

Abstract
Introduction
Cystathionine gamma-lyase (CSE) produces H2S via enzymatic conversion of L-cysteine and plays a critical role in cardiovascular homeostasis. We investigated the effects of genetic modulation of CSE and exogenous H2S therapy in the setting of pressure overload-induced heart failure.
Methods and Results
Transverse aortic constriction (TAC) was performed in wild-type (WT), CSE knockout (KO), and cardiac specific CSE transgenic (CS-CSE Tg) mice. In addition, C57BL/6J or CSE KO mice received a novel–H2S donor (SG-1002). Mice were followed for 12 weeks using echocardiography. We observed a >60% reduction in myocardial and circulating H2S levels following TAC. CSE KO mice exhibited cardiac dilatation and dysfunction significantly greater than WT mice following TAC and CS-CSE Tg mice maintained cardiac structure and function following TAC. H2S therapy with SG-1002 resulted in cardioprotection during TAC via upregulation of the VEGF-Akt-eNOS-nitric oxide-cGMP pathway with preserved mitochondrial function, attenuated oxidative stress, and increased myocardial vascular density.
Conclusions
Our results demonstrate that H2S levels are decreased in mice in the setting of heart failure. Moreover, CSE plays a critical role in the preservation of cardiac function in heart failure and oral H2S therapy prevents the transition from compensated to decompensated heart failure in part via upregulation of endothelial nitric oxide synthase (eNOS) and increased NO bioavailability.
Keywords: nitric oxide (NO), cyclic guanosine monophosphate (cGMP), vascular endothelial growth factor (VEGF), left ventricular function, hypertrophy
Introduction
Cardiac hypertrophy is independently related to cardiovascular events and death.1 Pressure overload initially induces hypertrophy to preserve cardiac function (compensated cardiac hypertrophy), while sustained pressure overload and pathological cardiac hypertrophy lead to cardiac remodeling with cardiac dilatation and loss of contractile function.2
Historically, hydrogen sulfide (H2S) has only been considered as a poisonous gas that contributes to morbidity and mortality in various industrial settings.3 However, H2S has recently emerged as a critical physiological gaseous signaling molecule that is produced enzymatically in all mammalian species at low micromolar levels via the action of cysteine metabolic enzymes: cystathionine-γ-lyase (CSE), cystathionine-β-synthase (CBS), and 3-mercaptopyruvate sulfutransferase (3-MST).4 Recently, our laboratory has demonstrated that H2S protects against acute myocardial ischemia/reperfusion (I/R) injury via anti-apoptotic effects mediated by PI3K/Akt, PKC and ERK 1/2 pathway as well as antioxidant actions via the activation and translocation of Nrf-2 to the nucleus including an increase in antioxidant response element-related antioxidants.5 Moreover, previous experimental studies suggest that H2S augments angiogenesis under ischemic conditions both in vitro and in vivo.6-8 H2S also exhibits potent anti-inflammatory actions9 and modulates mitochondrial respiration in part by reversible inhibition of cytochrome c oxidase.10
Recent studies provide strong evidence that H2S derived from CSE modulates cardioprotection in the setting of myocardial I/R injury. Specifically, cardiac-restricted overexpression of CSE results in increased H2S bioavailability and cardioprotection in response to both acute myocardial I/R injury10 and ischemia-induced heart failure.11 In contrast, genetic deficiency of CSE significantly attenuates H2S, bioavailability and results in exacerbated myocardial I/R injury.12,13 Recent clinical evidence suggests that total plasma sulfide is negatively related to severity of congestive heart failure and low plasma sulfide predicts a higher mortality.14 The precise role of H2S in the pathogenesis of pressure-induced cardiac hypertrophy has not yet been established. Specifically, it is currently unknown if myocardial or circulating levels of H2S are altered and what if any role CSE plays in the progression of cardiac hypertrophy and heart failure in the setting of transverse aortic constriction (TAC).
To clarify these issues, we measured myocardial and blood levels of free H2S and the H2S metabolite, sulfane sulfur, in a murine model of pressure overload-induced cardiac hypertrophy and failure. In addition, we also investigated the effects of both genetic deficiency and overexpression of CSE as well as the effects of exogenous H2S therapy on cardiac pathology in the setting of TAC-induced heart failure.
Methods
Experimental Animals
CSE deficient (KO) mice (C57/Sv129 background) and cardiac restricted (αMHC) CSE Tg mice (C57BL/6J background) were developed as described.10,12 Male C57BL/6J mice and endothelial nitric oxide synthase deficient mice (eNOS KO) were purchased from The Jackson Laboratory (Bar Harbor, ME) at 8-10 weeks of age. All experimental protocols were approved by the Institute for Animal Care and Use Committee at Emory University School of Medicine and conformed to the Guide for the Care and Use of Laboratory Animals, published by the National Institutes of Health (NIH Publication No. 86-23, revised 1996), and with federal and state regulations.
Transverse Aortic Constriction (TAC) Protocol
To create pressure overload, the thoracic aorta was tied between the right inominate and the left carotid arteries against a 27-gauge needle with 7-0 silk suture followed by removal of the needle. Echocardiography was performed using a VisualSonics Vevo®-2100 ultrasound system as previously described.15
Hydrogen Sulfide Donor
A recently developed, orally active H2S releasing compound (SG-1002) was provided by Sulfagenix (Cleveland, OH). See below for chemical structure and description. SG-1002 was administered in the diet to mice to achieve dosages of 20 mg/kg/day in C57BL/6J mice or 40 mg/kg/day in CSE KO mice at one week prior to TAC procedure and was continued up to 12 weeks following TAC. In addition, some C57BL/6J mice receiving SG-1002 diet were placed on the control diet at 1 week or 3 weeks following TAC.
Measurement of Hydrogen Sulfide and Sulfane Sulfur
Hydrogen sulfide and sulfane sulfur levels were measured in heart and blood according to previously described methods.16
Western Blot Analysis
Western blot analysis was performed as described previously.15
Myocardial Measurement of NO Metabolites
Nitrite (NO2-) analysis of cardiac tissue was performed as previously described.15
Serum Measurements of VEGF and BNP
Serum levels of VEGF (VEGF ELISA kit, R&D Systems) and brain natriuretic peptide (BNP) (BNP EIA kit, Phoenix Pharmaceuticals, Inc.) were determined by ELISA at 6 and/or 12 weeks following TAC.
Cardiac Mitochondrial Respiration Assay
Myocardial mitochondria were isolated and mitochondrial respiratory capacity was assessed as previously described.16
8-Isoprostane Assay
Concentrations of 8-isoprostane in the plasma and heart were determined by 8-isoprostane EIA kit according to manufacture’s instruction (Cayman Chemicals, Michigan).
Cardiac cGMP
Concentrations of cGMP in the heart were determined with a commercially available kit according to manufacture’s instruction (Abcam).
Histology and Immunohistochemistry
Hearts were collected at the indicated times, fixed in 10% buffered formalin, embedded in paraffin stained with Masson’s trichrome and Picrosirius Red (to detect fibrosis). Digital images were analyzed using ImageJ. Immunohistochemistry was performed to visualize vascular density with a commercially available kit (Blood Vessel Staining Kit, Miilipore). Primary antibody against CD31 (abcam; 1:50) was used. Digital images were obtained with a microscope at a magnification of 400X. CD31 positive vessels numbers were counted using ImageJ and vessels number per mm2 was calculated to evaluate the number of vessels per field.
Powder X-Ray Diffraction and Mass Spectrometry
In order to determine the precise chemical structure of SG-1002 we performed a combination of powder X-Ray diffraction and mass spectrometry. Both powder x-ray diffraction and Powder diffraction (XRD) experiments were performed using a Bruker D8 DIFFRAC powder diffractometer (Co K-alpha radiation) with a VANTECH detector in theta-theta mode. The samples were microcrystalline powders. Theta-theta scans were performed with a step width of 0.01 degrees and scan time of 1 s/step. Analysis was done using the Bruker-AXS EVA software package, by comparing the diffraction pattern with known diffraction patterns filed in the PDF-2 2006 database.
For mass spectrometry water was added to the sample and the supernatant was analyzed using static nanospray on a Thermo Scientific LTQ Fourier Transform Ultra mass spectrometer. A nanospray voltage of -1.5 KV was used to ionize the sample. The analysis of the ions was done in the Fourier Transform-Ion Cyclotron Resonance portion of the instrument with the resolution set to 100,000 at 400 m/z. Data was acquired using Xcaliber software from Thermo Scientific.
Statistical analysis
All data are expressed as mean ± SEM. Statistical significance was evaluated using unpaired Student’s t test for comparison between 2 means and ANOVA (1-way or 2-way) for comparison among 3 or more means using Prism 5 (GraphPad Software Inc). For the ANOVA, if a significant result was found, the Tukey (1-way ANOVA) or Bonferroni (2-way ANOVA) test was used as the posthoc analysis. Kaplan-Meier survival curves were compared by use of a log-rank (Mantel-Cox) test. For all data, a value of p<0.05 denotes statistical significance.
Results
Sulfide Levels Decrease After TAC-Induced Heart Failure
We examined the effects of TAC-induced heart failure on the myocardial expression of the three known H2S-producing enzymes, as well as the levels of circulating and myocardial sulfide levels at 6 weeks of TAC. Our analysis revealed that the expression of CBS was unaltered (Figure 1A-B). However, CSE expression was upregulated in the vehicle mice compared to the sham (Figure 1A-B; p<0.001), whereas myocardial 3-MST expression was significantly downregulated compared to Sham levels (Figure 1A-B; p<0.01). Interestingly, free H2S and sulfane sulfur levels were significantly lower in the blood (p<0.01) and heart (p<0.001) of TAC+Vehicle mice when compared to sham-operated, control mice (Figure 1E-H).
Figure 1.
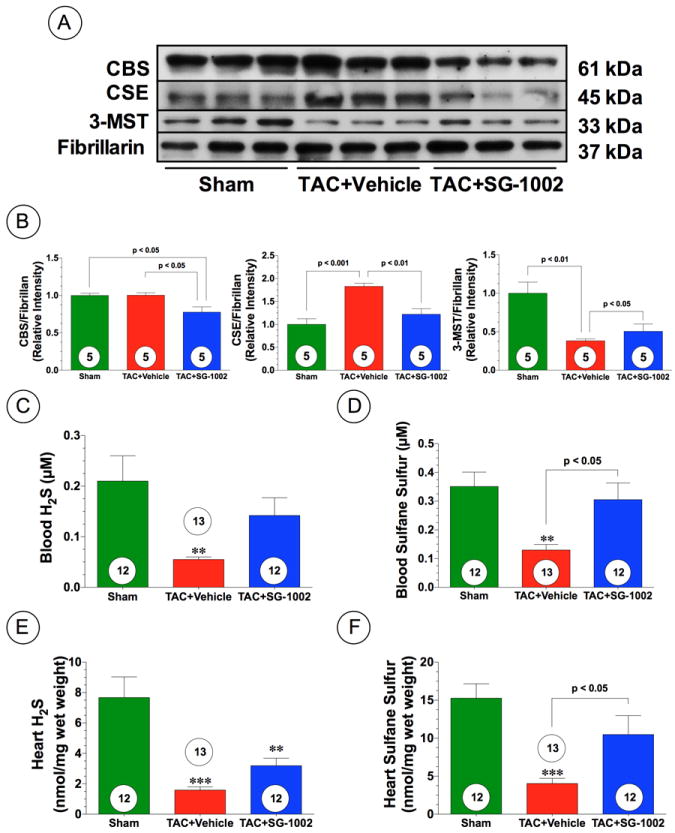
Heart failure reduces sulfide levels in mice. (A-B) Representative immunoblots and densitometric analysis of cystathionine gamma lyase (CSE), cystathionine beta synthase (CBS), and 3-mercaptopyruvate sulfutransferase (3-MST) in the hearts of Sham, TAC+Vehicle, and TAC+SG-1002 treated mice at 6 weeks of TAC. (C-D) Circulating levels of free H2S and sulfane sulfure after 6 weeks of pressure overload-induced heart failure (TAC) in groups of mice maintained on a standard chow (TAC+Vehicle) or maintained on a chow containing the H2S donor SG-1002 (TAC+SG-1002, 20 mg/kg/day). (E-F) Myocardial levels of free H2S and sulfane sulfur in the experimental groups. Results are expressed as mean ± SEM. Numbers in bars represent the sample size. **p<0.01 and ***p<0.001 vs. Sham.
CSE Deficiency Exacerbates Cardiac Dysfunction Following TAC
To investigate the role of endogenous H2S in pressure overload, we performed TAC surgery in CSE KO mice and evaluated cardiac structure and function using echocardiography. Initially, we confirmed that CSE KO mice exhibited lower free H2S and sulfane sulfur levels in the blood and heart compared to WT mice (Supplemental Figure 1; p<0.05). CSE KO mice exhibited significantly greater cardiac enlargement and pulmonary edema at 12 weeks following TAC compared to WT mice (Figure 2A). CSE KO mice exhibited significant LV cavity dilatation, as seen by increases in LVESD and LVEDD and exhibited exacerbated cardiac dysfunction from 3 weeks to 12 weeks following TAC compared to WT mice (Figure 2B-C and Supplemental Figure 2A). Despite the increased cardiac structure and functional changes in the CSE KO mice, no statistically significant difference in the mortality was observed after TAC when compared to the WT mice (Supplemental Figure 3A).
Figure 2.
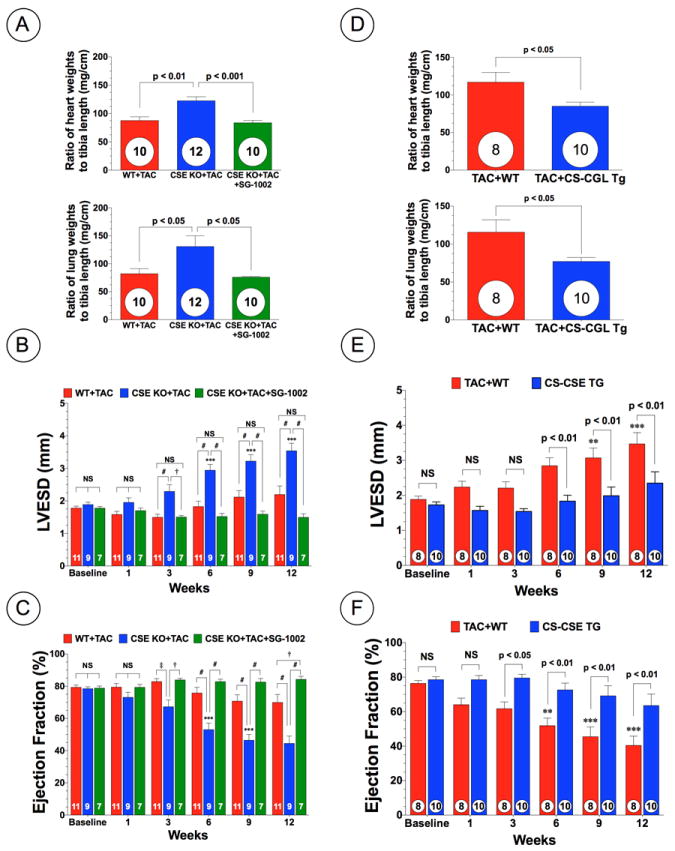
Deficiency of cystathionine gamma lyase (CSE) exacerbates cardiac dysfunction following TAC, whereas overexpression of CSE attenuates cardiac dysfunction. (A) Myocardial weights (mg/cm) and lung weights (mg/cm) expressed as ratio of tibia length at 12 weeks following TAC in wild-type (WT+TAC) mice, CSE deficient (CSE KO+TAC) mice, and CSE KO mice treated with SG1002 (CSE KO+TAC+SG-1002). (B) LV end-systolic diameter (LVESD in mm) and (C) LV ejection fraction (%) following TAC. (D) Myocardial weights (mg/cm) and lung weights (mg/cm) expressed as ratio of tibia length at 12 weeks of TAC in wild-type (WT+TAC) and cardiac specific CSE transgenic mice (CS-CSE Tg+TAC). (E) LVESD and (F) LV ejection fraction from 1 week to 12 weeks of TAC in WT and CS-CSE Tg mice. Results are expressed as mean ± SEM. †p<0.05, ‡p<0.01 and #p<0.001 vs. WT. *p<0.05, **p<.01, and ***p<0.001 vs. Baseline.
Myocardial Overexpression of CSE Attenuates Cardiac Dysfunction without Preventing Cardiac Hypertrophy Following TAC
Overexpression of CSE has been shown to increase H2S production in the heart without alteration in CBS expression.10 We also confirmed no alterations in cardiac CBS expression in CS-CSE Tg mice but CS-CSE Tg mice exhibited less 3-MST expression compared to WT mice (Supplemental Figure 4). We next examined whether overexpression of CSE specifically within the cardiac myocyte would attenuate cardiac hypertrophy and/or dysfunction following TAC using CS-CSE Tg mice. CS-CSE Tg mice exhibited significantly less cardiac enlargement and pulmonary edema, as assessed by the ratio of heart and lung weights to tibia length (mg/cm) when compared to WT controls (Figure 2D). Furthermore, echocardiography analysis revealed that CS-CSE Tg mice exhibited less cardiac dilatation and dysfunction from 6 weeks to 12 weeks following TAC (Figure 2E-F and Supplemental Figure 2B). Again, no statistically significant difference in mortality was observed between the two groups (Supplemental Figure 3B).
Administration of Exogenous H2S Prevents Cardiac Enlargement, Preserves LV Function, and Reduces Fibrosis Following TAC
Next, we examined the effects of administration of oral H2S therapy on pressure overload-induced cardiac hypertrophy and dysfunction in wild-type C57BL/6J mice. For these experiments we administered SG-1002 (20 mg/kg/day) in the chow. SG-1002 (Figure 3A) is a novel, sulfur-containing compound consisting primarily of alpha sulfur (approximately 92%) with lesser amounts of long-chain sodium polythionates (approximately 8%). Our initial studies found that SG-1002 treatment partially restored free H2S and significantly restored sulfane sulfur levels in the blood (Figure 1C-D; p<0.05 vs. TAC+Vehicle) and heart (Figure 1E-F; p<0.05 vs. TAC+Vehicle) following TAC. Gross morphologic analysis at 12 weeks following TAC, revealed that hearts from vehicle mice enlarged to a greater extent compared to SG-1002 treated mice (Figure 3B). This was confirmed by heart weight/tibia length ratios, which found that the hearts of both vehicle and SG-1002 treated mice, were significantly increased compared to Sham mice at 6 and 12 weeks following TAC (Figure 3C; p<0.001). However, SG-1002 treated mice showed significantly less of an increase compared to vehicle mice (p<0.001). In addition, SG-1002 treated mice displayed significantly less pulmonary edema when compared to vehicle mice. (Figure 3D). Moreover, we evaluated circulating BNP levels as an indication of heart failure severity following TAC. BNP levels increased significantly (p<0.01) in vehicle mice at 6 and 12 weeks compared to sham mice, but SG-1002 treatment significantly inhibited BNP (p<0.01 vs. TAC+Vehicle) levels following TAC (Figure 3E). Echocardiography analysis revealed that SG-1002 treatment prevented cardiac dilatation (Figure 3F and Supplemental Figure 2C; p<0.01 vs. TAC+Vehicle) and cardiac contractile dysfunction (Figure 3G; p<0.001 vs. TAC+Vehicle) from 6 weeks to 12 weeks following TAC. Histological analysis of Masson’s Trichrome and Picrosirius Red stained sections at 12 weeks following TAC revealed extensive areas of intermuscular and perivascular fibrosis in hearts from TAC+Vehicle mice (Figure 4; p<0.01 vs. Sham). Although fibrosis was evident in the sections taken from TAC+SG-1002 heart, it was significantly less when compared to the TAC+Vehicle hearts (p<0.001 for Masson’s Trichrome and p<0.01 for Picrosirius Red). Finally, SG-1002 treated mice exhibited improved, but not statistically significant improved survival rate compared to vehicle mice (80% vs. 61%, p=0.23) (Supplemental Figure 3C).
Figure 3.
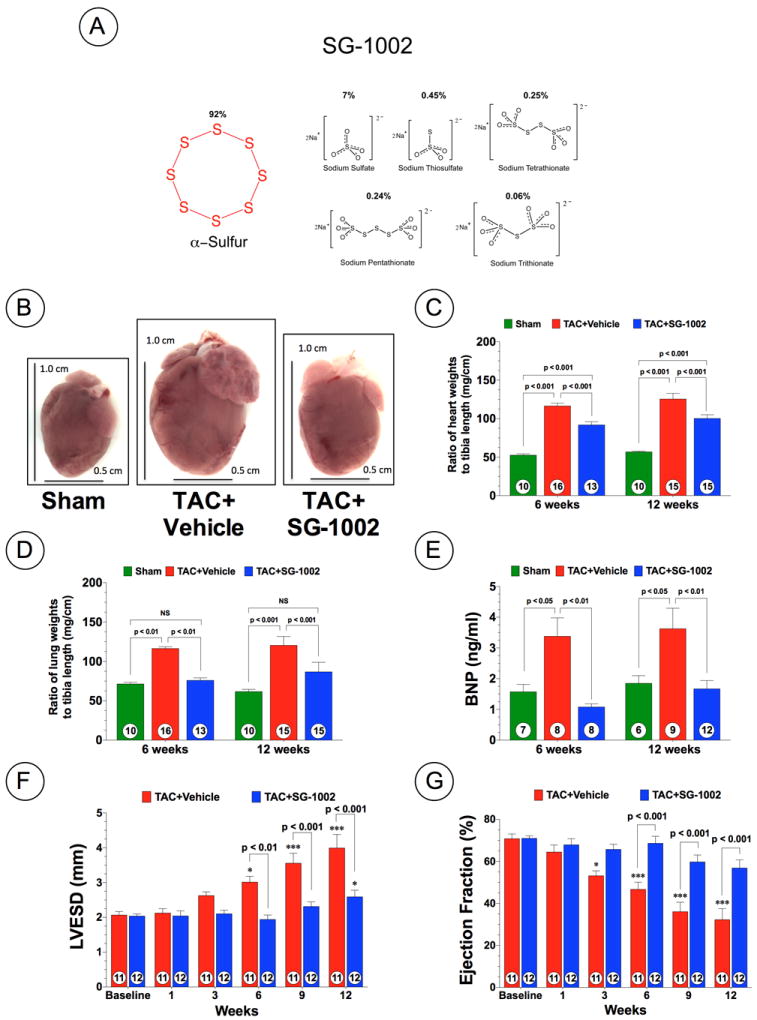
Exogenous H2S therapy prevents cardiac dilatation and dysfunction following TAC. (A) Chemical Structure of SG-1002. (B) Representative heart pictures of Sham, Vehicle (TAC+Vehicle), and SG-1002 (TAC+SG-1002) treated mice at 12 weeks of TAC. (C) The ratio of heart weight to tibia lengths. (D) The ratio of lung weight/tibia lengths. (E) Circulating BNP levels (ng/ml) at 6 and 12 weeks of TAC. (F) LVESD and (H) ejection fraction from 1 week to 12 weeks TAC. Results are expressed as mean ± SEM. *p<0.05 and ***p<0.001 vs. Baseline.
Figure 4.
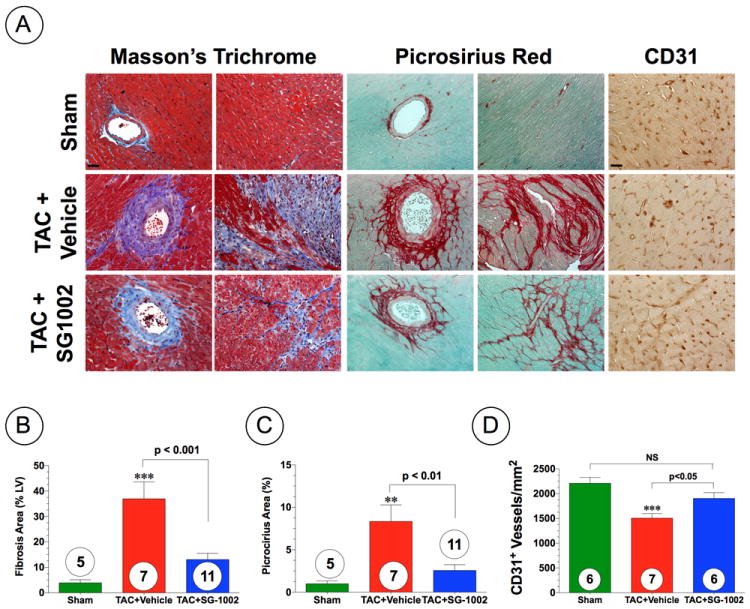
H2S attenuates the intermuscular and perivascular fibrosis following TAC. (A) Representative photomicrographs of Masson’s Trichrome, Picrosirius Red, and CD31 stained heart sections depicting intermuscular and perivascular fibrosis and vascular density in hearts from Sham, TAC+Vehicle, and TAC+SG-1002 treated mice at 6 weeks of TAC. (B) Summary of fibrosis area as % of the LV as calculated from Masson’s Trichrome sections. (C) Summary of fibrosis area as % of the LV calculated from the Picrosirius Red sections. (D) Summary of CD31+ vessels per area (mm2). Results are expressed as mean ± SEM. **p<0.01, and ***p<0.001 vs. Sham.
Further analysis revealed that the administration of SG-1002 to CSE KO mice slightly, but not significantly, increased free H2S levels in the blood and heart, whereas administration of SG-1002 did significantly increase sulfane sulfur levels in both the blood (p<0.001) and the heart (p<0.05) as compared to CSE KO mice fed a control diet (Supplemental Figure 1). The administration of SG-1002 also completely diminished LV cavity dilatation in CSE KO mice when compared to CSE KO mice fed a control diet (Figure 2B; p<0.05). Interestingly, SG-1002 treated CSE KO mice maintained cardiac ejection fraction following TAC as compared to not only control diet-fed CSE KO mice but also WT mice at 12 weeks following TAC (Figure 2C; p<0.001 vs. CSE KO+TAC and p<0.05 vs. WT+TAC). However, no statistically significant difference in mortality was observed between the CSE KO groups (Supplemental Figure 3A).
Together, the results indicate that endogenous H2S bioavailability is markedly attenuated in heart failure following pressure overload even though CSE and CBS expression levels are maintained or upegulated. Moreover, augmentation of H2S levels by genetic or pharmacological approaches prevents the transition from compensated to decompensated cardiac hypertrophy.
Withdrawal of SG-1002 Leads to Development of Cardiac Dilatation and Dysfunction
Experiments were then conducted to determine how withdrawal of SG-1002 from the chow would affect the development of cardiac dilatation and dysfunction after TAC. For these experiments, we administered SG-1002 in the chow for 1 week and then subjected different groups of mice to 6 weeks of TAC: (1) Mice received SG-1002 in the chow for 6 weeks following TAC, (2) Mice received SG-1002 in the chow for 1 week following TAC and then received normal chow for 5 weeks; (3) Mice received SG-1002 in the chow for 3 weeks following TAC and then received normal chow for 3 weeks. Echocardiography analysis revealed that withdrawal of SG-1002 after 1 week of TAC resulted in a larger increase in LVESD and LVEDD, as well as a larger decrease in ejection fraction at 6 weeks of TAC when compared to the non-withdrawal group (Supplemental Figure 5A-C; p<0.01 vs. SG-1002). Withdrawing SG-1002 at 3 weeks of TAC resulted in a non-significant increase in both of these parameters at 6 weeks of TAC when compared to the non-withdrawal group. These data indicate that the withdrawal of SG-1002 early following the onset of pressure-overload does not prevent the development of cardiac dilatation and dysfunction, suggesting that the benefits of SG-1002 are achieved when the diet is maintained throughout the follow-up period.
H2S Therapy Augments VEGF-Akt-eNOS-Nitric Oxide Signaling Following TAC
The serine/threonine kinase Akt regulates cardiac growth, myocardial angiogenesis, and survival in cardiac myocytes.17 We investigated whether SG-1002 treatment activated Akt phosphorylation in the heart following TAC (Figure 5). Representative Western blots for Akt phosphorylation status in the heart at 6 weeks following TAC are shown in Figure 5A. SG-1002 treatment did not alter total Akt expression in the heart (Figure 5B) but did significantly increase the expression of phosphorylated Akt at threonine residue 308 (Akt-PThr308) (p<0.001) and serine residue 473 (Akt-PSer473) when compared to vehicle mice (Figure 5C; p<0.001). We next investigated whether SG-1002 treatment upregulated VEGF, a potent angiogenic and cytoprotective cytokine in the myocardium. At 6 weeks following TAC, SG-1002 treated mice showed significantly greater VEGF protein expression levels in the heart (Figure 5D; p<0.01 vs. Sham and p<0.05 vs. TAC+Vehicle), but not in the systemic circulation (Supplemental Figure 6A). Further analysis revealed that 6 weeks of TAC caused a significant decrease in the number of CD31+ vessels in the hearts of vehicle-treated mice (Figure 4A and D; p<0.001 vs. Sham). However, treatment with SG-1002 significantly attenuated the observed vessel dropout (p<0.05 vs. TAC+Vehicle).
Figure 5.
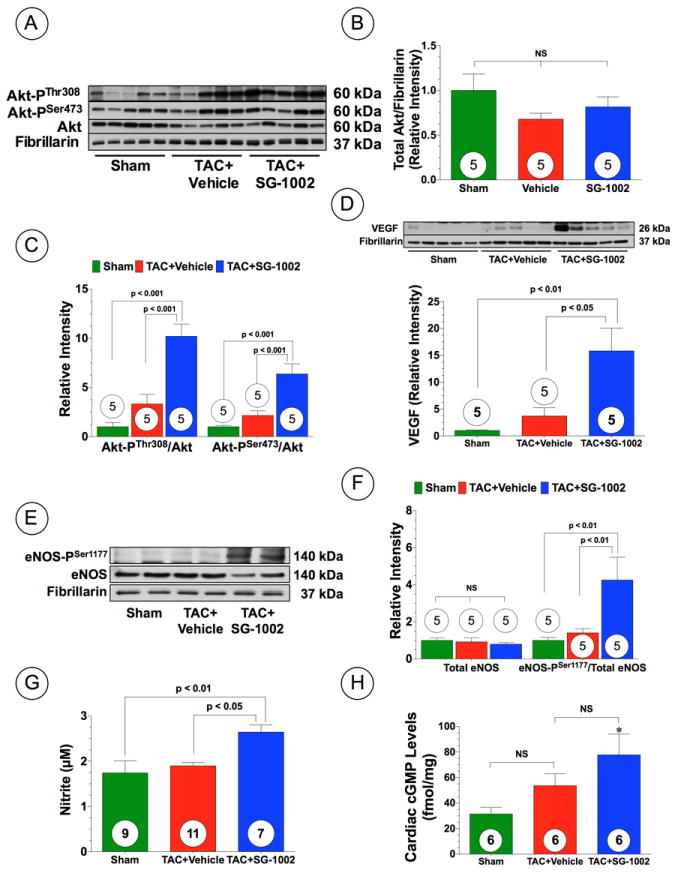
H2S upregulates Akt phosphorylation, VEGF expression, and activates the eNOS-NO pathway following TAC. (A) Representative immunoblots and densitometric analysis of (A-C) total Akt, Akt-PSer473, and Akt-PThr308, (D) VEGF (E-F) total eNOS and eNOS-PSer1177 in hearts from Sham, TAC+Vehicle, and TAC+SG-1002 at 6 weeks of TAC. (G) Nitrite and (H) cGMP levels in the hearts of the experimental groups at 6 weeks of TAC. Results are expressed as mean ± SEM.
Nitric oxide (NO) generated from endothelial nitric oxide synthase (eNOS) is known to promote vascular and myocardial cell cytoprotection during ischemic conditions18. To investigate the potential involvement of eNOS in SG-1002 induced cardioprotection following TAC, the expression and the phosphorylation status of eNOS at serine residue 1177 (eNOS-PSer1177) were assessed by Western blot analysis in the hearts of Sham, vehicle and SG-1002 treated mice. There were no differences in total eNOS expression in the heart among all groups (Figure 5E-F). However, the eNOS activation site (eNOS-PSer1177) exhibited significantly greater phosphorylation following SG-1002 when compared to Sham and TAC+Vehicle mice (Figure 5E-F; p<0.01). Furthermore, SG-1002 treatment increased cardiac nitrite and cGMP levels, following TAC compared to Sham mice (Figure 5G-H; p<0.05), which is indicative of increased NO bioavailability following H2S therapy. We also investigated myocardial expression of both nNOS and iNOS in mice subjected to TAC that received either vehicle or SG-1002 (Supplemental Figure 6B-D). nNOS expression in the both vehicle and SG-1002 treated mice trended to be higher than the Sham, but did not reach statistical significance. Interestingly, iNOS expression in the TAC+Vehicle group was upregulated compared to the Sham group (p<0.01), but SG-1002 mice diminished this upregulation (p < 0.01 vs. TAC+Vehicle). We next investigated if eNOS was critical for the protection afforded by SG-1002. Mice deficient in eNOS (eNOS KO) were subjected to TAC and administered normal chow (eNOS KO+TAC+Vehicle) or SG-1002 (eNOS KO+TAC+SC-1002). Analysis at 6 weeks following TAC, revealed that hearts from both groups of mice were enlarged compared to sham mice, as evidenced by an increase in heart weight/tibia length ratios (Figure 6A) and an increase in circulating BNP levels (Figure 6B). Echocardiography analysis also revealed that both groups of mice displayed cardiac dilatation and cardiac contractile dysfunction from 3 to 6 weeks following TAC (Figure 6C-D and Supplemental Figure 2D; p<0.01 vs. Baseline). Importantly, SG-1002 did not reduce cardiac enlargement, cardiac dilatation, or improve cardiac contractile function, indicating that eNOS is critical for SG-1002 to provide its protective effects.
Figure 6.
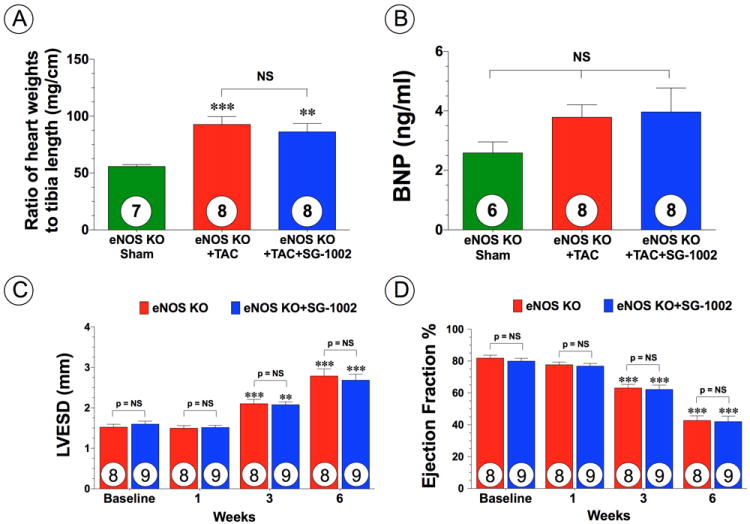
SG-1002 does not provide protection in eNOS deficient mice. (A) Myocardial weights expressed as ratio of tibia length and (B) circulating BNP levels at 6 weeks of TAC in eNOS deficient (eNOS KO) sham, eNOS KO mice subjected to TAC (eNOS KO+TAC) and eNOS KO mice treated with SG-1002 (eNOS KO+TAC+SG-1002). (C) LVESD and (D) ejection fraction from 1 week to 6 weeks TAC. Results are expressed as mean ± SEM. **p<0.01 and ***p<0.001 vs. eNOS KO sham or Baseline.
H2S Therapy Attenuates Mitochondrial Respiratory Dysfunction and Oxidative Stress Following TAC
Mitochondrial energetic failure is considered one of the central pathological mechanisms in heart failure resulting from cardiac hypertrophy19,20. Therefore, we investigated respiratory function of isolated mitochondria obtained from mouse hearts at 6 weeks following TAC. A significant decrease in State 3 respiration rates (Supplemental Figure 7A; p<0.01) and RCR (Supplemental Figure 7B; p<0.001) was observed in the TAC+Vehicle mice compared to the Sham mice. However, SG-1002 treatment preserved mitochondrial respiratory function when compared to TAC+Vehicle mice (p<0.05 for State 3 and p<0.01 for RCR). No difference in State 4 respiration was observed among any of the study groups (Supplemental Figure 7A).
Mitochondrial dysfunction leads to impaired ATP production and increased reactive oxygen species (ROS) generation that can result in increased apoptosis21. We therefore examined 8-isoprostane levels as a marker of antioxidant deficiency and oxidative stress in both the plasma and heart at 6 weeks following TAC. Both the TAC+Vehicle and TAC+SG-1002 treated mice exhibited higher plasma levels of 8-isoprostane compared to sham mice (Supplemental Figure 7C; p<0.05). However, TAC+Vehicle mice exhibited significantly higher 8-isoprostane levels in the heart compared to sham mice (p<0.001), whereas the administration of SG-1002 attenuated the TAC-induced increase in 8-isoprostane levels (Supplemental Figure 7D; p<0.05 vs. TAC+Vehicle). Next, we checked cardiac Nox4 expression as another marker of oxidative stress. At 6 weeks following TAC, myocardial NADPH oxidase 4 (Nox4) expression was significantly upregulated in the TAC+Vehicle mice compared to Sham mice (Supplemental Figure 7E; p<0.01). However, SG-1002 treatment significantly inhibited the upregulation of Nox4 (p<0.01 vs. TAC+Vehicle). Additional analysis revealed that SG-1002 treatment resulted in an upregulation in the expression of the antioxidant heme oxygenase 1 (HO1) in the heart following TAC (Supplemental Figure 7F; p<0.01 vs. Sham and TAC+Vehicle).
Discussion
Previous studies suggest that both exogenous and endogenously derived H2S exhibit potent cytoprotective effects in models of acute myocardial I/R and ischemia-induced heart failure.11,22 However, the role of endogenous H2S in pressure overload-induced heart failure has not been fully elucidated. In the current study, we have identified a number of novel findings regarding the role of CSE-derived H2S on the severity of heart failure following TAC and have also provided important insights into the mechanism by which oral H2S therapy attenuates TAC-induced heart failure.
Recently, a clinical study reported lower circulating sulfide levels in patients suffering from congestive heart failure correlated negatively with the severity of the disease.14 While this a small patient study with a limited sample size, it does provide a preliminary indication that sulfide levels are decreased and may be an important predictor of heart failure severity. The current study provides several lines of evidence to support this idea. First, we provide data that this is mirrored in an experimental model of pressure overload-induced heart failure, as evidenced by the finding that both myocardial and circulating levels of free H2S and sulfane sulfur are significantly reduced following TAC. Second, we have clearly demonstrated that a deficiency in endogenous H2S results in an exacerbation of cardiac dysfunction following TAC, whereas genetic overexpression of CSE and increased H2S bioavailability significantly preserved left ventricular function. Finally, chronic oral administration of a novel sulfur-containing compound that augments H2S levels provides protection against the adverse remodeling associated with TAC by increasing circulating and cardiac sulfide levels. While the mechanisms responsible for the heart failure-induced decline in sulfide levels are currently not known, this finding strongly suggests that a deficiency of H2S may contribute to the pathophysiology and progression of heart failure. These findings also suggest that increasing the bioavailabililty of H2S with an oral H2S donating agent significantly preserves cardiac function in the setting of heart failure.
One of the main findings of the current study is that administration of SG-1002 significantly preserved cardiac function following TAC. Given that H2S is a physiological gas that freely diffuses into multiple intracellular compartments, it can be postulated that H2S targets multiple pathological cascades simultaneously. In this study, we demonstrated that SG-1002 treatment activated a VEGF-Akt-eNOS-NO-cGMP signaling pathway at 6 weeks following the induction of TAC (a time point when cardiac hypertrophy and left ventricular dysfunction are significant). VEGF is a very potent angiogenic and cytoprotective cytokine. Givvimani et al. previously reported that sodium hydrogen sulfide (NaHS) in the drinking water augmented angiogenesis via increasing VEGF expression and inhibition of antiangiogenic factors (angiostatin and endostatin).23 The serine/threonine protein kinase Akt regulates cardiac growth, glucose metabolism, contractile function, and cell death, and is also an important factor for VEGF-mediated angiogenesis.24 In the current study, we found that SG-1002 prevented TAC-induced decrease in CD31+ vessels. Given that VEGF and Akt were increased in the SG-1002-treated hearts, one could argue that it is possible that SG-1002 is inducing angiogenesis.25,26 On the other hand, this observation could be a result of less injury in these hearts. Regardless of the mechanism, the data suggests that SG-1002 prevents vessel dropout after TAC. Therefore, the activation of VEGF and Akt by SG-1002 could contribute to the observed attenuation of cardiac dysfunction by regulating hypertrophy, cell death, and/or vascular density.27,28 Additional studies are certainly warranted to investigate the mechanisms by which SG-1002 are activating VEGF and if SG-1002 induces angiogenesis.
It has been generally thought that H2S and NO exert their biological effects via independent signaling pathways. Recent experimental evidence suggests that there is crosstalk between the H2S and NO signaling pathways, which could provide synergistic and additional regulatory effects. For example, H2S upregulates NO production in endothelial cells through the activation of eNOS in an Akt-dependent manner.29 Likewise, NO has been shown to enhance the production of H2S from vascular tissue30 and more recently, Coletta et al demonstrated that NO and H2S are mutually required for the control of vascular function and angiongenesis.31 Therefore, another major finding of the current study is the evidence that exogenous H2S therapy very potently activates eNOS and increases NO bioavailability within the myocardium. This is important for two reasons: (1) it further corroborates the evidence that there is crosstalk between the H2S and NO signaling pathways under in vivo pathological conditions and (2) provides strong evidence that H2S-mediated cardioprotection is dependent upon at least in part increased bioavailability of NO in an in vivo model of disease. Furthermore, the finding that SG-1002 does not provide protection in mice deficient in eNOS indicates that the activation of eNOS by SG-1002 is required for the observed protective effects against TAC.
In terms of its effects on hypertrophy, NO produced from eNOS has been shown to have antihypertrophic effects in the heart as evidenced by the findings that eNOS KO mice have hypertension and cardiac hypertrophy32 and exhibit exacerbated cardiac dysfunction due to pressure overload induced hypertrophy compared to WT mice.33 Moreover, cardiac specific overexpression of eNOS prevents isoproterenol induced cardiac hypertrophy.34 However, in sharp contrast, Takimoto et al. suggested that pressure overload results in eNOS-uncoupling resulting in increased myocardial oxidant production and exacerbated cardiac function.35 In spite of this, physicians have been successfully using drugs which are able to activate eNOS, (i.e. ACE-I, ARB, and beta-blockers) in the treatment of heart failure.36 Therefore, controversy still remains in regards to the utility and effectiveness of NO-based therapies in the treatment of heart failure, which warrants further investigation to resolve these issues. Additionally, both NO and H2S are known to increase HO-1 levels, an enzyme that produces carbon monoxide (CO).5,37 This suggests that the activation of one of the endogenously produced gases can lead to the activation of the other two. Under these conditions, the three gases have the ability to synergize to produce antiapoptotic, antioxidant, anti-inflammatory, and antihypertrophic effects, which ultimately can lead to cardioprotection.
An increase in oxidative stress and/or a deficiency in the endogenous antioxidant reserve can also cause contractile dysfunction.38 The cardioprotective effects of H2S against myocardial I/R are mediated by antioxidant signaling.5 In addition, H2S directly scavenges reactive oxygen species (ROS) in vitro.39 Therefore, endogenous H2S may directly and/or indirectly contribute to modulation of oxidative stress in the setting of pressure overload-induced hypertrophy. Here, we demonstrate that H2S attenuates the TAC-induced increase in oxidative stress, as evidenced by the finding that SG-1002 decreases cardiac 8-isoprostane levels. In terms of mechanism, we found that SG-1002 attenuates the TAC-induced upregulation of Nox4, a member of the NADPH oxidase family that is a major source of ROS-related cardiac dysfunction in the setting of pressure overload.40 We also found that SG-1002 upregulated the expression of HO-1 and preserved mitochondrial respiratory function. Since, mitochondrial respiratory dysfunction in the heart leads to metabolic remodeling, deficit cardiac energetics, and increased oxidative stress19,41, the preserved mitochondrial respiratory function observed in the current study could be an additional mechanism to explain the inhibition of oxidative stress by H2S following TAC.
The findings of the current study indicate that preserving sulfide levels during the development of pressure overload-induced heart failure preserves cardiac function and prevents the transition from compensated to decompensated cardiac hypertrophy. Furthermore, the current study indicates that administration of a novel oral H2S donor facilitates these protective effects by activating a VEGF-Akt-eNOS-NO-cGMP signaling pathway significantly increasing NO bioavailability in (Figure 7). This cardioprotective signaling cascade ultimately results in the inhibition of oxidative stress, attenuated cardiac fibrosis, prevention of vessel dropout, preservation of mitochondrial respiration, and preserved left ventricular function. Our study suggests that endogenously produced H2S plays an important role in the preservation of cardiac function in heart failure and that oral H2S therapy may be a therapeutic option for the treatment of LV dysfunction in the setting of pressure overload-induced hypertrophy.
Figure 7.
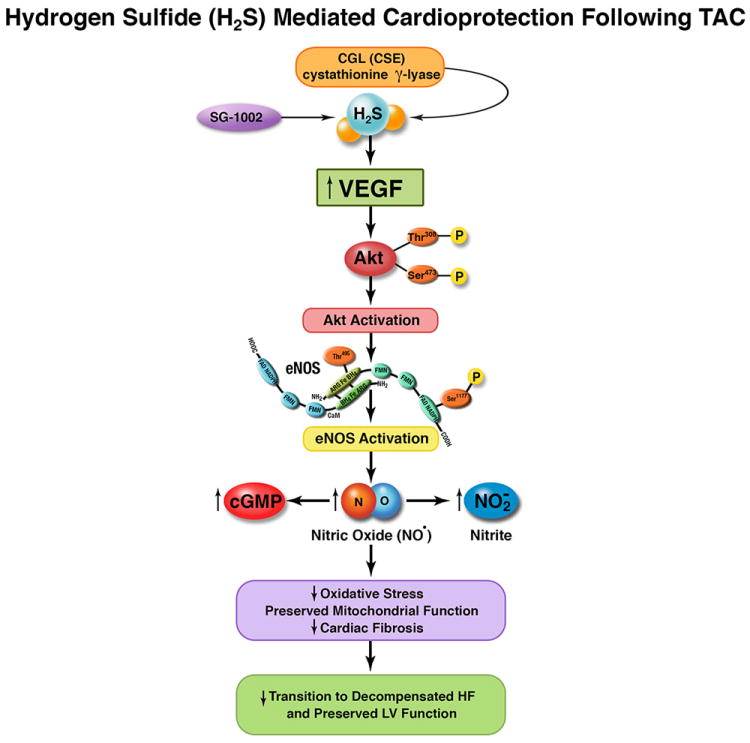
Schematic diagram highlighting the proposed mechanism by which cystathionine gama lyase (CSE) or exogenous hydrogen sulfide protects the heart following transverse aortic constriction (TAC). Our data suggest (CSE) or hydrogen suflide donor therapy with SG-1002 activates vascular endothelial growth factor (VEGF) and subsequently phosphorylates Akt. Akt activation results in phosphorylation and activation of eNOS. Following eNOS activation nitric oxide (NO) and nitrite (NO2) bioavailability are increased in conjunction with increases in myocardial cGMP. These molecular signals result in reduced myocardial oxidative stress and injury, improvements in mitochondrial respiration, and decreased cardiac fibrosis. Ultimately, these cytoprotective actions prevent the transition from compensated to decompensated heart failure and left ventricular (LV) ejection fraction is preserved.
Supplementary Material
Commentary-Clinical Perspective.
Despite decades of research heart failure continues to be a major health problem as evidenced by a rise in the number of hospitalizations for heart failure, the number of deaths attributed to heart failure, and the ever-increasing costs associated with care. Hydrogen sulfide (H2S) is a recently identified endogenous gaseous signaling molecule that modulates diverse physiological signals and protects the myocardium during ischemia/reperfusion. Recent clinical evidence suggests that circulating H2S levels are decreased in patients with heart failure and that the severity of heart failure is inversely correlated with H2S bioavailability. In the present study, we examined the effects of H2S deficiency as well as H2S therapy on the severity of cardiac hypertrophy and heart failure in mice following transverse aortic banding. Mice deficient in a key H2S generating enzyme exhibited exacerbated heart failure while mice with cardiac-restricted overexpression of this enzyme were protected against heart failure. We also demonstrated that a novel H2S donor significantly attenuates adverse left ventricular (LV) modeling and preserves LV function when administered prior to the onset of heart failure. Key actions of H2S involved reductions in oxidative stress and myocardial fibrosis coupled with increased myocardial capillary density. Additional experiments determined that the cardioprotective effects of H2S therapy were mediated via upregulation of endothelial nitric oxide synthase (eNOS) and increased nitric oxide bioavailabilty. Together, these findings further support the emerging concept that H2S therapy may be of clinical importance in the treatment of cardiovascular disease and may have a practical clinical use.
Acknowledgments
We thank David Polhemus and Claire Pearce for their expert technical assistance during the course of these studies. We are also grateful to John W. Elrod, Ph.D. for all of his assistance during the course of these studies.
Funding Sources: This work was supported by grants from the National Heart, Lung, and Blood Institute (National Institutes of Health; 5R01HL092141, 5R01HL093579, 1U24HL 094373, and 1P20HL113452 to D.J.L. and 5R01HL098481 to J.W.C.). This work was also supported by a Discovery Grant from Natural Sciences and Engineering Research Council of Canada to Rui Wang. We are also grateful for the generous funding support from the Carlyle Fraser Heart Center of Emory University Hospital Midtown.
Conflict of Interest Disclosures: David J. Lefer, Ph.D., John W. Elrod, Ph.D., Gabriel Gojon, Sr., Ph.D., and Gabriel Gojon, Jr. are cofounders of Sulfagenix, Inc. Sulfagenix is currently developing hydrogen sulfide based therapeutics for cardiovascular disease states. In addition, Gabriel Gojon, Sr. and Gabriel Gojon, Jr. are inventors on several United States Patent applications for the use of H2S based therapeutics for a variety of disease conditions including cardiovascular diseases.
References
- 1.Levy D, Garrison RJ, Savage DD, Kannel WB, Castelli WP. Prognostic implications of echocardiographically determined left ventricular mass in the framingham heart study. N Engl J Med. 1990;322:1561–1566. doi: 10.1056/NEJM199005313222203. [DOI] [PubMed] [Google Scholar]
- 2.Heineke J, Molkentin JD. Regulation of cardiac hypertrophy by intracellular signalling pathways. Nat Rev Mol Cell Biol. 2006;7:589–600. doi: 10.1038/nrm1983. [DOI] [PubMed] [Google Scholar]
- 3.Ramazzini B. Diseases of workers - de morbis artificum diatriba - 1713. New York: C. Hafner Publishing Co Inc; 1964. [Google Scholar]
- 4.Li L, Rose P, Moore PK. Hydrogen sulfide and cell signaling. Annu Rev Pharmacol Toxicol. 2011;51:169–187. doi: 10.1146/annurev-pharmtox-010510-100505. [DOI] [PubMed] [Google Scholar]
- 5.Calvert JW, Jha S, Gundewar S, Elrod JW, Ramachandran A, Pattillo CB, Kevil CG, Lefer DJ. Hydrogen sulfide mediates cardioprotection through nrf2 signaling. Circ Res. 2009;105:365–374. doi: 10.1161/CIRCRESAHA.109.199919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cai WJ, Wang MJ, Moore PK, Jin HM, Yao T, Zhu YC. The novel proangiogenic effect of hydrogen sulfide is dependent on akt phosphorylation. Cardiovasc Res. 2007;76:29–40. doi: 10.1016/j.cardiores.2007.05.026. [DOI] [PubMed] [Google Scholar]
- 7.McCarthy PJ, Pattillo CB, Hood JL, Lefer DJ, Kevil CG. Abstract: Hydrogen sulfide increases blood flow in a model of peripheral artery disease. Free Radic Biol and Med. 2008;45:S88–S88. [Google Scholar]
- 8.Wang MJ, Cai WJ, Li N, Ding YJ, Chen Y, Zhu YC. The hydrogen sulfide donor NaHS promotes angiogenesis in a rat model of hind limb ischemia. Antioxid Redox Signal. 2010;12:1065–1077. doi: 10.1089/ars.2009.2945. [DOI] [PubMed] [Google Scholar]
- 9.Sodha NR, Clements RT, Feng J, Liu Y, Bianchi C, Horvath EM, Szabo C, Stahl GL, Sellke FW. Hydrogen sulfide therapy attenuates the inflammatory response in a porcine model of myocardial ischemia/reperfusion injury. J Cardiovasc Pharmacol. 2009;138:977–984. doi: 10.1016/j.jtcvs.2008.08.074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Elrod JW, Calvert JW, Morrison J, Doeller JE, Kraus DW, Tao L, Jiao X, Scalia R, Kiss L, Szabo C, Kimura H, Chow CW, Lefer DJ. Hydrogen sulfide attenuates myocardial ischemia-reperfusion injury by preservation of mitochondrial function. Proc Natl Acad Sci USA. 2007;104:15560–15565. doi: 10.1073/pnas.0705891104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Calvert JW, Elston M, Nicholson CK, Gundewar S, Jha S, Elrod JW, Ramachandran A, Lefer DJ. Genetic and pharmacologic hydrogen sulfide therapy attenuates ischemia-induced heart failure in mice. Circulation. 2010;122:11–19. doi: 10.1161/CIRCULATIONAHA.109.920991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yang G, Wu L, Jiang B, Yang W, Qi J, Cao K, Meng Q, Mustafa AK, Mu W, Zhang S, Snyder SH, Wang R. H2s as a physiologic vasorelaxant: Hypertension in mice with deletion of cystathionine gamma-lyase. Science. 2008;322:587–590. doi: 10.1126/science.1162667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Calvert JW, Coetzee WA, Lefer DJ. Novel insights into hydrogen sulfide--mediated cytoprotection. Antioxid Redox Signal. 2010;12:1203–1217. doi: 10.1089/ars.2009.2882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kovacic D, Glavnik N, Marinsek M, Zagozen P, Rovan K, Goslar T, Mars T, Podbregar M. Total plasma sulfide in congestive heart failure. J Card Fail. 2012;18:541–548. doi: 10.1016/j.cardfail.2012.04.011. [DOI] [PubMed] [Google Scholar]
- 15.Aragon JP, Condit ME, Bhushan S, Predmore BL, Patel SS, Grinsfelder DB, Gundewar S, Jha S, Calvert JW, Barouch LA, Lavu M, Wright HM, Lefer DJ. Beta(3)-adrenoreceptor stimulation ameliorates myocardial ischemia-reperfusion injury via endothelial nitric oxide synthase and neuronal nitric oxide synthase activation. J Am Coll Cardiol. 2011;58:2683–2691. doi: 10.1016/j.jacc.2011.09.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Predmore BL, Kondo K, Bhushan S, Zlatopolsky MA, King AL, Aragon JP, Grinsfelder DB, Condit ME, Lefer DJ. The polysulfide, diallyl trisulfide, protects the ischemic myocardium by preservation of endogenous hydrogen sulfide and increasing nitric oxide bioavailability. Am J Physiol Heart Circ Physiol. 2012;302:H2410–2418. doi: 10.1152/ajpheart.00044.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dimmeler S, Zeiher AM. Akt takes center stage in angiogenesis signaling. Circ Res. 2000;86:4–5. doi: 10.1161/01.res.86.1.4. [DOI] [PubMed] [Google Scholar]
- 18.Calvert JW, Lefer DJ. Clinical translation of nitrite therapy for cardiovascular diseases. Nitric Oxide. 2010;22:91–97. doi: 10.1016/j.niox.2009.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rosca MG, Hoppel CL. Mitochondria in heart failure. Cardiovasc Res. 2010;88:40–50. doi: 10.1093/cvr/cvq240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sharov VG, Todor AV, Silverman N, Goldstein S, Sabbah HN. Abnormal mitochondrial respiration in failed human myocardium. J Mol Cell Cardiol. 2000;32:2361–2367. doi: 10.1006/jmcc.2000.1266. [DOI] [PubMed] [Google Scholar]
- 21.Yu E, Mercer J, Bennett M. Mitochondria in vascular disease. Cardiovasc Res. 2012;95:173–82. doi: 10.1093/cvr/cvs111. [DOI] [PubMed] [Google Scholar]
- 22.Predmore BL, Lefer DJ. Development of hydrogen sulfide-based therapeutics for cardiovascular disease. J Cardiovasc Transl Res. 2010;3:487–498. doi: 10.1007/s12265-010-9201-y. [DOI] [PubMed] [Google Scholar]
- 23.Givvimani S, Munjal C, Gargoum R, Sen U, Tyagi N, Vacek JC, Tyagi SC. Hydrogen sulfide mitigates transition from compensatory hypertrophy to heart failure. J Appl Physiol. 2011;110:1093–1100. doi: 10.1152/japplphysiol.01064.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chaanine AH, Hajjar RJ. Akt signalling in the failing heart. Eur J Heart Fail. 2011;13:825–829. doi: 10.1093/eurjhf/hfr080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Papapetropoulos A, Pyriochou A, Altaany Z, Yang G, Marazioti A, Zhou Z, Jeschke MG, Branski LK, Herndon DN, Wang R, Szabo C. Hydrogen sulfide is an endogenous stimulator of angiogenesis. Proc Natl Acad Sci USA. 2009;106:21972–21977. doi: 10.1073/pnas.0908047106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Szabo C, Papapetropoulos A. Hydrogen sulphide and angiogenesis: Mechanisms and applications. Br J Pharmacol. 2011;164:853–865. doi: 10.1111/j.1476-5381.2010.01191.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shiojima I, Walsh K. Role of akt signaling in vascular homeostasis and angiogenesis. Circ Res. 2002;90:1243–1250. doi: 10.1161/01.res.0000022200.71892.9f. [DOI] [PubMed] [Google Scholar]
- 28.DeBosch B, Treskov I, Lupu TS, Weinheimer C, Kovacs A, Courtois M, Muslin AJ. Akt1 is required for physiological cardiac growth. Circulation. 2006;113:2097–2104. doi: 10.1161/CIRCULATIONAHA.105.595231. [DOI] [PubMed] [Google Scholar]
- 29.Predmore BL, Julian D, Cardounel AJ. Hydrogen sulfide increases nitric oxide production from endothelial cells by an akt-dependent mechanism. Front Physiol. 2011;2:104. doi: 10.3389/fphys.2011.00104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhao W, Zhang J, Lu Y, Wang R. The vasorelaxant effect of h(2)s as a novel endogenous gaseous k(atp) channel opener. EMBO J. 2001;20:6008–6016. doi: 10.1093/emboj/20.21.6008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Coletta C, Papapetropoulos A, Erdelyi K, Olah G, Modis K, Panopoulos P, Asimakopoulou A, Gero D, Sharina I, Martin E, Szabo C. Hydrogen sulfide and nitric oxide are mutually dependent in the regulation of angiogenesis and endothelium-dependent vasorelaxation. Proc Natl Acad Sci USA. 2012;109:9161–9166. doi: 10.1073/pnas.1202916109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Huang PL, Huang Z, Mashimo H, Bloch KD, Moskowitz MA, Bevan JA, Fishman MC. Hypertension in mice lacking the gene for endothelial nitric oxide synthase. Nature. 1995;377:239–242. doi: 10.1038/377239a0. [DOI] [PubMed] [Google Scholar]
- 33.Ichinose F, Bloch KD, Wu JC, Hataishi R, Aretz HT, Picard MH, Scherrer-Crosbie M. Pressure overload-induced lv hypertrophy and dysfunction in mice are exacerbated by congenital nos3 deficiency. Am J Physiol Heart Circ Physiol. 2004;286:H1070–1075. doi: 10.1152/ajpheart.00940.2003. [DOI] [PubMed] [Google Scholar]
- 34.Ozaki M, Kawashima S, Yamashita T, Hirase T, Ohashi Y, Inoue N, Hirata K, Yokoyama M. Overexpression of endothelial nitric oxide synthase attenuates cardiac hypertrophy induced by chronic isoproterenol infusion. Circ J. 2002;66:851–856. doi: 10.1253/circj.66.851. [DOI] [PubMed] [Google Scholar]
- 35.Takimoto E, Champion HC, Li M, Ren S, Rodriguez ER, Tavazzi B, Lazzarino G, Paolocci N, Gabrielson KL, Wang Y, Kass DA. Oxidant stress from nitric oxide synthase-3 uncoupling stimulates cardiac pathologic remodeling from chronic pressure load. J Clin Invest. 2005;115:1221–1231. doi: 10.1172/JCI21968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Clark AL. Almanac 2011: Heart failure. The national society journals present selected research that has driven recent advances in clinical cardiology. Heart. 2011;97:1643–1649. doi: 10.1136/heartjnl-2011-300897. [DOI] [PubMed] [Google Scholar]
- 37.Li Q, Guo Y, Ou Q, Cui C, Wu WJ, Tan W, Zhu X, Lanceta LB, Sanganalmath SK, Dawn B, Shinmura K, Rokosh GD, Wang S, Bolli R. Gene transfer of inducible nitric oxide synthase affords cardioprotection by upregulating heme oxygenase-1 via a nuclear factor-{kappa}b-dependent pathway. Circulation. 2009;120:1222–1230. doi: 10.1161/CIRCULATIONAHA.108.778688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dhalla AK, Hill MF, Singal PK. Role of oxidative stress in transition of hypertrophy to heart failure. J Am Coll Cardiol. 1996;28:506–514. doi: 10.1016/0735-1097(96)00140-4. [DOI] [PubMed] [Google Scholar]
- 39.Whiteman M, Armstrong JS, Chu SH, Jia-Ling S, Wong BS, Cheung NS, Halliwell B, Moore PK. The novel neuromodulator hydrogen sulfide: An endogenous peroxynitrite ‘scavenger’? J Neurochem. 2004;90:765–768. doi: 10.1111/j.1471-4159.2004.02617.x. [DOI] [PubMed] [Google Scholar]
- 40.Kuroda J, Ago T, Matsushima S, Zhai P, Schneider MD, Sadoshima J. Nadph oxidase 4 (nox4) is a major source of oxidative stress in the failing heart. Proc Natl Acad Sci USA. 2010;107:15565–15570. doi: 10.1073/pnas.1002178107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Foo RS, Mani K, Kitsis RN. Death begets failure in the heart. J Clin Invest. 2005;115:565–571. doi: 10.1172/JCI24569. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.