

Abstract
Intracellular pathogens commonly invade and replicate inside of intestinal cells and exit from these cells is a crucial step in pathogen transmission. For convenience, studies of intracellular pathogens are often conducted using in vitro cell culture systems, which unfortunately lack important features of polarized, intact intestinal epithelial cells. The nematode C. elegans provides a tractable system to study intracellular pathogens in vivo, where features of differentiated epithelial cells are easily visualized. In a recent paper, we used C. elegans as a host organism to study the exit strategy of Nematocida parisii, a naturally occurring intracellular pathogen in the microsporidia phylum. We showed that N. parisii remodels the C. elegans host cytoskeleton, and then exits host cells in an actin-dependent, non-lytic fashion. These findings illuminate key details about the transmission of microsporidia, which are poorly understood but ubiquitous pathogens. More generally, these findings have implications for exit strategies used by other intracellular pathogens that also infect epithelial cells.
Keywords: C. elegans, actin, host cell exit, host-pathogen interactions, intermediate filaments, intestinal infection, intracellular pathogen, microsporidia, terminal web
Introduction
By residing inside of host cells, intracellular pathogens have the advantages of protection from extracellular antimicrobials and access to host nutrients and energy stores. However, intracellular replication requires that pathogens eventually exit from their host cell in order to spread to new cells or to new hosts. While some intracellular pathogens such as Toxoplasma gondii, Leishmania species and Plasmodium falciparum exit by lysing host cells (Fig. 1A), other intracellular pathogens have evolved strategies that maintain host cell integrity during exit.4 Understanding the mechanisms of host cell exit could aid in the development of treatments to prevent intracellular pathogen transmission. In a recent study, we showed that microsporidia, a phylum of pathogens closely related to fungi, exits the C. elegans gut epithelia in a non-lytic manner involving actin.5
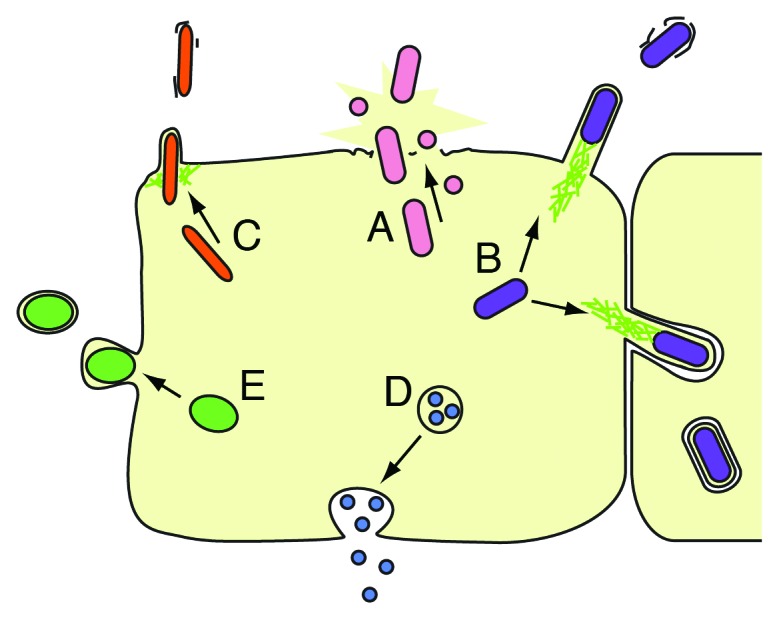
Figure 1. Exit strategies of intracellular pathogens. (A) Lytic exit from host cells through activation of “pyroptosis,” secretion of membrane pore-forming toxins, or secretion of proteases. (e.g., S. flexneri, Salmonella spp, L. pneumophila, L. monocytogenes, F. tularensis, Chlamydia spp, P. falciparum, Leishmania spp, T. gondii). (B) Exit by actin ‘comet tails,’ which protrude into host membrane to induce engulfment by neighboring cells, sometimes resulting in a double membrane (used by L. monocytogenes, S. flexneri, R. rickettsii, R. conorii, Burkholderia, Pseudomallei). (C) Exit through an actin-rich, pore-like “ejectosome” that is inserted in the host membrane (used by M. marinum1). (D) Exit by exocytosis (used by C. neoformans, C. albicans2). (E) Exit by budding out of the host cell coated in host membrane, leaving the host cell intact (used by Chlamydia spp, O. tsutsugamushi, P. berghei3) (adapted with permission from Hybiske and Stephens, 20084).
Actin and pathogen egress
Many intracellular pathogens exploit host actin in order to exit.6 For example, Listeria and Rickettsiae species both secrete actin-nucleation promoting factors that interact with host Arp2/3 actin polymerization machinery. Actin polymerization forms comet-like actin “tails” on the pathogens that are used for motility and jetting between and exiting from host cells. Shigella species exit host cells by activating formin-mediated actin polymerization to generate protrusions from the host cell, which are engulfed by neighboring cells to facilitate pathogen exit and spreading (Fig. 1B).7 Another actin-dependent exit strategy is employed by Mycobacterium species and involves an actin-rich structure called an “ejectosome,” which is a pore-like structure in the host membrane through which pathogens can exit the host cell (Fig. 1C).1 In addition to the above-mentioned bacteria, the eukaryotic pathogen C. neoformans also uses actin to non-lytically exit from host cells. The exit process used by C. neoformans, as well as C. albicans (another pathogenic yeast), appears similar to exocytosis, although the underlying mechanisms are poorly understood (Fig. 1D).2,8,9 A summary of these five major pathogen exit mechanisms is illustrated in Figure 1. Based on the prevalence of actin-related exit strategies used by the diverse microbes shown in this figure, host actin appears to be an ideal resource exploited by pathogens to facilitate exit.6
The lysis or preservation of host membrane can have important implications for virulence of the exiting pathogen. In particular, a pathogen that lyses its host cell inflicts damage on its host, thus harming the host’s ability to support future pathogen growth. In the case of organisms with non-renewing tissues, such as C. elegans, and especially with unicellular organisms, cell lysis can have a substantial impact on host survival. It has been hypothesized that over time intracellular pathogens evolve reduced virulence in order to maximize replication.10,11 Thus, it would be expected that pathogens that have coevolved with their hosts would trend toward non-lytic exit strategies.
Microsporidia are natural pathogens of C. elegans and provide a convenient in vivo system for the study of intracellular pathogens
Most studies of intracellular pathogen exit have been conducted in tissue culture cells or in unicellular hosts that lack important features of in vivo metazoan tissue structure. As such, findings in tissue culture may differ from findings in vivo, as exemplified by a recent study of Listeria infection by Nikitas et al.12 The authors performed microscopy of whole-tissue mounts to characterize Listeria intracellular trafficking through intestinal cells. Surprisingly, they found that in vivo trafficking differs substantially from the well-studied in vitro pathway mentioned above, in which Listeria escapes from the internalization vacuole and then induces actin tail polymerization to force its way into new host cells.13-16 In vivo, the authors found that Listeria remains membrane-bound as it transits from the apical to the basolateral side of intestinal epithelial cells, and then exits via exocytosis at the basolateral side of cells to disseminate systematically.12 Interestingly, this in vivo transcytosis pathway does not require the well-described Listeria factors identified by in vitro studies. These contrasting results from in vitro vs. in vivo studies highlight the importance of studying intestinal infections in vivo.
A key feature of metazoan intestinal epithelial cells is their apical-basolateral polarity, which is not necessarily maintained in in vitro studies.17 The apical surface of these cells is decorated with actin-rich microvilli that protrude into the intestinal lumen where they can absorb nutrients. These microvilli are anchored into a cytoskeletal structure called the terminal web (Fig. 2A). Although the terminal web is a prominent feature that was noted long ago in electron micrograph (EM) images of vertebrate intestinal cells, little is known about how this structure is first assembled and then remodeled to allow for vesicle passage.18-24 A major challenge in addressing these questions is the relative inaccessibility of this tissue in vertebrate systems. Fortunately, key features of intestinal cells are shared between humans and the nematode C. elegans, including actin-rich microvilli anchored into a terminal web made of actin and intermediate filaments (Fig. 2A). This conservation, together with the convenience and transparency of nematodes, make C. elegans an excellent in vivo model system to study the exit of intracellular pathogens from intestinal cells.

Figure 2. Intestinal cell morphology and microsporidia life cycle. (A) Electron micrograph of C. elegans intestinal epithelial cell. The microvilli brush border (arrows) lining the lumen is prominent on the apical surface of the cell. Microvilli are anchored to the cell with the terminal web (bracket), which is visible as a line below the microvilli. Both of these morphological features are conserved with human intestinal cells. (B) Phases of N. parisii exit strategy from C. elegans host intestinal cells. Green is ACT-5 and orange is IFB-2. Pathogen cells are depicted in red and nuclei depicted in black. During Phase I of infection, microsporidia spores fire their polar tubes, and inject their nucleus and sporoplasm into the host cell. This material develops into a multinucleated meront, and ACT-5 appears basolaterally, where it can form filament-like structures. During the final step of Phase I, gaps form in IFB-2 expression along the apical part of the cell. During Phase II, spores mature at approximately 40hpi and exit in a non-lytic, actin-dependent fashion from the host cell into the intestinal lumen.
In 2008, we discovered the first intracellular pathogen of C. elegans through identification of a natural pathogen that infects the intestinal cells. We named this pathogen Nematocida parisii, or “nematode killer from Paris,” and found it defines a new genus and species of microsporidia.25 Microsporidia comprise a large phylum of over 1200 species of eukaryotic intracellular pathogens.26-28 The phylogenetic placement of microsporidia is controversial, but their closest relatives are fungi. Microsporidia can infect a wide variety of animal hosts, commonly infecting the intestine. Fourteen species of microsporidia can infect humans and some can lead to lethal diarrhea in AIDS patients. Due to the problems they cause in immunocompromised patients and the lack of treatments, microsporidia have been deemed priority pathogens by the US. National Institutes of Health.29 Microsporidia also cause significant agricultural problems in fisheries and have been implicated in colony collapse disorder in honeybees.30-33 These obligate intracellular pathogens are challenging to study because they can only replicate inside of host cells.
Figure 2B illustrates the N. parisii life cycle within C. elegans intestinal cells, which is similar to the life cycle of microsporidia species that infect other hosts. Upon ingestion, microsporidia spores germinate and pierce the host cell membrane with a polar tube, through which the spore nucleus and sporoplasm are injected into the host cell. Once inside the cell, the pathogen produces multinucleated structures called meronts, which then replicate and redifferentiate into mature spores. In C. elegans, microsporidian spores then exit from the apical side of cells into the lumen, and are defecated out to transmit the infection to new hosts. N. parisii infection in C. elegans offers a unique system to obtain insights into these poorly understood pathogens. In addition, it provides the rare opportunity to leverage the powerful tools of the C. elegans model system on co-evolved host-pathogen interactions in polarized epithelial cells in vivo.
Microsporidia Infection Causes Reorganization of the Host Cytoskeletal Terminal Web
We had previously observed in EM images that the terminal web was restructured in N. parisii-infected animals, resulting in terminal web “gaps.”25 The C. elegans terminal web is comprised of an intestinal-specific actin isoform called ACT-5, and several intermediate filament proteins, including IFB-2. In uninfected animals these proteins are apically localized within intestinal cells, with ACT-5 localized to both the terminal web and microvilli, and IFB-2 localized exclusively to the terminal web. By infecting a C. elegans strain expressing YFP::ACT-5 and IFB-2::CFP, we were able to track IFB-2 localization in live animals to determine when the host terminal web is remodeled during N. parisii infection, and how this relates to ACT-5 localization. During the replicative, or “meront,” phase of infection, we found that ACT-5 relocalizes to the basolateral side of the cell, where it is not normally found in uninfected animals. After this relocalization, we observed gaps in IFB-2::CFP expression, which appears similar to the restructuring of the terminal web observed by EM (Fig. 2B). Interestingly, we found that IFB-2::CFP gap formation precedes spore exit and that all contagious animals have IFB-2::CFP gaps. Furthermore, after the initial bout of gap formation, the number of gaps does not increase over the course of infection. These results imply that terminal web gap formation is part of an orchestrated exit strategy, rather than damage that occurs as the spores exit from the apical side of the cell.
Because terminal web gaps form after ACT-5 relocalization to the basolateral side of cells, we hypothesized that depletion of ACT-5 from the apical side might trigger gap formation. To test this idea, we reduced ACT-5 levels with RNA interference and found that this knock-down was sufficient to induce gaps in IFB-2::CFP expression in the absence of infection. This result suggests that depletion of ACT-5 at the apical side promotes terminal web weakening in preparation for microsporidia exit. Terminal web weakening occurs immediately prior to spore formation, which would be the ideal time to remove a potential barrier to exit. We have termed this actin-mediated preparation for spore exit “Phase I” to provide contrast to a different role for actin later in infection that is part of “Phase II,” described below. (Fig. 2B). Such seemingly careful preparation for spore exit implies the existence of precisely timed molecular interactions between N. parisii and C. elegans, as might be expected in a host/pathogen pair that has co-evolved. Indeed, Nematocida strains have been found infecting wild-caught Caenorhabditis nematodes in many parts of the world, consistent with this possibility of co-evolution (Fèlix MA, unpublished data).25
Thousands of Microsporidia Spores Exit Host Cells in a Process that Requires ACT-5
If ACT-5 relocalization induces terminal web gap formation to facilitate exit, we would expect that depleting ACT-5 in infected animals would increase gap number and increase pathogen exit. To test this hypothesis, we developed an assay to measure the number of spores exiting from the intestinal cells of infected animals. We found that a single infected animal could produce thousands of spores in one hour, and this production rate could continue for dozens of hours. This finding is especially striking considering that C. elegans has a total of only 20 intestinal cells! With this assay, we compared wild-type animals to those defective for act-5, either caused by treatment with RNAi or by being heterozygous for an act-5 deletion allele (act-5 homozygous mutants are dead). Unexpectedly, we found that a reduction of ACT-5 resulted in a dose-dependent reduction in spore excretion. This observation indicated a complex role for act-5 and led to our two-phase pathogen exit model. In Phase I of this model, actin inhibits exit by maintaining terminal web integrity, and in Phase II, actin promotes exit by an unknown mechanism (Fig. 2B). A further test of Phase I in this model would be to determine whether IFB-2 gaps are required for exit, but we have not yet identified a method to block gap formation. In any case, our results indicate that actin plays a multi-faceted role in the microsporidian exit strategy, with many outstanding questions to address (see below).
N. parisii Exits into the Lumen Free of Host Plasma Membrane, and Without Causing Lysis
Our results indicated that C. elegans intestinal cells produce large numbers of N. parisii spores while animals are still alive, suggesting that host membranes are left intact. To investigate membrane topology during N. parisii exit, we visualized host apical plasma membrane in a strain expressing a PGP-1::GFP membrane marker, which localizes to the apical side of intestinal cells. We found that spores exited into the lumen free of PGP-1::GFP, indicating that microsporidia do not take host membrane with them while exiting the cell. Furthermore, we tested for cell lysis upon spore exit by feeding contagious animals propidium iodide, a dye that cannot diffuse freely into intact cells. Using this assay, we found that host cell integrity was maintained and thus N. parisii exit is non-lytic, as perhaps would be expected for an obligate intracellular pathogen of an animal with non-renewing cells.
Outlook
Our findings indicate that C. elegans host cell restructuring is precisely coordinated with N. parisii pathogen development so that physical barriers are removed at the proper time to enable non-lytic exit. These findings highlight key events of the infection cycle that would not be observed in less natural experimental systems. Similar themes emerged in studies of the bacterial pathogen Listeria as mentioned above, as well as studies of the eukaryotic pathogen Toxoplasma gondii. In human fibroblasts, it was shown that T. gondii exits host cells using a different mechanism when exit occurs naturally than when exit is artificially stimulated by permeabilization of the host cell, which is a common experimental technique in the field.34 By studying N. parisii infection in its natural host, which is a powerful model organism, we expect to uncover meaningful host-pathogen interactions for a group of obligate intracellular pathogens that have been historically very difficult to study.
In future studies, we aim to determine how N. parisii spores exit without lysing the host cell. Orchestrating a non-lytic exit with membrane-free spores released into the lumen likely requires that some membrane surrounds spores prior to exit (Fig. 1). However, EM images show that N. parisii appears to be in direct contact with the cytoplasm during its early replicative stage.25 Therefore, it is possible that N. parisii steals intracellular host membrane as it forms spores in order to enter host trafficking pathways. These membrane-coated spores could then fuse with the plasma membrane to exit the cell free of membrane. It will be interesting to determine which host proteins regulate this process and the level at which N. parisii directs this regulation.
In future studies, it will also be interesting to investigate whether terminal web remodeling is a conserved exit strategy used by other species of microsporidia, such as the human-infecting species Encephalitozoon intestinalis, Encephalitozoon cuniculi, and Enterocytozoon bieneusi. And beyond the microsporidia phylum, it seems certain that other intracellular pathogens must cross terminal web barriers to exit. The terminal web is found not only in intestinal cells but also in many differentiated epithelial cells, such as those found in lung and urogenital epithelia, which are also subject to intracellular infection by a variety of pathogens. Deciphering how intracellular pathogens remodel host tissues in vivo to make a discreet exit will lead to a better understanding of pathogen transmission.
Acknowledgments
We would like to thank Andrew Chisholm, Robert Luallen, and Kevin Hybiske for reading earlier drafts and providing helpful suggestions. This work was supported by NIH predoctoral training grant T32 GM008666 to S.C.S. and NIAID R01 AI087528, the Searle Scholars Program, Ray Thomas Edwards Foundation, David and Lucille Packard Foundation fellowship to E.R.T.
Footnotes
Previously published online: www.landesbioscience.com/journals/worm/article/20501
References
- 1.Hagedorn M, Rohde KH, Russell DG, Soldati T. Infection by tubercular mycobacteria is spread by nonlytic ejection from their amoeba hosts. Science. 2009;323:1729–33. doi: 10.1126/science.1169381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bain JM, Lewis LE, Okai B, Quinn J, Gow NA, Erwig LP. Non-lytic expulsion/exocytosis of Candida albicans from macrophages. Fungal Genet Biol. 2012 doi: 10.1016/j.fgb.2012.01.008. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sturm A, Amino R, van de Sand C, Regen T, Retzlaff S, Rennenberg A, et al. Manipulation of host hepatocytes by the malaria parasite for delivery into liver sinusoids. Science. 2006;313:1287–90. doi: 10.1126/science.1129720. [DOI] [PubMed] [Google Scholar]
- 4.Hybiske K, Stephens RS. Exit strategies of intracellular pathogens. Nat Rev Microbiol. 2008;6:99–110. doi: 10.1038/nrmicro1821. [DOI] [PubMed] [Google Scholar]
- 5.Estes KA, Szumowski SC, Troemel ER. Non-lytic, actin-based exit of intracellular parasites from C. elegans intestinal cells. PLoS Pathog. 2011;7:e1002227. doi: 10.1371/journal.ppat.1002227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Stevens JM, Galyov EE, Stevens MP. Actin-dependent movement of bacterial pathogens. Nat Rev Microbiol. 2006;4:91–101. doi: 10.1038/nrmicro1320. [DOI] [PubMed] [Google Scholar]
- 7.Heindl JE, Saran I, Yi CR, Lesser CF, Goldberg MB. Requirement for formin-induced actin polymerization during spread of Shigella flexneri. Infect Immun. 2010;78:193–203. doi: 10.1128/IAI.00252-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chrisman CJ, Alvarez M, Casadevall A. Phagocytosis of Cryptococcus neoformans by, and nonlytic exocytosis from, Acanthamoeba castellanii. Appl Environ Microbiol. 2010;76:6056–62. doi: 10.1128/AEM.00812-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Johnston SA, May RC. The human fungal pathogen Cryptococcus neoformans escapes macrophages by a phagosome emptying mechanism that is inhibited by Arp2/3 complex-mediated actin polymerisation. PLoS Pathog. 2010;6:e1001041. doi: 10.1371/journal.ppat.1001041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Anderson RM, May RM. Coevolution of hosts and parasites. Parasitology. 1982;85:411–26. doi: 10.1017/S0031182000055360. [DOI] [PubMed] [Google Scholar]
- 11.Frank SA. Models of parasite virulence. Q Rev Biol. 1996;71:37–78. doi: 10.1086/419267. [DOI] [PubMed] [Google Scholar]
- 12.Nikitas G, Deschamps C, Disson O, Niault T, Cossart P, Lecuit M. Transcytosis of Listeria monocytogenes across the intestinal barrier upon specific targeting of goblet cell accessible E-cadherin. J Exp Med. 2011;208:2263–77. doi: 10.1084/jem.20110560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tilney LG, DeRosier DJ, Tilney MS. How Listeria exploits host cell actin to form its own cytoskeleton. I. Formation of a tail and how that tail might be involved in movement. J Cell Biol. 1992;118:71–81. doi: 10.1083/jcb.118.1.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Myers JT, Tsang AW, Swanson JA. Localized reactive oxygen and nitrogen intermediates inhibit escape of Listeria monocytogenes from vacuoles in activated macrophages. J Immunol. 2003;171:5447–53. doi: 10.4049/jimmunol.171.10.5447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kocks C, Hellio R, Gounon P, Ohayon H, Cossart P. Polarized distribution of Listeria monocytogenes surface protein ActA at the site of directional actin assembly. J Cell Sci. 1993;105:699–710. doi: 10.1242/jcs.105.3.699. [DOI] [PubMed] [Google Scholar]
- 16.Henry R, Shaughnessy L, Loessner MJ, Alberti-Segui C, Higgins DE, Swanson JA. Cytolysin-dependent delay of vacuole maturation in macrophages infected with Listeria monocytogenes. Cell Microbiol. 2006;8:107–19. doi: 10.1111/j.1462-5822.2005.00604.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pukkila-Worley R, Ausubel FM. Immune defense mechanisms in the Caenorhabditis elegans intestinal epithelium. Curr Opin Immunol. 2012;24:3–9. doi: 10.1016/j.coi.2011.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bossinger O, Fukushige T, Claeys M, Borgonie G, McGhee JD. The apical disposition of the Caenorhabditis elegans intestinal terminal web is maintained by LET-413. Dev Biol. 2004;268:448–56. doi: 10.1016/j.ydbio.2004.01.003. [DOI] [PubMed] [Google Scholar]
- 19.Fath KR, Burgess DR. Microvillus assembly. Not actin alone. Curr Biol. 1995;5:591–3. doi: 10.1016/S0960-9822(95)00117-5. [DOI] [PubMed] [Google Scholar]
- 20.Hirokawa N, Tilney LG, Fujiwara K, Heuser JE. Organization of actin, myosin, and intermediate filaments in the brush border of intestinal epithelial cells. J Cell Biol. 1982;94:425–43. doi: 10.1083/jcb.94.2.425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ku NO, Zhou X, Toivola DM, Omary MB. The cytoskeleton of digestive epithelia in health and disease. Am J Physiol. 1999;277:G1108–37. doi: 10.1152/ajpgi.1999.277.6.G1108. [DOI] [PubMed] [Google Scholar]
- 22.Mooseker MS. Organization, chemistry, and assembly of the cytoskeletal apparatus of the intestinal brush border. Annu Rev Cell Biol. 1985;1:209–41. doi: 10.1146/annurev.cb.01.110185.001233. [DOI] [PubMed] [Google Scholar]
- 23.Takemura R, Masaki T, Hirokawa N. Developmental organization of the intestinal brush-border cytoskeleton. Cell Motil Cytoskeleton. 1988;9:299–311. doi: 10.1002/cm.970090403. [DOI] [PubMed] [Google Scholar]
- 24.Thomas GH. Spectrin: the ghost in the machine. Bioessays. 2001;23:152–60. doi: 10.1002/1521-1878(200102)23:2<152::AID-BIES1022>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- 25.Troemel ER, Félix MA, Whiteman NK, Barrière A, Ausubel FM. Microsporidia are natural intracellular parasites of the nematode Caenorhabditis elegans. PLoS Biol. 2008;6:2736–52. doi: 10.1371/journal.pbio.0060309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Keeling PJ, Fast NM. Microsporidia: biology and evolution of highly reduced intracellular parasites. Annu Rev Microbiol. 2002;56:93–116. doi: 10.1146/annurev.micro.56.012302.160854. [DOI] [PubMed] [Google Scholar]
- 27.Texier C, Vidau C, Viguès B, El Alaoui H, Delbac F. Microsporidia: a model for minimal parasite-host interactions. Curr Opin Microbiol. 2010;13:443–9. doi: 10.1016/j.mib.2010.05.005. [DOI] [PubMed] [Google Scholar]
- 28.Williams BA. Unique physiology of host-parasite interactions in microsporidia infections. Cell Microbiol. 2009;11:1551–60. doi: 10.1111/j.1462-5822.2009.01362.x. [DOI] [PubMed] [Google Scholar]
- 29.Didier ES, Weiss LM. Microsporidiosis: current status. Curr Opin Infect Dis. 2006;19:485–92. doi: 10.1097/01.qco.0000244055.46382.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bromenshenk JJ, Henderson CB, Wick CH, Stanford MF, Zulich AW, Jabbour RE, et al. Iridovirus and microsporidian linked to honey bee colony decline. PLoS One. 2010;5:e13181. doi: 10.1371/journal.pone.0013181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Higes M, Martín-Herńndez R, Botías C, Bailón EG, González-Porto AV, Barrios L, et al. How natural infection by Nosema ceranae causes honeybee colony collapse. Environ Microbiol. 2008;10:2659–69. doi: 10.1111/j.1462-2920.2008.01687.x. [DOI] [PubMed] [Google Scholar]
- 32.Shaw RWKM. Fish Microsporidia. In: Wittner M WL, ed. The Microsporidia and Microsporidiosis: ASM, 1999. [Google Scholar]
- 33.Troemel ER. New models of microsporidiosis: infections in Zebrafish, C. elegans, and honey bee. PLoS Pathog. 2011;7:e1001243. doi: 10.1371/journal.ppat.1001243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lavine MD, Arrizabalaga G. Exit from host cells by the pathogenic parasite Toxoplasma gondii does not require motility. Eukaryot Cell. 2008;7:131–40. doi: 10.1128/EC.00301-07. [DOI] [PMC free article] [PubMed] [Google Scholar]