects protein stability on the basis of the N-terminal amino acid. Substrates of the N-end rule are recognized by the UBR1 E3-ubiquitin ligase25 and the increase in STAT1 and STAT3 activity that occurs following its knockdown suggests that a positively acting pathway components may represent UBR1 substrates. Although the overall level of STAT proteins does not appear to be directly affected by knockdown of UBR1 (Table 2) it would be interesting to test other JAK-STAT pathway components and other elements of the N-end rule machinery to examine a potential link between these two cellular processes.
One of the key aspects of the STAT1 and STAT3 transcriptional assay development was the decision to avoid transiently transfected reporter assays in preference for the direct measurement of pathway target gene mRNA expressed by endogenous promoters from within their normal chromatin context (Fig. 2). This is especially relevant in the light of recent reports from Drosophila, which suggest that chromatin structure may be directly linked to JAK-STAT pathway activity via a direct physical interaction of STAT92E with Heterochromatin Protein 1 (HP1).26,27 Consistent with this, human JAK2 has also recently been suggested to modify chromatin via direct phosphorylation of Histone H3, so interfering with HP1 association.28 Consistent with such interactions, this screen has also identified a number of putative regulators of heterochromatin (Fig. 3C). These include SUPT16H, a component of the FACT complex required for transcription from chromatinized DNA29 as well as RBBP4, a member of the histone deacetylase complex30 and RBBP5 a key component of the H3K4 methyltransferase complex.31 Although preliminary, it will be intriguing to determine the significance of these interactions in the context of STAT-mediated transcription and chromatinization in the future.
Although this screen has identified several intriguing interactions, it is not generally feasible to individually verify the depletion of individual proteins following siRNA-mediated knockdown in large scale screens. However, the high proportion of loci with STAT-modulator phenotypes suggests that most siRNA pools are indeed effective. While the efficiency of siRNA targeting STAT1 and STAT3 has been directly demonstrated (Fig. 1B), it remains possible that false negatives, caused by a failure to reduce protein levels, or false positives caused by off-target effects, may be present. Ultimately, further biological characterization of the interactors identified will be required. In addition, while both STAT phosphorylation and transcriptional activity have been widely used as proxies for pathway activity in the past,5,17,32,33 our experience suggests that neither is necessarily consistent in isolation. Indeed, we have identified genes whose knockdown appears to modulate phosphorylation and transcription in different directions, genes that modulate phosphorylation without changing transcription, and genes that affect transcription without any apparent effect on STAT phosphorylation. Thus, while both approaches are valid, more detailed analysis of protein function will be required before mechanistic interactions can be proven.
In summary, we have demonstrated that whole genome screening approaches in low complexity animal models are able to generate potential lead candidates highly enriched for genuine regulators of the corresponding vertebrate pathway. Knowledge of the potential molecular mechanisms by which these human genes mediate their effects will be important in understanding and ultimately treating the human diseases that arise from the misregulation of this signaling pathway.
Materials and Methods
Selection of homologs
The 90 previously identified Drosophila modulators13 were systematically screened for potential homologs using HomoloGene, Inparanoid and best BLAST homologs parsed from the Flight database20 (http://flight.licr.org).
Cell culture
HeLa SS6 cells (a kind gift from Jens Gruber and Mary Osborn) were maintained in D-MEM [including 4.5 mg/ml glucose, L-glutamine and pyruvate (Gibco)] with 10% heat inactivated fetal bovine serum (PAA) and penicillin-streptomycin (Invitrogen). Cells were grown at 37°C in a humidified incubator with 5% CO2 at subconfluent densities. HeLa cells were stimulated with recombinant human IFN-γ (R&D Systems, 40 ng/ml) or OSM (R&D Systems, 20 ng/ml).
siRNAs
‘Smart pool’ siRNAs (Dharmacon RNA Technologies) targeting the transcripts of each locus (Table 1) consisted of a pool of four independent dsRNAs designed to maximize the chance of effective knockdown while minimizing potential off-target effects. siRNA transfections of HeLa SS6 cells were performed with Oligofectamine (Invitrogen) with 50 nM siRNA in 96-well plates.
Detection of target gene activity
Gene expression levels in human cells were quantitatively measured using either a branched DNA assay (QuantiGene, Panomics) or quantitative PCR (qPCR) approaches, both of which gave comparable results. HeLa cells were grown to confluence in 96-well plates and lysed in 100 µl proprietary lysis buffer. Seventy microliters of lysate were used for determination of all mRNA levels except for β-ACTIN, where only 10 µl were used. Mixed probe sets were added to the lysates according to the manufacturer’s instructions and hybridized in sealed capture plates. Following overnight hybridization at 56°C, capture plates were washed before incubation with 100 µl per well label extender for 1 h at 56°C. After further washing, plates were incubated with 100 µl per well amplifier for 1 h at 56°C, washed three times and finally incubated with 100 µl per well substrate for 30 min. Plates were cooled down at room temperature for 10 min, and luminescence was detected for 0.2 sec per well on a luminometer (Wallac Victor Light 1420 Luminescence Counter, PerkinElmer).
HeLa cells were plated at a density of 5 × 103 cells per well in a 96-well plate. Next day, these cells were treated with 50 nm concentration of different siRNAs. After 3 d, the cells were than treated with human IFN-γ (R&D Systems, 40 ng/ml) or OSM (R&D Systems, 20 ng/ml) for 6 h, lysed in 100 μl of lysis buffer according to the manufacturer’s instructions (Qiagen RNeasy Mini kit). Total RNA was prepared and first strand cDNA was synthesized using First Strand Synthesis Verso cDNA kit (ABgene).
Real-time PCR was performed in 96-well plates using ABsolute QPCR ROX mix and a GeneAmp 7700 sequence detector (Applied Biosystems) and analyzed by ΔΔCT.GBP1 and SOCS3 levels were normalized to β-actin levels. Primers and probes were designed using Primer Express Software (Applied Biosystems). BLAST searches (www.ncbi.nlm.nih.gov) were performed to confirm specificity of the nucleotide sequences. Primers and probes were synthesized by Sigma Aldrich and were from 5′ to 3′ as follows: For GBP1: GCCAGGCCACATCCTAGTTCT and GGCGAAGATCCAGGAGTCATT, probe TGGACACCGAGGGTCTGGGAGATGT. For SOCS3: AGCTGGTCTCCTTTTCCTACTCATACTA and GGTGAAAGATGTCCCGTCTCC, probe TGGGTGGATGGAGCGGGAGGA and for β-actin: ATCATTGCTCCTCCTGAGCG and GACAGCGAGGCCAGGATG, probe TACTCCGTGTGGATCGGCGGCT.
Detection of STAT post-translational modifications
HeLa cells were lysed in ‘Mammalian Cell Lysis Buffer’ (Perbio) and normalized levels of protein were protein gel blotted using standard protocols. Primary antibodies against human β-ACTIN, STAT1, pSTAT1, STAT3, pSTAT3, STAT5 and pSTAT5 were purchased from Cell Signaling Technologies and used at a 1:1000 dilution. The activity of HRP-conjugated secondary antibodies (Jackson Labs) was determined using ECL protein gel blotting substrate (Pierce). Chemiluminescence was detected using either film or a Luminescent Image Analyzer LAS-1000 (FUJIFILM) and Intelligent Dark Box II (FUJIFILM).
Quantitative protein gel blot analysis
To quantitatively determine the fold change in STAT and pSTAT after siRNA treatment, bands were identified by thresholding a region of interest comprising all bands using the “Triangle” algorithm34 that is based on normalization of height and dynamic range of the image intensity histogram, as implemented in ImageJ.35 For STAT3 and pSTAT3 protein gel blots and their corresponding β-ACTIN controls, thresholds determined using the “Triangle” algorithm were used directly, whereas for STAT1 and pSTAT1 protein gel blots and their corresponding β-ACTIN controls, 15 a.u. were subtracted from the threshold determined by the Triangle algorithm due to lower signal intensities. All pixels with intensities above the threshold were assigned a value of 1, and all pixels with intensities below the threshold were assigned a value of 0. The original image was divided by this mask to prevent pixels with intensities below the threshold from being considered in further calculations. The average intensity in a smaller region of interest around individual bands was calculated by summing the intensities of the identified pixels (i.e., the total intensity of the band) and dividing this sum by the number of pixels (i.e., the area occupied by the band). We then calculated the ratio of STAT and pSTAT protein levels relative to β-ACTIN levels and normalized these values to those from control samples on the same protein gel blot to determine fold change.
Acknowledgments
The authors wish to thank Jens Gruber, Mary Osborn and Marysia Placzek for reagents and access to equipment as well as Herbert Jäeckle for support. Sabine Häder, Iris Plischke, Cristiana Hentea, Anna Lena Ernst and Kirsty Johnstone provided technical assistance. Financial support to M.Z. was provided by the Deutsche Forschungsgemeinschaft, Cancer Research UK and a Max Planck Society Verfügungsrahmen für Technologie-Transfer-Vorhaben award. D.P. was supported by a Cancer Research UK postdoctoral position and P.M. by the German National Academic Foundation (Studienstiftung). M.Z. is a Senior Cancer Research Fellow of Cancer Research-UK and a member of the MRC Centre for Developmental and Biomedical Genetics.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Footnotes
Previously published online: www.landesbioscience.com/journals/jak-stat/article/18006
References
- 1.Pires-daSilva A, Sommer RJ. The evolution of signalling pathways in animal development. Nat Rev Genet. 2003;4:39–49. doi: 10.1038/nrg977. [DOI] [PubMed] [Google Scholar]
- 2.Levy DE, Darnell JE., Jr Stats: transcriptional control and biological impact. Nat Rev Mol Cell Biol. 2002;3:651–62. doi: 10.1038/nrm909. [DOI] [PubMed] [Google Scholar]
- 3.Yamaoka K, Saharinen P, Pesu M, Holt VE, 3rd, Silvennoinen O, O'Shea JJ. The Janus kinases (Jaks) Genome Biol. 2004;5:253. doi: 10.1186/gb-2004-5-12-253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schindler C, Darnell JE., Jr Transcriptional responses to polypeptide ligands: the JAK-STAT pathway. Annu Rev Biochem. 1995;64:621–52. doi: 10.1146/annurev.bi.64.070195.003201. [DOI] [PubMed] [Google Scholar]
- 5.Ehret GB, Reichenbach P, Schindler U, Horvath CM, Fritz S, Nabholz M, et al. DNA binding specificity of different STAT proteins. Comparison of in vitro specificity with natural target sites. J Biol Chem. 2001;276:6675–88. doi: 10.1074/jbc.M001748200. [DOI] [PubMed] [Google Scholar]
- 6.Shankaran V, Ikeda H, Bruce AT, White JM, Swanson PE, Old LJ, et al. IFNgamma and lymphocytes prevent primary tumour development and shape tumour immunogenicity. Nature. 2001;410:1107–11. doi: 10.1038/35074122. [DOI] [PubMed] [Google Scholar]
- 7.Durbin JE, Hackenmiller R, Simon MC, Levy DE. Targeted disruption of the mouse Stat1 gene results in compromised innate immunity to viral disease. Cell. 1996;84:443–50. doi: 10.1016/S0092-8674(00)81289-1. [DOI] [PubMed] [Google Scholar]
- 8.Bowman T, Garcia R, Turkson J, Jove R. STATs in oncogenesis. Oncogene. 2000;19:2474–88. doi: 10.1038/sj.onc.1203527. [DOI] [PubMed] [Google Scholar]
- 9.Minamoto S, Ikegame K, Ueno K, Narazaki M, Naka T, Yamamoto H, et al. Cloning and functional analysis of new members of STAT induced STAT inhibitor (SSI) family: SSI-2 and SSI-3. Biochem Biophys Res Commun. 1997;237:79–83. doi: 10.1006/bbrc.1997.7080. [DOI] [PubMed] [Google Scholar]
- 10.Mukherjee T, Hombria JC, Zeidler MP. Opposing roles for Drosophila JAK/STAT signalling during cellular proliferation. Oncogene. 2005;24:2503–11. doi: 10.1038/sj.onc.1208487. [DOI] [PubMed] [Google Scholar]
- 11.Nicola NA, Greenhalgh CJ. The suppressors of cytokine signaling (SOCS) proteins: important feedback inhibitors of cytokine action. Exp Hematol. 2000;28:1105–12. doi: 10.1016/S0301-472X(00)00525-7. [DOI] [PubMed] [Google Scholar]
- 12.Wormald S, Hilton DJ. Inhibitors of cytokine signal transduction. J Biol Chem. 2004;279:821–4. doi: 10.1074/jbc.R300030200. [DOI] [PubMed] [Google Scholar]
- 13.Müller P, Kuttenkeuler D, Gesellchen V, Zeidler MP, Boutros M. Identification of JAK/STAT signalling components by genome-wide RNA interference. Nature. 2005;436:871–5. doi: 10.1038/nature03869. [DOI] [PubMed] [Google Scholar]
- 14.Decker T, Kovarik P. Transcription factor activity of STAT proteins: structural requirements and regulation by phosphorylation and interacting proteins. Cell Mol Life Sci. 1999;55:1535–46. doi: 10.1007/s000180050393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Decker T, Kovarik P. Serine phosphorylation of STATs. Oncogene. 2000;19:2628–37. doi: 10.1038/sj.onc.1203481. [DOI] [PubMed] [Google Scholar]
- 16.Karsten P, Plischke I, Perrimon N, Zeidler MP. Mutational analysis reveals separable DNA binding and trans-activation of Drosophila STAT92E. Cell Signal. 2006;18:819–29. doi: 10.1016/j.cellsig.2005.07.006. [DOI] [PubMed] [Google Scholar]
- 17.Jankovic DL, Gibert M, Baran D, Ohara J, Paul WE, Theze J. Activation by IL-2, but not IL-4, up-regulates the expression of the p55 subunit of the IL-2 receptor on IL-2- and IL-4-dependent T cell lines. J Immunol. 1989;142:3113–20. [PubMed] [Google Scholar]
- 18.Jensen J, Galsgaard ED, Karlsen AE, Lee YC, Nielsen JH. STAT5 activation by human GH protects insulin-producing cells against interleukin-1beta, interferon-gamma and tumour necrosis factor-alpha-induced apoptosis independent of nitric oxide production. J Endocrinol. 2005;187:25–36. doi: 10.1677/joe.1.06086. [DOI] [PubMed] [Google Scholar]
- 19.Wellbrock C, Weisser C, Hassel JC, Fischer P, Becker J, Vetter CS, et al. STAT5 contributes to interferon resistance of melanoma cells. Curr Biol. 2005;15:1629–39. doi: 10.1016/j.cub.2005.08.036. [DOI] [PubMed] [Google Scholar]
- 20.Sims D, Bursteinas B, Gao Q, Zvelebil M, Baum B. FLIGHT: database and tools for the integration and cross-correlation of large-scale RNAi phenotypic datasets. Nucleic Acids Res. 2006;34:D479–83. doi: 10.1093/nar/gkj038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Birmingham A, Anderson EM, Reynolds A, Ilsley-Tyree D, Leake D, Fedorov Y, et al. 3′ UTR seed matches, but not overall identity, are associated with RNAi off-targets. Nat Methods. 2006;3:199–204. doi: 10.1038/nmeth854. [DOI] [PubMed] [Google Scholar]
- 22.Pereira-Leal JB, Seabra MC. Evolution of the Rab family of small GTP-binding proteins. J Mol Biol. 2001;313:889–901. doi: 10.1006/jmbi.2001.5072. [DOI] [PubMed] [Google Scholar]
- 23.Bucci C, Lutcke A, Steele-Mortimer O, Olkkonen VM, Dupree P, Chiariello M, et al. Co-operative regulation of endocytosis by three Rab5 isoforms. FEBS Lett. 1995;366:65–71. doi: 10.1016/0014-5793(95)00477-Q. [DOI] [PubMed] [Google Scholar]
- 24.Marchetti M, Monier MN, Fradagrada A, Mitchell K, Baychelier F, Eid P, et al. Stat-mediated signaling induced by type I and type II interferons (IFNs) is differentially controlled through lipid microdomain association and clathrin-dependent endocytosis of IFN receptors. Mol Biol Cell. 2006;17:2896–909. doi: 10.1091/mbc.E06-01-0076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hwang CS, Shemorry A, Auerbach D, Varshavsky A. The N-end rule pathway is mediated by a complex of the RING-type Ubr1 and HECT-type Ufd4 ubiquitin ligases. Nat Cell Biol. 2010;12:1177–85. doi: 10.1038/ncb2121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shi S, Larson K, Guo D, Lim SJ, Dutta P, Yan SJ, et al. Drosophila STAT is required for directly maintaining HP1 localization and heterochromatin stability. Nat Cell Biol. 2008;10:489–96. doi: 10.1038/ncb1713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yan SJ, Lim SJ, Shi S, Dutta P, Li WX. Unphosphorylated STAT and heterochromatin protect genome stability. FASEB J. 2011;25:232–41. doi: 10.1096/fj.10-169367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Griffiths DS, Li J, Dawson MA, Trotter MW, Cheng YH, Smith AM, et al. LIF-independent JAK signalling to chromatin in embryonic stem cells uncovered from an adult stem cell disease. Nat Cell Biol. 2011;13:13–21. doi: 10.1038/ncb2135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Orphanides G, Wu WH, Lane WS, Hampsey M, Reinberg D. The chromatin-specific transcription elongation factor FACT comprises human SPT16 and SSRP1 proteins. Nature. 1999;400:284–8. doi: 10.1038/22350. [DOI] [PubMed] [Google Scholar]
- 30.Nicolas E, Morales V, Magnaghi-Jaulin L, Harel-Bellan A, Richard-Foy H, Trouche D. RbAp48 belongs to the histone deacetylase complex that associates with the retinoblastoma protein. J Biol Chem. 2000;275:9797–804. doi: 10.1074/jbc.275.13.9797. [DOI] [PubMed] [Google Scholar]
- 31.Dou Y, Milne TA, Ruthenburg AJ, Lee S, Lee JW, Verdine GL, et al. Regulation of MLL1 H3K4 methyltransferase activity by its core components. Nat Struct Mol Biol. 2006;13:713–9. doi: 10.1038/nsmb1128. [DOI] [PubMed] [Google Scholar]
- 32.Zhong Z, Wen Z, Darnell JE., Jr Stat3: a STAT family member activated by tyrosine phosphorylation in response to epidermal growth factor and interleukin-6. Science. 1994;264:95–8. doi: 10.1126/science.8140422. [DOI] [PubMed] [Google Scholar]
- 33.Shuai K, Stark GR, Kerr IM, Darnell JE., Jr A single phosphotyrosine residue of Stat91 required for gene activation by interferon-gamma. Science. 1993;261:1744–6. doi: 10.1126/science.7690989. [DOI] [PubMed] [Google Scholar]
- 34.Zack GW, Rogers WE, Latt SA. Automatic measurement of sister chromatid exchange frequency. J Histochem Cytochem. 1977;25:741–53. doi: 10.1177/25.7.70454. [DOI] [PubMed] [Google Scholar]
- 35.Abramoff MD, Magalhaes PJ, Ram SJ. Image Processing with ImageJ. Biophotonics International. 2004;11:36–42. [Google Scholar]