

Abstract
STAT2 is unique among the STAT family of transcription factors in that its activation is driven predominantly by only two classes of cell surface receptors: Type I and III interferon receptors. As such, STAT2 plays a critical role in host defenses against viral infections. Viruses have evolved to target STAT2 by either inhibiting its expression, blocking its activity, or by targeting it for degradation. Consequently, these viral onslaughts have driven remarkable divergence in the STAT2 gene across species that is not observed in other STAT family members. Thus, the evolution of STAT2 may preserve its activity and protect each species in the face of an ever-changing viral community.
Keywords: interferon, signal transducer and activator of transcription, STAT2, viral antagonism, comparative genomics, interferon stimulated gene
Viral infections trigger a cascade of intracellular events that lead to the secretion of pro-inflammatory and anti-viral cytokines. For example, ssRNAs from viruses such as influenza activate the intracellular sensors TLR7, RIG-I-like receptors (RLRs) and nucleotide-binding domain and leucine-rich-repeat-containing proteins (NLRs).1 Collectively, these sensors promote the transcription and translation of two classes of cytokines that mobilize cells of the innate and adaptive immune system and create a state of viral resistance in neighboring cells and tissues. Pro-inflammatory cytokines, such as IL-1β, TNF-α and IL-8, recruit and activate innate cells such as neutrophils and macrophages that act early in the response to limit the infection. The anti-viral cytokines IFN-α/β and IFN-λ (type I and III interferons, respectively) act locally to prevent viral replication and spread. In professional antigen presenting cells, such as plasmacytoid dendritic cells, TLR7 activation drives expression of IFN-β along with various members of the IFN-α subtype genes.2,3 These cells act both locally and regionally within lymph nodes to prime T cells and drive the adaptive immune response. In parallel, somatic cells, such as lung epithelial cells, secrete both type I and III interferons that inhibit viral replication within infected cells and create a state of resistance in non-infected cells, effectively limiting viral spread.
Type I and III interferons, while signaling through distinct receptors, share STAT2 as a common signaling intermediate.4,5 Over 300 genes have been identified as direct transcriptional targets of STAT2 activation, many of which remain to be characterized.6 Many of the IFN-induced genes that have been characterized play an almost exclusive role in the anti-viral response and remain silenced in the absence of infection. Thus, STAT2 is a pivotal regulator of the interferon-regulated anti-viral response, and the lack of redundancy in this pathway creates an “Achilles’ heel” that the host, as a species, must protect.
Receptor-Mediated STAT2 Activation and the Interferon Response
In humans, type I interferons are encoded by ~16 α genes and individual genes encoding β, κ, ω and ε. Their gene products are highly structurally related and all bind to a single receptor (IFNAR) consisting of heterodimeric R1 and R2 subunits.7 In contrast, IFN-λ consists of 3 genes, IFN-λ1, 2 and 3 (formally, IL-29, IL-28a and IL-28b) that are more related to the IL-10 family than to interferon.8,9 IFN-λ binds a receptor (IFNLR) composed of a unique IFNLR1 and a shared IL-10R2 subunit. Both the IFNAR and IFNLR recruit and activate STATs 1, 2 and 3. While STATs 1 and 3 are promiscuously activated by a variety of other cytokines and growth factor receptors, STAT2 is selectively recruited to the IFNAR and IFNLR. STAT2 recruitment and activation by both receptors involves tyrosine phosphorylation of STAT2 by JAK kinases and subsequent oligomerization with STAT1 and IRF-9.10,11 Although a fraction of STAT1:STAT2 heterodimers can translocate to the nucleus and bind atypical gamma activated sequence (GAS)-like elements,12 the canonical interferon-stimulated gene factor-3 (ISGF3) complex of STAT2:STAT1:IRF-9 regulates a large fraction of the interferon pathway genes.13 Thus, STAT2 is central to the induction of these genes in response to both IFN-α/β and IFN-λ. In some cases, type II interferon, IFN-γ, can mobilize the antiviral response in a STAT2-dependent manner.14 Although there is no evidence for direct recruitment of STAT2 to the IFN-γR, IFN-γ signaling can drive the formation of an ISGF3-like complex containing STAT2,14 inhibit viral replication15 and induce expression of IFN-α/β target genes,15,16 perhaps through the pairing of phosphorylated STAT1 with latent STAT2. Regardless of its mode and route of activation, STAT2 acts as the gatekeeper to the antiviral response, which is underscored by the severe susceptibility of STAT2 knockout mice to viruses ranging from influenza to dengue.17-19
Type I and III interferon, while produced in different amounts by distinct cell populations, regulate the expression of an overlapping set of interferon stimulated genes (ISGs). Some examples include 2–5′-oligoadenylate synthase (OAS), which decorates viral RNAs with branched polyadenosine, and RNA endonuclease L, which promptly degrades RNAs containing these polyadenosine modifications.20 Most ISGs, however, have not been well characterized, and some of these genes are so enigmatic that they do not contain any canonical secondary structures that could aid in predicting their function. Over 300 ISGs have been identified by microarray and genomic analysis,6,21 which is curious given the magnitude of antiviral activity exhibited by select individual ISGs. For example, MxA potently inhibits the replication of a wide range of viruses and can do so in the absence of other ISGs or interferon signaling if ectopically expressed.22 Not all ISGs exhibit such robust effects individually; nonetheless, when combined with other ISGs, the host can mount an impressive arsenal of anti-viral proteins that could conceptually block every step in the viral life cycle.23 However, like OAS and MxA, most ISG expression requires interferon signaling through STAT2. Thus, while there are three major nodes to drive interferon secretion (TLRs, RIG-I and NLRs) and two pathways available to drive STAT2 activation (IFN-α/β and IFN-λ), there is only one STAT2.
In addition to its role in interferon mediated antiviral effect, STAT2 also confers anti-proliferative and apoptotic activities on a variety of cell types in response to both type I and III interferon.24 This activity is perhaps one of the most fundamental actions of interferon to inhibit viral spread by blocking the replication of virally infected cells. However, much attention has been focused on the use of IFN-α/β to inhibit cancer outgrowth and metastasis given its unique ability to inhibit proliferation of a variety of tumor cell types.25-27 STAT2 is a critical signaling intermediate that regulates much of the apoptotic and anti-proliferative effects of IFN-α/β and IFN-λ. Further, mutations within STAT2 that protract its ligand-mediated phosphorylation can drive cells down the apoptotic pathway.4,28
A variety of genetic mutations have been identified in humans that affect IFN-α/β and IFN-λ production, including mutations in NEMO, TYK2 and TLR3.29-31 Further, functional defects in the human UNC93B1 gene, which controls signaling via TLR3, 7, 8 and 9, results in dramatic reductions in IFN-α/β and IFN-λ secretion in response to ligands that activate these receptors.32 However, these subjects are susceptible to only a few types of viral infections, mainly encephalitis caused by herpes simplex virus. This restriction in viral susceptibility may reflect the inherent redundancy in driving IFN-α/β and IFN-λ production through alternative pathways such as RIG-I and NLRs. In contrast, to date, no mutations have been found that block responsiveness to type I or III interferons in humans, particularly within either the IFN-α/β receptors or STAT2. Yet, defects in a variety of genes that affect both IFN-γ production and responsiveness have been characterized including IFNGR1, IFNGR2, STAT1 and IL-12RB1.33,34 Thus, it is interesting to speculate that mutations that affect IFN-γ responsiveness, while debilitating, can be tolerated; yet, mutations that cripple IFN-α/β or IFN-λ responsiveness such as STAT2 are perhaps lethal.
STAT2 in Inflammation
A cascade of cytokines regulate processes of inflammation that drive both local and systemic activation of immune cells, and this process can be initiated by both pathogen sensing as well as sterile tissue injury. When discussing the role of STATs in inflammation, it is perhaps more accurate to discuss their function in the context of distinct cytokine receptors, as most of the STAT family members can be activated by multiple cytokines. For example, STAT3 can be activated by both IL-6 and IL-10, yet their biological activities are diametrically opposed and counterregulatory with IL-6 contributing to inflammation and IL-10 blocking inflammation.35,36 While IFN-α/β can induce local inflammation under certain circumstances, it does not mediate the intense forms of inflammation seen with TNF-α or IL-1β.
Initially, IFN-α/β was thought to act similarly to IL-12 in driving Th1 development through the recruitment and activation of STAT4, thereby driving Th1-mediated inflammation.37 Indeed, STAT2 was shown to be involved in recruiting STAT4 to the human IFNAR.38 However, our recent findings demonstrated that although STAT4 is transiently activated by the human IFNAR, it is not sufficient to promote Th1 development.39 Further, distinct IFN-α subtypes can drive STAT4 activation to the murine IFNAR,40 which can occur in mouse cells41 even in the absence of STAT2,42 yet still does not promote either Th1 or Tc1 development compared with the effects of IL-12.39,40,43-47 Nonetheless, IFN-α/β may regulate some aspects of inflammation that play a more supportive rather than direct role. For example, IFN-α/β (as well as IFN-γ) is a potent regulator of the chemokine CXCL10 (IP-10), which recruits activated effector T and NK cells to sites of infection. Thus, direct pathogen sensing by epithelial and endothelial cells can lead to recruitment of inflammatory cells via autocrine responses to IFN-α/β. IFN-α/β also activates NK cells directly by driving lytic activity and IFN-γ secretion. Further, recent studies by Tracey and colleagues demonstrated that the IFN-α/β-induced kinase, PKR, regulates an alternative pathway for inflammasome activation.48 As a result, the induction of PKR by IFN-α/β can lead to potent secretion of HMGB1 and other leaderless cytokines that are dependent upon inflammasome-mediated cleavage. Although IFN-α/β regulates these processes, a formal test for the direct role STAT2 in driving these responses has not been performed in STAT2 knockout mice. However, a recent study by Gamero and colleagues demonstrated reduced skin and colonic chemokine and cytokine expression in STAT2 knockout mice in two models of chemically induced carcinogenesis.49 These results suggest that STAT2 may directly contribute to carcinogenesis by promoting expression of proinflammatory cytokines and chemokines. In summary, STAT2 participates in some aspects of inflammation by autocrine responses to IFN-α/β and downstream induction of inflammatory chemokines and cytokines.
Viruses Target STAT2
Given the critical nature of the interferon pathway in viral defense, it is not surprising that viruses have evolved to antagonize the antiviral state induced by type I and type III interferons. The different antagonistic mechanisms include inhibition of interferon production, interferon signaling and inhibition of interferon induced antiviral proteins. STAT2 in particular is a target of many viral proteins, which in some cases determines viral host range. Here we discuss the viruses that have particularly acquired the ability to subvert the interferon signaling pathway by targeting STAT2 (summarized in Table 1).
Table 1. STAT2 antagonism by viral proteins.
| Virus | Viral protein | Mode of STAT2 antagonism | Species | References |
|---|---|---|---|---|
| Human parainfluenza virus 2 (hPIV2) |
V protein |
Protein degradation |
Human, mouse |
65 |
| Parainfluenza virus 5 (hPIV5), previously Simian virus 5 (SV5) |
V protein |
STAT2 dependent STAT1 antagonism |
Human, mouse STAT2 unaffected |
41 and 67 |
| Sendai virus (SeV) |
C protein |
Inhibit phosphorylation/activation by associating with STAT1 |
Rodent pathogen, Experiments on human cell lines |
64 |
| Hendra virus |
V protein |
Complex formation and cytoplasmic sequestration |
Equine, Human |
59 |
| Nipah virus |
V protein |
Complex formation and cytoplasmic sequestration |
Human |
57 and 58 |
| Measles virus |
V protein |
Complex formation and cytoplasmic sequestration, suppression of DC activation |
Human |
52 and 54 |
| Respiratory syncytial virus (RSV) |
NS1, NS2 |
Proteasomal degradation of complex |
Human |
61 and 63 |
| Dengue virus (DENV) |
NS5 |
Protein degradation, blocking phosphorylation |
Human, mouse STAT2 unaffected |
69–72 |
| Japanese encephalitis virus (JEV) |
unknown |
Blocking tyrosine phosphorylation |
Human, non-human primate |
83 |
| Langat virus |
NS5 |
Blocking tyrosine phosphorylation |
non-human primate |
84 |
| Herpes simplex virus 2 (HSV2) |
unknown |
mRNA and protein degradation, blocking phosphorylation |
Human |
50 |
| Lymphocytic choriomeningitis virus (LCMV) |
unknown |
Suppression of DC development via STAT1 independent STAT2 dependent IFN signaling |
Mouse |
52 |
| Mouse cytomegalovirus (MCMV) | M27 | Downregulation of STAT2 protein | Mouse | 51 |
Viruses from a broad array of genera have developed unique ways to subvert the interferon response, and such examples include herpes simplex virus 2 (HSV-2),50 mouse cytomegalovirus (MCMV)51 and lymphocytic choreomeningitis virus (LCMV).52 However, the paramyxoviridae family of viruses contains perhaps the most replete examples of how viruses have subverted the interferon response pathway by targeting STAT2.53,54 The Henipavirus (Nipah and Hendra virus) are newly emerging zoonotic pathogens causing acute respiratory disease in both human and other animal populations. Nipah virus, Hendra virus and measles virus all belong to the paramyxoviridae family. Each of these viruses along with other members of the paramyxoviridae family express a V protein, which is encoded by a polycistronic gene segment (P gene) that utilizes unique overlapping reading frames.55,56 The V protein from measles, Nipah and Hendra viruses can bind to STAT2 and prevent the interferon response via sequestration of STAT2 in the cytoplasm.53 However, the modes of interaction of the V protein from measles mediate this interference by distinct mechanisms from that of Henipahviruses. While the V proteins of these viruses contain a conserved C-terminal domain (CTD), only the measles virus requires the CTD for binding and targeting STAT2 as means of interference with interferon signaling.54,55 In contrast, the Nipah virus V protein can also bind STAT2 in a STAT1 dependent manner through distinct regions of the V protein mapping outside the boundary of the CTD.57 These complexes are unable to translocate to the nucleus, thus sequestering STAT2 to the cytoplasm and inhibiting efficient interferon signaling.57-59 The Hendravirus V protein also operates in a similar manner to Nipah virus, although the distinct interaction with STATs have not been mapped.59 In addition, the Nipah virus V protein has been shown to contain a nuclear export signal (NES) that enables the V protein to shuttle between the cytoplasm and nucleus. Nonetheless, binding of STAT1:STAT2 complexes by Nipah V protein is sufficient to prevent IFN-α/β signaling even in the absence of the NES.58 Therefore viruses belonging to the same family have evolved to target STAT2 using diverse mechanisms in order to evade interferon response in the host.
Respiratory syncytial virus (RSV) is a member of the paramyxovirus family and causes severe flu and cold like symptoms in humans, particularly in children. RSV antagonizes the interferon-signaling pathway through two virally encoded nonstructural proteins, NS1 and NS2.60-62 Together, NS1/2 form a complex with STAT2 and drive ubiquitin-mediated proteasomal degradation. Binding of STAT2 with this complex requires the C-terminus of NS2,61 while NS1 acts as an E3 ligase that targets STAT2 for degradation.63 In a similar manner to RSV, the rodent pathogen Sendai virus (SeV) encodes a C protein that antagonizes IFN-α/β signaling at all phases of the infection process by interacting with STAT1 and blocking activation of STAT2.64
Finally, the human parainfluenza virus 2 (HPIV2) is an RNA virus of the paramyxoviridae family that causes upper and lower respiratory infections. In human, HPIV2 blocks type I interferon signaling by selectively inducing degradation of STAT2 but not STAT1.65,66 The degradation of STAT2 is independent of IFN-α/β signaling, and like other members of this family of viruses, the V protein mediates the degradation of STAT2. Post-translational degradation of STAT2 protein by HPIV2 V protein was blocked by proteasome inhibitors indicating a role for proteasomal degradation.65 In contrast, the V protein of parainfluenza virus 5 [PIV5, previously known as simian virus V (SV5)], another member of the same family of viruses, preferentially induces STAT1 degradation in a STAT2-dependent manner in human. The C-terminal domain of this V protein is highly conserved across the members of rubulavirus genus of the paramyxoviridae family that includes both HPIV2 and PIV5. Although PIV5 can target human STAT1 via STAT2, mouse STAT2 does not interact with the PIV5 V protein.67 Thus, mouse STAT2 restricts PIV5 V protein interaction leaving the interferon response pathway intact. As such, PIV5 is unable to efficiently replicate in mice due to an intact interferon response. Transgenic mice expressing human STAT2 restores the ability of PIV5 V protein to mediated degradation of STAT1.41 Likewise, expression of human STAT2 in mouse embryonic fibroblasts permits antagonism of endogenous murine IFN-α/β signaling by both PIV5 and HPIV2 V proteins.67 Thus, the requirement for STAT2 interaction for antagonism of interferon response limits the host range of PIV5.
Dengue virus (DENV) is another example of how host-pathogen co-evolution has become a host range-determining factor. Dengue virus is a mosquito-borne RNA virus that causes dengue fever, dengue hemorrhagic fever and dengue shock syndrome in humans. DENV belongs to the genus flavivirus and has been shown to replicate efficiently in a variety of human immune cells including monocytes, macrophages, B cells and dendritic cells (reviewed in ref. 68). Initial studies with DENV replicons expressing all of the non-structural proteins demonstrated reduced IFN-α stimulated genes concomitant with a reduction in endogenous STAT2 protein levels.69 In later studies, DENV NS5 was found to directly bind to and promote ubiquitin-mediated degradation of human, but not mouse STAT2.70,71 Mouse STAT2 expression in human cell lines restored the IFN-dependent block in virus production. NS5 interaction with human STAT2 was mapped to the coiled-coil domain of human STAT2 containing a species-specific sequence between residues 181–301. As NS5 failed to bind mouse STAT2, replacement of this region of mouse sequence with the human counterpart restored NS5 binding to the chimerized mouse molecule.71 In parallel studies, DENV NS5 was found to specifically antagonize IFN-α, but not IFN-γ signaling by binding to and inhibiting STAT2 phosphorylation.72 Recently, Schoggins et al. have demonstrated that overexpression of STAT2 in STAT1-deficient fibroblast can limit DENV replication without any significant ISG expression.73 This suggests that overexpression of STAT2 can stoichiometrically overcome viral NS5 activity. Collectively these studies demonstrate that DENV is able to establish infection in human due to its ability to surmount human antiviral response via STAT2 degradation and blocking phosphorylation that may be circumvented in humans through augmented STAT2 expression. However, murine STAT2 has evolved to disengage this mechanism of STAT2 antagonism by DENV and thus restricting DENV host range to humans.
STAT2 Evolution
Given the central role of STAT2 in the induction of ISGs, it is perhaps surprising that only a restricted subset of viruses have adapted their mechanism of innate subversion to target STAT2. For some viruses, STAT2 determines the host range across species, and this is particularly evident with dengue virus, discussed above. However, closer examination of the STAT2 gene and protein product reveals some important insights into how viruses shape the evolution of their hosts. In this section, we will explore how STAT2 has emerged as a highly divergent member of the STAT family while retaining its ability to exclusively regulate the interferon response in each species.
The STAT family can be traced as far back to rudimentary STAT-like genes that exist in slime molds (D. discoideum),74 thus suggesting that the current members that exist in mammals (STATs 1, 2, 3, 4 5A, 5B and 6) arose from a common ancestral gene through duplication.75 In mammals, the seven STAT family members are located as clusters in 3 genomic locations: in humans, STATs 1 and 4 on Chr. 2, STATs 3, 5A and 5B on Chr. 17 and STATs 2 and 6 on Chr. 12. Considering their syntenic arrangement across species, how conserved are the various STAT members in mammals? Within the Ensemble database, there are seven species, including human, for which the full-length sequences are available for six of the seven STAT family members. An alignment of these sequences reveals remarkable similarity (> 95% for most pair-wise comparisons) for each of the STAT genes except STAT2 (Fig. 1). STAT3 is the most highly conserved member, retaining > 98% sequence similarity comparing human with both mouse and rat. However, human STAT2, while being relatively well conserved with macaque and chimpanzee, diverges significantly when paired with mouse, rat, horse and elephant. A phylogenetic analysis further reveals the degree to which STAT2 has diverged even within the non-human primate species when compared with the human counterpart (Fig. 2). Moreover, STAT2’s dimerization partner STAT1 displays roughly 5-fold greater sequence similarity than STAT2 within primates (based on sequence identity within the phylogenetic analysis in Fig. 2), making STAT2 the most divergent member of the STAT family.
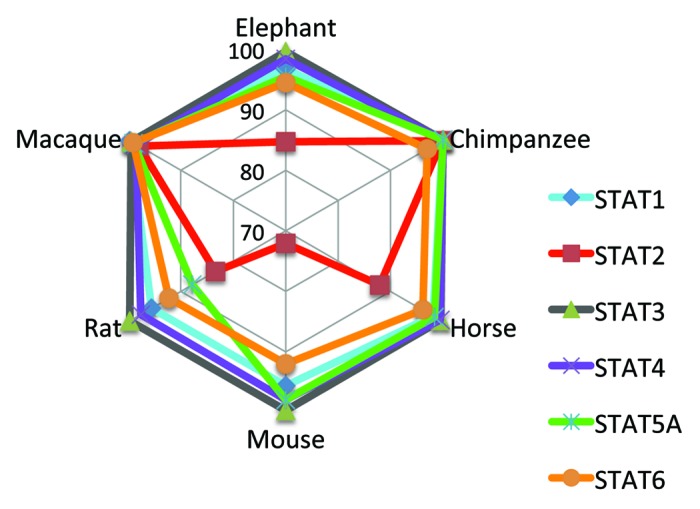
Figure 1. Sequence similarity of STAT family members among select mammalian species. The full length protein sequences of STATs 1, 2, 3, 4, 5A and 6 were aligned against the human sequence for each of the species shown in the radar plot. The Clustal alignment algorithm within MacVector was used for the alignment, and the percent sequence similarity to human in pair-wise comparisons is plotted in the graph.
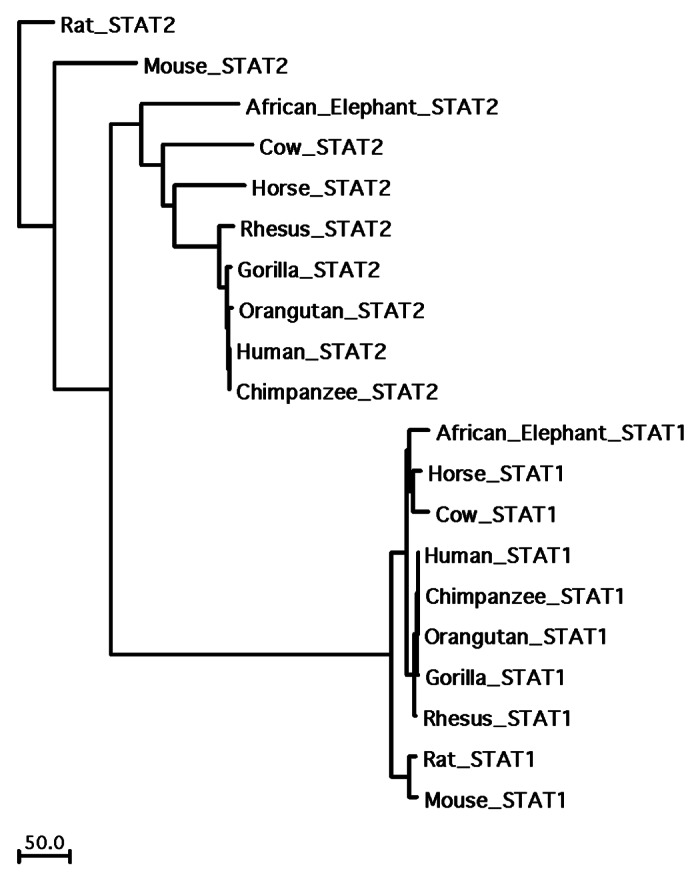
Figure 2. Phylogenetic relationship between STAT1 and STAT2 among select mammals and non-human primates. The full length sequences of STAT1 and STAT2 for each species shown were aligned with the Clustal algorithm within MacVector. Phylogenetic analysis was performed with this alignment, and the scale indicates the absolute numbers of sequence differences across the length of the dendrogram.
Within each STAT family member, the SH2 domain is the most highly conserved segment of the gene across species, and STAT2 is no exception to this (Fig. 3). However, just beyond the SH2 domain, the C-terminal transactivation domain of STAT2 diverges so significantly that only small segments of conservation are retained at the distal end of the protein. Most striking is the disruption of the structure and insertion of a “mini-satellite” segment within the C-terminus of mouse STAT2 that contains a repetitive element PAPQVLLE. This element has no significant similarity to any other protein in the database, and mouse is the only species that harbors this sequence.

Figure 3. The STAT2 C-terminus is highly divergent across species. The sequences of STAT2 from the indicated species were aligned beginning with residue 651 of human STAT2. The SH2 and C-terminus of STAT2 is indicated along with the conserved tyrosine residue (Y690), which is phosphorylated in responses to IFN-α/β signaling.
The divergence in STAT2 was first appreciated when mouse STAT2 was cloned and sequenced over 12 years ago.76 At that time, it was known that STAT2 interacted with other transcription factors to regulate expression of various ISGs, and one of those factors was CBP.77 Park et al. demonstrated that while the C-terminus of mouse was significantly different than human, CBP could still interact with the C-terminus of both mouse and human STAT2 and functionally cooperate to regulate transcription.76 In addition, STAT2 was also found to be required for recruitment of STAT4 to the human, but not mouse IFNAR.38 Further, the human STAT2 C-terminus was found to be required for this recruitment in human cells, and the significant divergence in this region partially explained the difference in signaling between the two species.78 However, the interaction was perhaps indirect79 as replacement of the mouse C-terminus with the human counterpart via knock-in failed to restore significant IFN-α/β-dependent STAT4 activation in mouse T cells.80 Nonetheless, these animals still responded normally to IFN-α/β by activating ISGF3 and by driving the expression of ISGs.47
These types of domain-swapping experiments reveal some very interesting aspects of STAT2 biology. Despite drastic differences within the C-terminus, this domain still maintains a physical interaction with CBP and perhaps many other transcription factors whose evolution has remained relatively constant compared with STAT2. Indeed, the C-terminus of STAT2 is relatively unstructured compared with other domains within the protein.81 However, when bound to the transcriptional activator zinc binding (TAZ) domain of CBP, the human STAT2 C-terminus undergoes dynamic folding to create a stable tertiary structure, thereby stabilizing its bound conformation to the TAZ domain. Unfortunately, the structure of the C-terminus from other species such as mouse with the CBP TAZ domain has not been solved. Nonetheless, STAT2 displays remarkable plasticity in both its proximal receptor activation and downstream gene transactivation capabilities regardless of the remarkable structural differences that exist, particularly within the C-terminus. Thus, even though STAT2 has diverged at a much greater rate than other STAT family members, the interactions that STAT2 must maintain to mediate its function are still preserved and presumably contain enough inherent flexibility to accommodate large changes in STAT2’s structure.
Considering the central role that STAT2 plays in regulating the intracellular anti-viral response, it represents an important target for viral interference. While speculative, it is likely that lethal pandemic viruses drive STAT2 evolution, in some cases, by targeting STAT2 directly. Perhaps the unique haplotypes that have been identified in STAT2 among East Asians and shared with Neanderthal point to recent selection events in human evolution.82 Regardless, the survival of each species, including humans, to viral assaults thus depends upon the flexibility of STAT2 to maintain distinct physical interactions, which preserve its function in driving the interferon response.
Acknowledgments
The authors would like to thank Sarah Gonzales for helpful discussions and for critically reading the manuscript. This work was supported by the National Institutes of Health (Bethesda, MD): F.C.Z. supported by AI005284 and AI56222 awarded to J.D.F.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Footnotes
Previously published online: www.landesbioscience.com/journals/jak-stat/article/23633
References
- 1.O’Neill LA, Bowie AG. Sensing and signaling in antiviral innate immunity. Curr Biol. 2010;20:R328–33. doi: 10.1016/j.cub.2010.01.044. [DOI] [PubMed] [Google Scholar]
- 2.Colonna M, Trinchieri G, Liu YJ. Plasmacytoid dendritic cells in immunity. Nat Immunol. 2004;5:1219–26. doi: 10.1038/ni1141. [DOI] [PubMed] [Google Scholar]
- 3.Barchet W, Cella M, Colonna M. Plasmacytoid dendritic cells--virus experts of innate immunity. Semin Immunol. 2005;17:253–61. doi: 10.1016/j.smim.2005.05.008. [DOI] [PubMed] [Google Scholar]
- 4.Maher SG, Sheikh F, Scarzello AJ, Romero-Weaver AL, Baker DP, Donnelly RP, et al. IFNalpha and IFNlambda differ in their antiproliferative effects and duration of JAK/STAT signaling activity. Cancer Biol Ther. 2008;7:1109–15. doi: 10.4161/cbt.7.7.6192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhou Z, Hamming OJ, Ank N, Paludan SR, Nielsen AL, Hartmann R. Type III interferon (IFN) induces a type I IFN-like response in a restricted subset of cells through signaling pathways involving both the Jak-STAT pathway and the mitogen-activated protein kinases. J Virol. 2007;81:7749–58. doi: 10.1128/JVI.02438-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sadler AJ, Williams BR. Interferon-inducible antiviral effectors. Nat Rev Immunol. 2008;8:559–68. doi: 10.1038/nri2314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Stark GR, Kerr IM, Williams BR, Silverman RH, Schreiber RD. How cells respond to interferons. Annu Rev Biochem. 1998;67:227–64. doi: 10.1146/annurev.biochem.67.1.227. [DOI] [PubMed] [Google Scholar]
- 8.Gad HH, Dellgren C, Hamming OJ, Vends S, Paludan SR, Hartmann R. Interferon-lambda is functionally an interferon but structurally related to the interleukin-10 family. J Biol Chem. 2009;284:20869–75. doi: 10.1074/jbc.M109.002923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dellgren C, Gad HH, Hamming OJ, Melchjorsen J, Hartmann R. Human interferon-lambda3 is a potent member of the type III interferon family. Genes Immun. 2009;10:125–31. doi: 10.1038/gene.2008.87. [DOI] [PubMed] [Google Scholar]
- 10.Bluyssen HA, Levy DE. Stat2 is a transcriptional activator that requires sequence-specific contacts provided by stat1 and p48 for stable interaction with DNA. J Biol Chem. 1997;272:4600–5. doi: 10.1074/jbc.272.7.4600. [DOI] [PubMed] [Google Scholar]
- 11.Horvath CM, Stark GR, Kerr IM, Darnell JE., Jr. Interactions between STAT and non-STAT proteins in the interferon-stimulated gene factor 3 transcription complex. Mol Cell Biol. 1996;16:6957–64. doi: 10.1128/mcb.16.12.6957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ghislain JJ, Wong T, Nguyen M, Fish EN. The interferon-inducible Stat2:Stat1 heterodimer preferentially binds in vitro to a consensus element found in the promoters of a subset of interferon-stimulated genes. J Interferon Cytokine Res. 2001;21:379–88. doi: 10.1089/107999001750277853. [DOI] [PubMed] [Google Scholar]
- 13.Brierley MM, Fish EN. Stats: multifaceted regulators of transcription. J Interferon Cytokine Res. 2005;25:733–44. doi: 10.1089/jir.2005.25.733. [DOI] [PubMed] [Google Scholar]
- 14.Matsumoto M, Tanaka N, Harada H, Kimura T, Yokochi T, Kitagawa M, et al. Activation of the transcription factor ISGF3 by interferon-gamma. Biol Chem. 1999;380:699–703. doi: 10.1515/BC.1999.087. [DOI] [PubMed] [Google Scholar]
- 15.Davis AM, Hagan KA, Matthews LA, Bajwa G, Gill MA, Gale M, Jr., et al. Blockade of virus infection by human CD4+ T cells via a cytokine relay network. J Immunol. 2008;180:6923–32. doi: 10.4049/jimmunol.180.10.6923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Morrow AN, Schmeisser H, Tsuno T, Zoon KC. A novel role for IFN-stimulated gene factor 3II in IFN-γ signaling and induction of antiviral activity in human cells. J Immunol. 2011;186:1685–93. doi: 10.4049/jimmunol.1001359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Park C, Li S, Cha E, Schindler C. Immune response in Stat2 knockout mice. Immunity. 2000;13:795–804. doi: 10.1016/S1074-7613(00)00077-7. [DOI] [PubMed] [Google Scholar]
- 18.Jewell NA, Cline T, Mertz SE, Smirnov SV, Flaño E, Schindler C, et al. Lambda interferon is the predominant interferon induced by influenza A virus infection in vivo. J Virol. 2010;84:11515–22. doi: 10.1128/JVI.01703-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wu S, Metcalf JP, Wu W. Innate immune response to influenza virus. Curr Opin Infect Dis. 2011;24:235–40. doi: 10.1097/QCO.0b013e328344c0e3. [DOI] [PubMed] [Google Scholar]
- 20.Silverman RH. Viral encounters with 2′,5′-oligoadenylate synthetase and RNase L during the interferon antiviral response. J Virol. 2007;81:12720–9. doi: 10.1128/JVI.01471-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.de Veer MJ, Holko M, Frevel M, Walker E, Der S, Paranjape JM, et al. Functional classification of interferon-stimulated genes identified using microarrays. J Leukoc Biol. 2001;69:912–20. [PubMed] [Google Scholar]
- 22.Haller O, Kochs G. Human MxA protein: an interferon-induced dynamin-like GTPase with broad antiviral activity. J Interferon Cytokine Res. 2011;31:79–87. doi: 10.1089/jir.2010.0076. [DOI] [PubMed] [Google Scholar]
- 23.Schoggins JW, Rice CM. Interferon-stimulated genes and their antiviral effector functions. Curr Opin Virol. 2011;1:519–25. doi: 10.1016/j.coviro.2011.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Steen HC, Gamero AM. The role of signal transducer and activator of transcription-2 in the interferon response. J Interferon Cytokine Res. 2012;32:103–10. doi: 10.1089/jir.2011.0099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gamero AM, Potla R, Wegrzyn J, Szelag M, Edling AE, Shimoda K, et al. Activation of Tyk2 and Stat3 is required for the apoptotic actions of interferon-beta in primary pro-B cells. J Biol Chem. 2006;281:16238–44. doi: 10.1074/jbc.M509516200. [DOI] [PubMed] [Google Scholar]
- 26.Gamero AM, Potla R, Sakamoto S, Baker DP, Abraham R, Larner AC. Type I interferons activate apoptosis in a Jurkat cell variant by caspase-dependent and independent mechanisms. Cell Signal. 2006;18:1299–308. doi: 10.1016/j.cellsig.2005.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Maher SG, Romero-Weaver AL, Scarzello AJ, Gamero AM. Interferon: cellular executioner or white knight? Curr Med Chem. 2007;14:1279–89. doi: 10.2174/092986707780597907. [DOI] [PubMed] [Google Scholar]
- 28.Scarzello AJ, Romero-Weaver AL, Maher SG, Veenstra TD, Zhou M, Qin A, et al. A Mutation in the SH2 domain of STAT2 prolongs tyrosine phosphorylation of STAT1 and promotes type I IFN-induced apoptosis. Mol Biol Cell. 2007;18:2455–62. doi: 10.1091/mbc.E06-09-0843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Filipe-Santos O, Bustamante J, Haverkamp MH, Vinolo E, Ku CL, Puel A, et al. X-linked susceptibility to mycobacteria is caused by mutations in NEMO impairing CD40-dependent IL-12 production. J Exp Med. 2006;203:1745–59. doi: 10.1084/jem.20060085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Minegishi Y, Saito M, Morio T, Watanabe K, Agematsu K, Tsuchiya S, et al. Human tyrosine kinase 2 deficiency reveals its requisite roles in multiple cytokine signals involved in innate and acquired immunity. Immunity. 2006;25:745–55. doi: 10.1016/j.immuni.2006.09.009. [DOI] [PubMed] [Google Scholar]
- 31.Zhang SY, Jouanguy E, Ugolini S, Smahi A, Elain G, Romero P, et al. TLR3 deficiency in patients with herpes simplex encephalitis. Science. 2007;317:1522–7. doi: 10.1126/science.1139522. [DOI] [PubMed] [Google Scholar]
- 32.Casrouge A, Zhang SY, Eidenschenk C, Jouanguy E, Puel A, Yang K, et al. Herpes simplex virus encephalitis in human UNC-93B deficiency. Science. 2006;314:308–12. doi: 10.1126/science.1128346. [DOI] [PubMed] [Google Scholar]
- 33.Zhang SY, Boisson-Dupuis S, Chapgier A, Yang K, Bustamante J, Puel A, et al. Inborn errors of interferon (IFN)-mediated immunity in humans: insights into the respective roles of IFN-alpha/beta, IFN-gamma, and IFN-lambda in host defense. Immunol Rev. 2008;226:29–40. doi: 10.1111/j.1600-065X.2008.00698.x. [DOI] [PubMed] [Google Scholar]
- 34.Casanova JL, Holland SM, Notarangelo LD. Inborn errors of human JAKs and STATs. Immunity. 2012;36:515–28. doi: 10.1016/j.immuni.2012.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Murray PJ. Understanding and exploiting the endogenous interleukin-10/STAT3-mediated anti-inflammatory response. Curr Opin Pharmacol. 2006;6:379–86. doi: 10.1016/j.coph.2006.01.010. [DOI] [PubMed] [Google Scholar]
- 36.Hodge DR, Hurt EM, Farrar WL. The role of IL-6 and STAT3 in inflammation and cancer. Eur J Cancer. 2005;41:2502–12. doi: 10.1016/j.ejca.2005.08.016. [DOI] [PubMed] [Google Scholar]
- 37.Rogge L, D’Ambrosio D, Biffi M, Penna G, Minetti LJ, Presky DH, et al. The role of Stat4 in species-specific regulation of Th cell development by type I IFNs. J Immunol. 1998;161:6567–74. [PubMed] [Google Scholar]
- 38.Farrar JD, Smith JD, Murphy TL, Murphy KM. Recruitment of Stat4 to the human interferon-alpha/beta receptor requires activated Stat2. J Biol Chem. 2000;275:2693–7. doi: 10.1074/jbc.275.4.2693. [DOI] [PubMed] [Google Scholar]
- 39.Ramos HJ, Davis AM, George TC, Farrar JD. IFN-alpha is not sufficient to drive Th1 development due to lack of stable T-bet expression. J Immunol. 2007;179:3792–803. doi: 10.4049/jimmunol.179.6.3792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Berenson LS, Farrar JD, Murphy TL, Murphy KM. Frontline: absence of functional STAT4 activation despite detectable tyrosine phosphorylation induced by murine IFN-alpha. Eur J Immunol. 2004;34:2365–74. doi: 10.1002/eji.200324829. [DOI] [PubMed] [Google Scholar]
- 41.Kraus TA, Garza L, Horvath CM. Enabled interferon signaling evasion in an immune-competent transgenic mouse model of parainfluenza virus 5 infection. Virology. 2008;371:196–205. doi: 10.1016/j.virol.2007.10.001. [DOI] [PubMed] [Google Scholar]
- 42.Wang J, Pham-Mitchell N, Schindler C, Campbell IL. Dysregulated Sonic hedgehog signaling and medulloblastoma consequent to IFN-alpha-stimulated STAT2-independent production of IFN-gamma in the brain. J Clin Invest. 2003;112:535–43. doi: 10.1172/JCI18637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ramos HJ, Davis AM, Cole AG, Schatzle JD, Forman J, Farrar JD. Reciprocal responsiveness to interleukin-12 and interferon-alpha specifies human CD8+ effector versus central memory T-cell fates. Blood. 2009;113:5516–25. doi: 10.1182/blood-2008-11-188458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Huber JP, Farrar JD. Regulation of effector and memory T-cell functions by type I interferon. Immunology. 2011;132:466–74. doi: 10.1111/j.1365-2567.2011.03412.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chowdhury FZ, Ramos HJ, Davis LS, Forman J, Farrar JD. IL-12 selectively programs effector pathways that are stably expressed in human CD8+ effector memory T cells in vivo. Blood. 2011;118:3890–900. doi: 10.1182/blood-2011-05-357111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Athie-Morales V, Smits HH, Cantrell DA, Hilkens CM. Sustained IL-12 signaling is required for Th1 development. J Immunol. 2004;172:61–9. doi: 10.4049/jimmunol.172.1.61. [DOI] [PubMed] [Google Scholar]
- 47.Berenson LS, Gavrieli M, Farrar JD, Murphy TL, Murphy KM. Distinct characteristics of murine STAT4 activation in response to IL-12 and IFN-alpha. J Immunol. 2006;177:5195–203. doi: 10.4049/jimmunol.177.8.5195. [DOI] [PubMed] [Google Scholar]
- 48.Lu B, Nakamura T, Inouye K, Li J, Tang Y, Lundbäck P, et al. Novel role of PKR in inflammasome activation and HMGB1 release. Nature. 2012;488:670–4. doi: 10.1038/nature11290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gamero AM, Young MR, Mentor-Marcel R, Bobe G, Scarzello AJ, Wise J, et al. STAT2 contributes to promotion of colorectal and skin carcinogenesis. Cancer Prev Res (Phila) 2010;3:495–504. doi: 10.1158/1940-6207.CAPR-09-0105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kadeppagari R-K, Sanchez RL, Foster TP. HSV-2 inhibits type-I interferon signaling via multiple complementary and compensatory STAT2-associated mechanisms. Virus Res. 2012;167:273–84. doi: 10.1016/j.virusres.2012.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zimmermann A, Trilling M, Wagner M, Wilborn M, Bubic I, Jonjic S, et al. A cytomegaloviral protein reveals a dual role for STAT2 in IFN-gamma signaling and antiviral responses. J Exp Med. 2005;201:1543–53. doi: 10.1084/jem.20041401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hahm B, Trifilo MJ, Zuniga EI, Oldstone MBA. Viruses evade the immune system through type I interferon-mediated STAT2-dependent, but STAT1-independent, signaling. Immunity. 2005;22:247–57. doi: 10.1016/j.immuni.2005.01.005. [DOI] [PubMed] [Google Scholar]
- 53.Rodriguez JJ, Horvath CM. Host evasion by emerging paramyxoviruses: Hendra virus and Nipah virus v proteins inhibit interferon signaling. Viral Immunol. 2004;17:210–9. doi: 10.1089/0882824041310568. [DOI] [PubMed] [Google Scholar]
- 54.Ramachandran A, Parisien J-P, Horvath CM. STAT2 is a primary target for measles virus V protein-mediated alpha/beta interferon signaling inhibition. J Virol. 2008;82:8330–8. doi: 10.1128/JVI.00831-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ramachandran A, Horvath CM. Paramyxovirus disruption of interferon signal transduction: STATus report. J Interferon Cytokine Res. 2009;29:531–7. doi: 10.1089/jir.2009.0070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wang LF, Michalski WP, Yu M, Pritchard LI, Crameri G, Shiell B, et al. A novel P/V/C gene in a new member of the Paramyxoviridae family, which causes lethal infection in humans, horses, and other animals. J Virol. 1998;72:1482–90. doi: 10.1128/jvi.72.2.1482-1490.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Rodriguez JJ, Parisien J-P, Horvath CM. Nipah virus V protein evades alpha and gamma interferons by preventing STAT1 and STAT2 activation and nuclear accumulation. J Virol. 2002;76:11476–83. doi: 10.1128/JVI.76.22.11476-11483.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Rodriguez JJ, Cruz CD, Horvath CM. Identification of the nuclear export signal and STAT-binding domains of the Nipah virus V protein reveals mechanisms underlying interferon evasion. J Virol. 2004;78:5358–67. doi: 10.1128/JVI.78.10.5358-5367.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Rodriguez JJ, Wang LF, Horvath CM. Hendra virus V protein inhibits interferon signaling by preventing STAT1 and STAT2 nuclear accumulation. J Virol. 2003;77:11842–5. doi: 10.1128/JVI.77.21.11842-11845.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lo MS, Brazas RM, Holtzman MJ. Respiratory syncytial virus nonstructural proteins NS1 and NS2 mediate inhibition of Stat2 expression and alpha/beta interferon responsiveness. J Virol. 2005;79:9315–9. doi: 10.1128/JVI.79.14.9315-9319.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Swedan S, Musiyenko A, Barik S. Respiratory syncytial virus nonstructural proteins decrease levels of multiple members of the cellular interferon pathways. J Virol. 2009;83:9682–93. doi: 10.1128/JVI.00715-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ramaswamy M, Shi L, Varga SM, Barik S, Behlke MA, Look DC. Respiratory syncytial virus nonstructural protein 2 specifically inhibits type I interferon signal transduction. Virology. 2006;344:328–39. doi: 10.1016/j.virol.2005.09.009. [DOI] [PubMed] [Google Scholar]
- 63.Elliott J, Lynch OT, Suessmuth Y, Qian P, Boyd CR, Burrows JF, et al. Respiratory syncytial virus NS1 protein degrades STAT2 by using the Elongin-Cullin E3 ligase. J Virol. 2007;81:3428–36. doi: 10.1128/JVI.02303-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Gotoh B, Takeuchi K, Komatsu T, Yokoo J. The STAT2 activation process is a crucial target of Sendai virus C protein for the blockade of alpha interferon signaling. J Virol. 2003;77:3360–70. doi: 10.1128/JVI.77.6.3360-3370.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Parisien JP, Lau JF, Rodriguez JJ, Sullivan BM, Moscona A, Parks GD, et al. The V protein of human parainfluenza virus 2 antagonizes type I interferon responses by destabilizing signal transducer and activator of transcription 2. Virology. 2001;283:230–9. doi: 10.1006/viro.2001.0856. [DOI] [PubMed] [Google Scholar]
- 66.Parisien J-P, Lau JF, Rodriguez JJ, Ulane CM, Horvath CM. Selective STAT protein degradation induced by paramyxoviruses requires both STAT1 and STAT2 but is independent of alpha/beta interferon signal transduction. J Virol. 2002;76:4190–8. doi: 10.1128/JVI.76.9.4190-4198.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Parisien J-P, Lau JF, Horvath CM. STAT2 acts as a host range determinant for species-specific paramyxovirus interferon antagonism and simian virus 5 replication. J Virol. 2002;76:6435–41. doi: 10.1128/JVI.76.13.6435-6441.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Morrison J, Aguirre S, Fernandez-Sesma A. Innate immunity evasion by Dengue virus. Viruses. 2012;4:397–413. doi: 10.3390/v4030397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Jones M, Davidson A, Hibbert L, Gruenwald P, Schlaak J, Ball S, et al. Dengue virus inhibits alpha interferon signaling by reducing STAT2 expression. J Virol. 2005;79:5414–20. doi: 10.1128/JVI.79.9.5414-5420.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ashour J, Laurent-Rolle M, Shi P-Y, García-Sastre A. NS5 of dengue virus mediates STAT2 binding and degradation. J Virol. 2009;83:5408–18. doi: 10.1128/JVI.02188-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ashour J, Morrison J, Laurent-Rolle M, Belicha-Villanueva A, Plumlee CR, Bernal-Rubio D, et al. Mouse STAT2 restricts early dengue virus replication. Cell Host Microbe. 2010;8:410–21. doi: 10.1016/j.chom.2010.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Mazzon M, Jones M, Davidson A, Chain B, Jacobs M. Dengue virus NS5 inhibits interferon-alpha signaling by blocking signal transducer and activator of transcription 2 phosphorylation. J Infect Dis. 2009;200:1261–70. doi: 10.1086/605847. [DOI] [PubMed] [Google Scholar]
- 73.Schoggins JW, Dorner M, Feulner M, Imanaka N, Murphy MY, Ploss A, et al. Dengue reporter viruses reveal viral dynamics in interferon receptor-deficient mice and sensitivity to interferon effectors in vitro. Proc Natl Acad Sci U S A. 2012;109:14610–5. doi: 10.1073/pnas.1212379109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kawata T, Shevchenko A, Fukuzawa M, Jermyn KA, Totty NF, Zhukovskaya NV, et al. SH2 signaling in a lower eukaryote: a STAT protein that regulates stalk cell differentiation in dictyostelium. Cell. 1997;89:909–16. doi: 10.1016/S0092-8674(00)80276-7. [DOI] [PubMed] [Google Scholar]
- 75.Wang Y, Levy DE. Comparative evolutionary genomics of the STAT family of transcription factors. JAK-STAT. 2012;1:23–33. doi: 10.4161/jkst.19418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Park C, Lecomte MJ, Schindler C. Murine Stat2 is uncharacteristically divergent. Nucleic Acids Res. 1999;27:4191–9. doi: 10.1093/nar/27.21.4191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Bhattacharya S, Eckner R, Grossman S, Oldread E, Arany Z, D’Andrea A, et al. Cooperation of Stat2 and p300/CBP in signalling induced by interferon-alpha. Nature. 1996;383:344–7. doi: 10.1038/383344a0. [DOI] [PubMed] [Google Scholar]
- 78.Farrar JD, Smith JD, Murphy TL, Leung S, Stark GR, Murphy KM. Selective loss of type I interferon-induced STAT4 activation caused by a minisatellite insertion in mouse Stat2. Nat Immunol. 2000;1:65–9. doi: 10.1038/76932. [DOI] [PubMed] [Google Scholar]
- 79.Tyler DR, Persky ME, Matthews LA, Chan S, Farrar JD. Pre-assembly of STAT4 with the human IFN-alpha/beta receptor-2 subunit is mediated by the STAT4 N-domain. Mol Immunol. 2007;44:1864–72. doi: 10.1016/j.molimm.2006.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Persky ME, Murphy KM, Farrar JD. IL-12, but not IFN-alpha, promotes STAT4 activation and Th1 development in murine CD4+ T cells expressing a chimeric murine/human Stat2 gene. J Immunol. 2005;174:294–301. doi: 10.4049/jimmunol.174.1.294. [DOI] [PubMed] [Google Scholar]
- 81.Wojciak JM, Martinez-Yamout MA, Dyson HJ, Wright PE. Structural basis for recruitment of CBP/p300 coactivators by STAT1 and STAT2 transactivation domains. EMBO J. 2009;28:948–58. doi: 10.1038/emboj.2009.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Mendez FL, Watkins JC, Hammer MF. A haplotype at STAT2 Introgressed from neanderthals and serves as a candidate of positive selection in Papua New Guinea. Am J Hum Genet. 2012;91:265–74. doi: 10.1016/j.ajhg.2012.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Lin RJ, Liao CL, Lin E, Lin YL. Blocking of the alpha interferon-induced Jak-Stat signaling pathway by Japanese encephalitis virus infection. J Virol. 2004;78:9285–94. doi: 10.1128/JVI.78.17.9285-9294.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Best SM, Morris KL, Shannon JG, Robertson SJ, Mitzel DN, Park GS, et al. Inhibition of interferon-stimulated JAK-STAT signaling by a tick-borne flavivirus and identification of NS5 as an interferon antagonist. J Virol. 2005;79:12828–39. doi: 10.1128/JVI.79.20.12828-12839.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]