

Abstract
Multiple lines of evidence suggest that the transcription factor STAT3 is linked to a protective and reparative response in the heart. Thus, increasing duration or intensity of STAT3 activation ought to minimize damage and improve heart function under conditions of stress. Two recent studies using genetic mouse models, however, report findings that appear to refute this proposition. Unfortunately, studies often approach the question of the role of STAT3 in the heart from the perspective that all STAT3 signaling is equivalent, particularly when it comes to signaling by IL-6 type cytokines, which share the gp130 signaling protein. Moreover, STAT3 activation is typically equated with phosphorylation of a critical tyrosine residue. Yet, STAT3 transcriptional behavior is subject to modulation by serine phosphorylation, acetylation, and redox status of the cell. Unphosphorylated STAT3 is implicated in gene induction as well. Thus, how STAT3 is activated and also what other signaling events are occurring at the same time is likely to impact on the outcome ultimately linked to STAT3. Notably STAT3 may serve as a scaffold protein allowing it to interact with other singling pathways. In this context, canonical gp130 cytokine signaling may function to integrate STAT3 signaling with a protective PI3K/AKT signaling network via mutual involvement of JAK tyrosine kinases. Differences in the extent of integration may occur between those cytokines that signal through gp130 homodimers and those through heterodimers of gp130 with a receptor α chain. Signal integration may have importance not only for deciding the particular gene profile linked to STAT3, but for the newly described mitochondrial stabilization role of STAT3 as well. In addition, disruption of integrated gp130-related STAT3 signaling may occur under conditions of oxidative stress, which negatively impacts on JAK catalytic activity. For these reasons, understanding the importance of STAT3 signaling to heart function requires a greater appreciation of the plasticity of this transcription factor in the context in which it is investigated.
Keywords: JAK1, JAK2, cardiac protection, gene expression, intracellular signaling, oxidative stress, preconditioning, redox, signal transduction
Introduction
Evidence suggests that overall the actions of the transcription factor STAT3 in the heart are beneficial. Some key studies involving genetic mouse models supporting a role for STAT3 in ischemic protection and preventing heart failure are listed in Table 1.1-9 For a comprehensive overview of this rather complicated topic, the reader is referred to several recent articles that chronicle the significant contribution that STAT3 plays in cardiac development, protection and remodeling.10-13 Of particular significance, this transcription factor has been implicated in the protection of cardiac myocytes that is provided by ischemic and pharmacological pre- and postconditioning, delayed ischemic preconditioning and post-infarct remodeling. Not surprisingly, many of the beneficial actions of STAT3 in the heart are ascribed to its transcriptional activity. STAT3 activation in the heart has been implicated (often based on circumstantial evidence) in the upregulation of anti-apoptotic (Bcl-xL),14,15 anti-oxidant (MnSOD and metallothioneins)16,17 and pro-angiogenic (VEGF and VE-cadherin)18 genes, as well as production of protective paracrine factors by endothelial cells.19 Some studies have also attributed anti-fibrotic and anti-inflammatory actions to STAT3 signaling in the heart through suppression of gene expression.20,21 STAT3 might suppress gene expression by well characterized means such as by competing with other transcription factors or cofactors. Alternatively, STAT3 might be linked to inhibition or induction of miRNAs that in turn determine the mRNA expression profile of cardiac cells.22
Table 1. Key genetic mouse models supporting a role for STAT3 in cardiac ischemia-reperfusion and heart failure.
|
Ischemia-reperfusion |
• Ischemic pre/post-conditioning lost in TNFα knockout,1 TNFα receptor knockout,2 IL-6 knockout3 or cardiac-myocyte STAT3 knockout mice4 • Cardiac myocyte STAT3-deficient mice show enhanced sensitivity to ischemia-reperfusion injury5 |
| Heart failure | • Cardiac myocyte STAT3-deficient mice show reduced myocardial capillary density and increased interstitial fibrosis within 4 mo, followed by dilated cardiomyopathy with impaired cardiac function and premature death due to heart failure5 |
| • Cardiac myocyte-targeted STAT3 knockout mice show greater sensitivity to inflammation, cardiac fibrosis and heart failure with advanced age6 | |
| • Cardiac myocyte-specific gp130 knockout mice develop heart failure in response to pressure overload accompanied by increased cardiac myocyte apoptosis7 | |
| • Mice with reduced STAT3 activity/levels have increased susceptibility to doxorubicin-induced heart failure and greater susceptibility to LPS-induced toxicity8 | |
| • Mice with cardiac myocyte-targeted STAT3 overexpression develop cardiac hypertrophy but are resistant to doxorubicin-induced cardiomyopathy9 |
Involvement of STAT3 in both early preconditioning and postconditioning would not by design involve gene expression. In this regard, recent evidence suggests that STAT3 has direct non-transcriptional actions at the level of the mitochondrion that are protective of cardiac function by limiting excessive reactive oxygen species (ROS) generation.23 These mitochondrial actions of STAT3 are poorly understood, but may have significance not only for both preconditioning and postconditioning, but heart failure as well. The nontranscriptional role of STAT3 may be related to the separate observations that STAT3 can serve as a scaffold protein and is redox-sensitive.24-26 The latter attribute of STAT3 may manifest itself by formation of higher order complexes that conceivably could affect its association with other proteins and subcellular distribution.
On the other hand, there is evidence that STAT3 activation in cardiac myocytes may be harmful in certain cases. STAT3 has been linked to pathological cardiac hypertrophy through both canonical (phosphorylation on Y705)9,27,28 and noncanonical (unphosphorylated STAT3 accumulation in the nucleus) means;29 although, as noted by others, convincing evidence linking STAT3 activation under normal levels of expression to actual physical hypertrophy of cardiac myocytes is scant.13 However, a recent study reported that uncontrolled STAT3 activation downstream of a mutant intractable gp130, the common receptor of interleukin-6 (IL-6) cytokines, is harmful to the heart in myocardial infarction by causing excessive inflammation (upregulation of IL-6 and complement-activating mannose-binding lectin C), ventricular rupture, and heart failure.30 A genetic reduction in cardiac myocyte STAT3 levels was able to rescue the mutant gp130 phenotype. In addition, hearts of mice with cardiac myocyte-targeted deletion of SOCS3, the STAT3-induced inhibitor of gp130, were characterized by development at about 25 weeks of age of cardiac contractile dysfunction, various ventricular arrhythmias and signs of heart failure that were preceded by abnormalities in Ca2+ handling and troponin I hypophosphorylation.31 Given previous reports linking STAT3 signaling to cardiac remodeling, it is notable that only “minimal histological abnormalities” were seen in SOCS3 cardiac knockout (KO) failing hearts, although cardiac myocyte hypertrophy was present. The SOCS3 KO phenotype was rescued by simultaneous cardiac-specific gp130 KO.
How can STAT3 activation be both beneficial and harmful to the heart? Unfortunately studies often approach the question of the role of STAT3 in the heart from the perspective that all STAT3 signaling is equivalent, particularly when it comes to signaling by the IL-6 type cytokines, which share the gp130 signaling protein. However, we propose that the context of how STAT3 is activated and also what other signaling events are occurring at the same time will impact on the outcome ultimately linked to STAT3 activation. As an extension of this proposition, a better understanding of the events that regulate STAT3 activation and its transcriptional and nontranscriptional (mitochondrial) behaviors is imperative. To complicate matters further, STAT3 “activation” for the purposes of transcription, and likely for its mitochondrial actions, may involve either tyrosine (Y705) or serine (S727) phosphorylation alone,32-36 both (more typically/canonically)37 or neither,38 and is now known to be impacted as well by acetylation and cellular redox status.13,39,40 In canonical signaling, S727 phosphorylation occurs in either the cytoplasm or nucleus on STAT3 that is likely already phosphorylated on Y705, as prior S727 phosphorylation seems to block Y705 phosphorylation.41 Interestingly S727 phosphorylation may play a critical role in the mitochondrial actions of STAT3.42 Thus, STAT3 S727 phosphorylation in the cytoplasm may function as a switch favoring the mitochondrial actions of STAT3 over its canonical nuclear actions, although this is conjecture and will need to be investigated.
Leukemia Inhibitory Factor (LIF) and LIF Receptor
A number of factors have been shown to induce STAT3 activation in the heart, including prolactin, granulocyte colony-stimulating factor (G-CSF), tumor necrosis factor-α (TNFα), erythropoietin (Epo), opioids, leptin, angiotensin II and insulin.13,43-47 Pre- and postconditioning-induced STAT3 activation is attributable to the release of paracrine factors, such as TNFα and IL-6.10-13,48-50 The increase in ROS that accompanies ischemia-reperfusion (IR) in the heart is also associated with marked STAT3 activation;51 whether ROS activates STAT3 via an effect on upstream JAK kinases, phosphatases or largely through the upregulation or release of paracrine factors is not known.52
Notably, cardiac IR is associated with the production of IL-6 cytokines, which are prominent activators of STAT3 signaling.10-13 The IL-6 family of cytokines signal through the common signaling subunit gp130.10-13 They can be further classified according to those that signal through gp130 homodimers (IL-6 and IL-11), gp130 heterodimers with the LIF receptor [LIF, cardiotrophin-1 (CT-1), ciliary neurotrophic factor (CNTF), cardiotrophin-like cytokine (CLC) and oncostatin M (OSM) in humans], or gp130 heterodimers with the OSM receptor (OSM). Additional ligand-binding proteins are needed for IL-6, IL-11, CNTF, CLC and probably CT-1. LIF likely binds first to the LIF receptor (LIFR/CD118), which then dimerizes with gp130.53 LIFR is ubiquitously expressed in the normal heart and unlike gp130 does not appear to be downregulated (at least appreciably) in human heart failure,54-57 although cardiac levels of LIF are increased in heart failure.58,59
The IL-6 family cytokines are frequently described as characterized by functional redundancy, particularly with regard to signaling in the heart.12 Although different actions among them have been noted, the differences are commonly explained away as due to differences in expression levels of receptors or duration of STAT3 activation. However, marked qualitative differences have been noted among the gp130-related cytokines as far as gene induction and functional impact, in particular between IL-6 and LIF, which are not amenable to a straightforward explanation based on the strength of their respective signaling responses. At an early time, it was noted that IL-6 and LIF differ as far as the induction of acute-phase genes.60 More recently, LIF and IL-6 have been shown to counter-regulate development of the T lymphocyte lineages with IL-6 coupling moreover to the activation of a gene response that downregulates LIF signaling.61
How IL-6 and LIF might exert dissimilar actions is not known. Based on conjecture, LIFR likely modifies the character of the gp130 signal, but how this occurs is not known. LIFR and gp130 are structurally very similar (Fig. 1).62,63 Both contain three box regions with the two membrane proximal ones being important for association with a JAK family kinase and the distal one playing a role in STAT3 and Src-family kinase Hck activation (and possibly reinforcing ERK activation at least for gp130).63,64 gp130 has four YXXQ motifs that upon tyrosine phosphorylation by a JAK family kinase serve as docking sites for STAT3 (and potentially STAT1); the cytoplasmic domain of LIFR is shorter and has three YXXQ motifs.
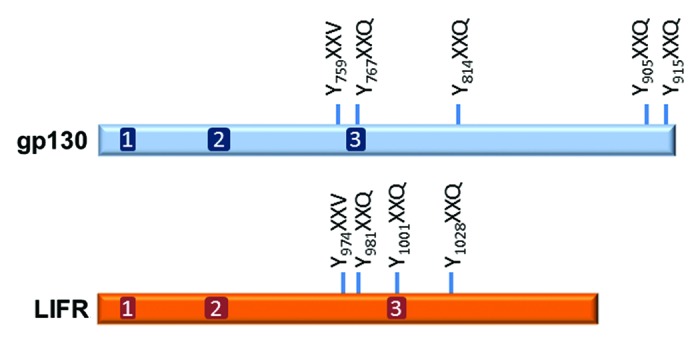
Figure 1. Schematic of the cytoplasmic regions of human gp130 and LIFR showing the relative locations of the three box motifs and the STAT3 YXXQ binding motifs. The YXXV domain is important for linking the receptors to PI3K/AKT and SHP2/MAPK signaling pathways and for termination of signaling by recruiting either SHP2 or SOCS3. SHP2 terminates signaling through its tyrosine phosphatase activity. SOCS3 terminates signaling by inhibiting JAK activity directly through the N-terminal kinase inhibitory region (KIR) or by facilitating the ubiquitination and proteasomal degradation of signaling components.
Both gp130 and LIFR contain a more membrane proximal YXXV motif that upon phosphorylation is important in signal termination by recruiting SHP2 or SOCS3.65 SHP2 terminates signaling through its tyrosine phosphatase activity, but also serves as a scaffold protein linking gp130 and LIFR to additional signaling pathways (Fig. 2).64,66-68 SOCS3 is induced by STAT3 and terminates LIF/IL-6 signaling by inhibiting JAK activity directly through the N-terminal kinase inhibitory region (KIR) or by facilitating ubiquitination and proteasomal degradation of the JAKs, other signaling components, or receptors.37 A significant difference between the YXXV site of LIFR and gp130 is that the former shows little affinity for SOCS3, while the latter binds both SOCS3 and SHP2.65 This might explain why LIF-induced STAT3 signaling is in general sustained (although at a reduced level from the initial increase), while IL-6 activation of STAT3 is brief (personal observation).

Figure 2. Basic signaling similarities and differences of gp130 (right, blue) and LIFR (left, orange). Gp130 has 4 STAT3 binding sites and LIFR has 3. Both receptors couple to ERK1/2 activation through SHP2 functioning as a scaffold protein. The tyrosine phosphatase activity of SHP2 is thought to contribute to termination of receptor signaling. The SHP2 site on gp130 also binds SOCS3, which terminates signaling by inhibiting JAK activity. The tyrosine phosphatase SHP1 associates with JAK1 and contributes as well to termination of gp1320 and LIFR signaling. gp130 contains an acidic domain (light blue) comprising amino acids 771 to 811 that binds the Src-family kinase Hck and couples to ERK1/2 and Pyk2 (not shown) activation.64 Phosphorylation of LIFR on S1044 by ERK1/2 was shown to promote receptor degradation.66 For gp130, S782 phosphorylation may regulate cell surface expression.67 At least for gp130, other phosphorylation events have been reported. PKCδ that is associated with STAT3 may phosphorylate gp130 on T890, helping to stabilize STAT3-gp130 association.68
In addition to JAK-STAT3 signaling, LIFR and gp130 couple to the activation of 2 major signaling pathways: SHP2/MAPK and phosphatidylinositol 3-kinase (PI3K)/AKT.10,12,69 Whether LIFR and gp130 couple equally well to both of these pathways is not known. The MAPK that have been best studied are ERK1/2 and ERK5. Tyrosine phosphorylation of SHP2 leads to its interaction with Grb2 (growth factor receptor bound protein) and SOS (son of sevenless), which triggers the Ras/RAF/MEK/ERK cascade (Fig. 2). Tyrosine phosphorylated SHP2 was linked to ERK5 activation via Grb2-associated binder-1 (Gab1), a scaffolding/docking protein which likely contributes to protective signaling as well (Fig. 3).70-75 ERK5 activation is thought to be responsible for the unique hypertrophic phenotype of longitudinal elongation produced by the IL-6 type cytokines on cardiac myocytes. Details on how LIFR and gp130 couple to PI3K/AKT signaling is not known, although the PI3K regulatory subunit p85 likely associates with SHP2 via Gab1 as well (Fig. 3). PI3K in turn leads to activation of AKT and a diverse series of signaling pathways. STAT3 appears to serve as a scaffold protein that helps assemble an activation module with JAKs activating STAT3 and indirectly, PI3K and AKT, and in turn PI3K “activating” STAT3 by phosphorylating S727.24,52,76

Figure 3. Coupling of gp130 (blue, right) to cardiac protective signaling. Similar events pertain to LIFR (orange, left), but for simplicity are not shown. The scaffold protein Gab1 forms a central point of a signaling complex linking JAK1-p85 (the regulatory subunit of PI3K), SHP2-p85, and STAT3-p85. The PI3K catalytic subunit p110 is activated resulting in creation of phosphorylated phosphatidylinositol binding sites for AKT. Once at the membrane, AKT is activated by phosphorylation by phosphoinositide dependent protein kinase 1 (PDPK1) and mammalian target of rapamycin complex 2 (mTORC2). AKT plays a role in both the trigger and mediator phases of pre- and postconditiong.70-73 The role of AKT in the mediator phase is illustrated here. AKT and ERK1/2 comprise the reperfusion injury salvage kinase (RISK) pathway. Both kinases phosphorylate and inhibit glycogen synthase kinase 3 β (GSK3β) leading to inhibition of mitochondrial permeability transition pore (MPTP) opening, which can cause cell death. ERK1/2 and AKT can inhibit GSK3β as well via nitric oxide synthase 3 (NOS3) activation. AKT also prevents MPTP opening by activating hexokinase II (HKII). Activation of STAT3 constitutes the survivor activating factor enhancement (SAFE) pathway for cardiac ischemic protection.71 Long-term STAT3 is thought to induce genes that are protective. A mitochondrial role for STAT3 has been proposed to explain short-term actions and may preferentially involve STAT3 phosphorylated on S727 (green circle). GAB1 and SHP2 are also linked to cardiac hypertrophy caused by the IL-6 type cytokines via ERK5 activation.
Notably, PI3K/AKT signaling is essential for conferring cardioprotection in response to ischemic pre- and postconditioning stimuli, being involved in both the trigger and mediator phases.73 Its involvement in the latter forms part of the reperfusion injury salvage kinase (RISK) pathway that confers protection by attenuating opening of the mitochondrial permeability transition pore (MPTP) through as yet undefined means (Fig. 3). A second set of intracellular signaling events that operates independently of the RISK pathway as a trigger for protection and also confers protection during reperfusion by targeting MPTP opening was recently identified and involves STAT3 activation. It was named the survivor activating factor enhancement (SAFE) pathway.76-78 Details on how the SAFE pathway works at the level of the mitochondrion are not known. Thus, the IL-6 family of cytokines have in theory the potential of conferring protection to cardiac myocytes from IR injury by activating both the SAFE and RISK pathways (Fig. 3). Indeed, the LIFR ligand CT-1 was shown to protect isolated cardiac myocytes and the adult rat heart from injury when added either just prior to ischemia or at reoxygenation/reperfusion.79,80 This was not the case with human heart muscle preparations which required longer exposure to CT-1 to confer protection indicating that gene expression was involved.81
A carboxyl-terminal acidic domain of gp130 was shown to couple to cellular proliferation and inhibition of stem cell differentiation through the binding and activation of Src family kinases, in particular Hck (Fig. 2).63,64 Hck represents an additional means by which gp130 may be linked to ERK activation. The relevance of this signaling pathway in the heart has not been explored. Neither is it known if Hck (or another Src family member) modulates gp130 signaling; however, such a scenario might explain the increased tyrosine phosphorylation of gp130 in the face of reduced tyrosine phosphorylation of JAK2 in hearts of patients with end-stage dilated cardiomyopathy.57
The focus of our research has been LIF, which has been shown to be produced by cardiac myocytes and to have protective effects on heart cells. Pretreatment of adult or neonatal cardiac myocytes with LIF protected against hypoxia-reoxygenation or doxorubicin-induced injury at a later (6–12 h) time point.15,16,82,83 LIF treatment was also shown to protect the heart from IR injury or myocardial infarction.84,85 These beneficial actions of LIF are attributed in part to the stimulation of angiogenesis and upregulation of MnSOD, Bcl-xl and VEGF.16,83-87 LIF was also shown to have effects on the growth, metabolism, contractility and Ca2+ handling of cardiac myocytes, which might overall be considered disadvantageous; however, because these studies mainly relied on cultured cells or isolated muscle the physiological significance of these effects is uncertain.74,75,88-95
Several recent observations support the conclusion that production of LIF by cardiac myocytes may have physiological importance under stress conditions in myocardial repair and regeneration beyond a protective action on the cells themselves. In the mouse, LIF was found to contribute to the homing of bone marrow-derived cardiac progenitors to the infarcted myocardium and the differentiation of resident cardiac stem cells into endothelial cells.96,97 Second, in a rat genetic model of heart failure, myocardium-produced LIF was shown to cause cholinergic transdifferentiation of cardiac sympathetic nerves, which might represent a means of protecting the heart from excessive sympathetic drive.59
STAT3—Too Much of a Good Thing?
Two recent studies seem to provide evidence supporting the old adage that too much of a good thing is harmful in the case of STAT3 activation in the injured or aging myocardium. Or do they? In one study, sustained activation of STAT3 in cardiac myocytes following a stress stimulus (MI) was achieved by expressing a mutant gp130 protein (Y757F) that does not bind SOCS3 in mice that also have a heart targeted deletion of gp130.30 Thus, the “rescued” gp130 signaling in cardiac myocytes of these mice was refractory to inhibition by SOCS3. Of note, the SOCS3 recruitment site is important for SHP2 binding and thus coupling of gp130 to MAPK, and possibly to PI3K/AKT, signaling would be expected to be impaired as well. Indeed, hearts harboring the mutant gp130 protein showed no increase over wild type mice in either ERK or AKT activation following MI, although STAT3 pY levels were markedly enhanced. The other study employed cardiac myocyte-targeted SOCS3 KO mice, which not only showed increased STAT3 pY levels, but increased AKT, ERK, and p38 MAPK activation.31 These signaling pathways were enhanced under basal conditions. The fact that STAT3 activation was enhanced under basal conditions in the SOCS3 KO model, but not the gp130 Y757F model, indicates that something was driving gp130 signaling (which could be characterized as complete) in the SOCS3 KO model in a feed-forward manner. Consequently, the SOCS3 KO mice developed cardiac hypertrophy and heart failure with age without additional stress. In these mice, imposition of stress by transverse aortic constriction further enhanced gp130 signaling and caused cardiac dysfunction. Together, both studies would seem to refute the oft-cited supposition that sustained STAT3 activation is beneficial and anti-inflammatory (as seen with IL-10), while brief STAT3 activation is harmful and pro-inflammatory (as seen with IL-6).98
STAT1 activation was not enhanced in either model. This is somewhat surprising as the genetic reprogramming of IL-6 signaling in SOCS3 KO macrophages was attributed to enhanced STAT1 activation, owing perhaps to enhanced recruitment of the two more membrane distal YXXQ STAT binding sites of gp130 that are capable of activating both STAT1 and STAT3.99,100 However, increased STAT1 activation was not found in the heart in either the SOCS3 KO (“preliminary observation”; personal communication, Dr Yajima) or the gp130 Y757F model.30 This would have neatly explained deleterious consequences of enhanced gp130 signaling as several lines of evidence have shown that STAT1 has pro-apoptotic actions in cardiac myocytes, both as a transcription factor and as a signaling molecule.101
Although both models were associated with increased STAT3 activation (tyrosine phosphorylation) there are marked differences in the phenotypes of the two models that are illuminating and indicate that prolonged STAT3 activation was not functionally equivalent in both (Table 2). Of note, the gp130 Y757F model was associated with enhanced cardiac inflammation, while the SOCS3 KO model was not (although inflammation in the gp130 Y757F model was assessed in hearts subjected to MI, whereas in the SOCS3 KO model inflammation was assessed under basal conditions). Since inflammation was observed in the instance where there was no concomitantly enhanced PI3K/AKT signaling, enhanced STAT3 signaling may not be inflammatory perhaps as long as it is balanced (both in magnitude and temporally) by enhanced PI3K signaling. Consistent with this possibility is the finding that cardiac myocyte-targeted STAT3 overexpression was associated with cardiac hypertrophy, but was protective against doxorubicin-induced heart failure.9
Table 2. Comparison of two genetic mouse models of sustained cardiac STAT3 activation.
| |
MODEL |
|||
|---|---|---|---|---|
| |
Y757F |
SOCS3 KO |
||
| Baseline* | Stress (MI) | Baseline | Stress (TAC) | |
| pY STAT3 |
≅ |
↑ (> > WT)** |
↑ |
↑ (> WT) |
| pS STAT3 |
nr |
nr |
nr |
nr |
| STAT3 |
≅ |
↑ (later times) |
≅ |
≅ |
| pYSTAT1 |
≅ |
≅ |
≅ (initial screening) |
nr |
| STAT1 |
≅ |
≅ |
≅ |
nr |
| PI3K/AKT |
≅ |
≅ (↓) |
↑ (AKT)† |
↑ (AKT) (> WT?) |
| SHP2/MAPK |
≅ |
≅ (modest ↑) |
↑ (ERK, p38)† |
↑ (ERK) (> WT?) |
| Phenotype | Normal cardiac function and morphology | ↑ mortality ↑ LV rupture ↑ cardiac Inflammation Heart failure |
Cardiac hypertrophy Heart failure (No cm disarray, necrosis, apoptosis, inflammation or interstitial fibrosis) |
Cardiac hypertrophy Cardiac dysfunction |
At 3 mo
Both level and duration
† At 15 weeks, but not 8 weeks. Symbols/abbreviations: ≅, no difference compared with wild type; cm, cardiac myocyte; nr, not reported; WT, wild type; ↑, increase; ↓, decrease.
While STAT3 signaling is important for the anti-inflammatory actions of IL-10, the basis for why IL-10-induced STAT3 signaling is anti-inflammatory is still unresolved; however, evidence was recently presented that a subset of STAT3-induced anti-inflammatory genes in macrophages in response to IL-10 treatment is dependent upon PI3K-mediated signaling.102 In this scenario, cytokine-induced activation of inflammatory genes by PI3K/AKT-mediated activation of NFκB (or by STAT3) may be offset by the actions of PI3K/AKT on STAT3 signaling (as long as the latter is sustained). Moreover, NFκB p65 and p50 are known to physically interact with functional consequences regardless of whether STAT3 is tyrosine and/or serine phosphorylated.103 Conceivably, different phosphorylation (or acetylation) profiles of STAT3 might be associated with different gene expression profiles linked to STAT-NFκB association.
PI3K signaling and JAK-STAT signaling could converge at multiple sites in both the cytoplasm and nucleus to affect expression of a particular gene, but of note the PI3K/AKT pathway also induces acetylation of lysine residues on STAT3 that affect dimerization and transcriptional behavior.39,40,104-106 In this regard, the gp130 Y757F model was associated with increased levels of cardiac STAT3, which may have been due to STAT3-induced STAT3 expression.38 Accumulating evidence shows that accumulation of unphosphorylated STAT3 in the nucleus can drive expression of a set of pro-inflammatory genes, including IL-6, which was in fact increased in the gp130 Y757F model.30,38,107 Thus, under certain conditions STAT3 activation sets into play a series of events that lead to a sustained inflammatory response.
Redox Sensitivity
Coupling of LIF and the other IL-6 family cytokines to a balanced PI3K-STAT3 response requires JAK activation. The increased STAT3 “activation” (i.e., increased tyrosine phosphorylation) that is observed with IR need not reflect a JAK-mediated event as protein tyrosine phosphatases are known to be inactivated by oxidative stress.108 In fact, we found that oxidative stress inhibits LIF-induced JAK1 and JAK2 activity in cardiac myocytes.109 Since then site-directed mutagenesis experiments were performed to demonstrate that two nearby cysteine residues in the amino-teminus region of JAK2’s catalytic domain act together as a redox-sensitive switch.110 The presence of this switch would thus permit the catalytic activity of JAK2 to be directly regulated by the redox state of the cell. Of note, these cysteine residues are highly conserved in both JAK1 and JAK2 among mammalian and most lower-order species.
STAT3 transcriptional response may also be directly affected by oxidative stress due to the presence of redox-sensitive cysteines.25,111 Oxidative stress undoubtedly impacts on STAT3 transcriptional signaling indirectly in multiple ways as well. We recently presented evidence that oxidative stress may attenuate LIF-induced gene expression in human microvascular endothelial cells by causing serine phosphorylation and degradation of STAT3's binding partner and transcriptional cofactor p300/CBP.112
Mitochondrial STAT3
Recent studies demonstrate STAT3 localizes to mitochondria of different cell types and regulates respiration.23,42,113,114 STAT3 deletion reduced respiration of cardiac myocyte mitochondria due to 50% decrease in activities of complexes I and II.42 Loss of mitochondrial STAT3 decreased ATP production and enhanced ROS generation.115 These findings are consistent with reports of increased ROS formation by complex I with decreased activity, as seen in the heart with ischemia or age.116,117 Besides regulating the electron transport chain, STAT3 associates with matrix-localized cyclophilin D, the target of mitochondrial permeability transition pore (MPTP) inhibitor cyclosporine.113 In fact, mitochondria from STAT3−/− hearts undergo MPTP opening at lower calcium levels, which has significance for ischemia-reperfusion injury.113 Recently, cardiac myocyte-specific overexpression of mitochondria-targeted STAT3 was found to partially block complexes I and II with no increased basal ROS production; however, there was no ischemia-induced ROS release from complex I and less reduction in complex I activity with ischemia.118 How STAT3 regulates complexes I/II is unknown, but some intermediary process is likely, since the ratio of complexes I and II to mitochondrial STAT3 is ~105.113,114 S727 phosphorylation would seem to be important in STAT3's mitochondrial actions. Impaired activity of complexes I and II of mitochondria from STAT3–/– pro-B cells could be restored by a mimetic of constitutively serine phosphorylated STAT3 (STAT3 Y705F/S727D), while STAT3 Y705F/S727A was ineffective.42 Also, a number of studies report that STAT3 pS727 is enriched in mitochondria compared with the cytoplasm;119-122 however, this may not be the case for all species.123 The question of whether ischemic pre- or post-conditioning dynamically regulate mitochondrial STAT3 levels or phosphorylation profile has not been determined.124 Conceivably, translocation of STAT3 to mitochondria could function as part of a feedback loop to control mitochondrial generation of ROS.
Conclusions and Future Directions
The transcription factor STAT3 has been implicated in a protective and reparative response in the heart. Thus, increasing duration or intensity of STAT3 activation ought to minimize damage and improve heart function under conditions of stress. However, given the many ways that STAT3 is post-translationally modified and its interaction with other signaling networks, the assumption that enhanced STAT3 activity is solely a reflection of increased tyrosine phosphorylation must be construed as a gross over-simplification. Understanding the beneficial importance of STAT3 signaling to heart function will require a greater appreciation of the context in which it is activated. In this regard, defining the differences in the impact on the heart between gp130 homodimer and heterodimer STAT3 activation will be illuminating.
Acknowledgments
This work was supported by a grant from NHLBI to G.W.B. (5R01HL088101-05) grants from the Lebanese University (MK-02-2011), the Lebanese National Council for Scientific Research (CNRS 05-10-09), and the COMSTECH-TWAS (09-122 RG/PHA/AF/AC_C) to M.K.
Footnotes
Previously published online: www.landesbioscience.com/journals/jak-stat/article/19776
References
- 1.Smith RM, Suleman N, McCarthy J, Sack MN. Classic ischemic but not pharmacologic preconditioning is abrogated following genetic ablation of the TNFalpha gene. Cardiovasc Res. 2002;55:553–60. doi: 10.1016/S0008-6363(02)00283-3. [DOI] [PubMed] [Google Scholar]
- 2.Flaherty MP, Guo Y, Tiwari S, Rezazadeh A, Hunt G, Sanganalmath SK, et al. The role of TNF-α receptors p55 and p75 in acute myocardial ischemia/reperfusion injury and late preconditioning. J Mol Cell Cardiol. 2008;45:735–41. doi: 10.1016/j.yjmcc.2008.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dawn B, Xuan YT, Guo Y, Rezazadeh A, Stein AB, Hunt G, et al. IL-6 plays an obligatory role in late preconditioning via JAK-STAT signaling and upregulation of iNOS and COX-2. Cardiovasc Res. 2004;64:61–71. doi: 10.1016/j.cardiores.2004.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Smith RM, Suleman N, Lacerda L, Opie LH, Akira S, Chien KR, et al. Genetic depletion of cardiac myocyte STAT-3 abolishes classical preconditioning. Cardiovasc Res. 2004;63:611–6. doi: 10.1016/j.cardiores.2004.06.019. [DOI] [PubMed] [Google Scholar]
- 5.Hilfiker-Kleiner D, Hilfiker A, Fuchs M, Kaminski K, Schaefer A, Schieffer B, et al. Signal transducer and activator of transcription 3 is required for myocardial capillary growth, control of interstitial matrix deposition, and heart protection from ischemic injury. Circ Res. 2004;95:187–95. doi: 10.1161/01.RES.0000134921.50377.61. [DOI] [PubMed] [Google Scholar]
- 6.Jacoby JJ, Kalinowski A, Liu MG, Zhang SS, Gao Q, Chai GX, et al. Cardiomyocyte-restricted knockout of STAT3 results in higher sensitivity to inflammation, cardiac fibrosis, and heart failure with advanced age. Proc Natl Acad Sci U S A. 2003;100:12929–34. doi: 10.1073/pnas.2134694100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hirota H, Chen J, Betz UA, Rajewsky K, Gu Y, Ross J, Jr., et al. Loss of a gp130 cardiac muscle cell survival pathway is a critical event in the onset of heart failure during biomechanical stress. Cell. 1999;97:189–98. doi: 10.1016/S0092-8674(00)80729-1. [DOI] [PubMed] [Google Scholar]
- 8.Shen Y, La Perle KM, Levy DE, Darnell JE., Jr Reduced STAT3 activity in mice mimics clinical disease syndromes. Biochem Biophys Res Commun. 2005;330:305–9. doi: 10.1016/j.bbrc.2005.02.154. [DOI] [PubMed] [Google Scholar]
- 9.Kunisada K, Negoro S, Tone E, Funamoto M, Osugi T, Yamada S, et al. Signal transducer and activator of transcription 3 in the heart transduces not only a hypertrophic signal but a protective signal against doxorubicin-induced cardiomyopathy. Proc Natl Acad Sci U S A. 2000;97:315–9. doi: 10.1073/pnas.97.1.315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fujio Y, Maeda M, Mohri T, Obana M, Iwakura T, Hayama A, et al. Glycoprotein 130 cytokine signal as a therapeutic target against cardiovascular diseases. J Pharmacol Sci. 2011;117:213–22. doi: 10.1254/jphs.11R05CR. [DOI] [PubMed] [Google Scholar]
- 11.Boengler K, Hilfiker-Kleiner D, Drexler H, Heusch G, Schulz R. The myocardial JAK/STAT pathway: from protection to failure. Pharmacol Ther. 2008;120:172–85. doi: 10.1016/j.pharmthera.2008.08.002. [DOI] [PubMed] [Google Scholar]
- 12.Fischer P, Hilfiker-Kleiner D. Survival pathways in hypertrophy and heart failure: the gp130-STAT axis. Basic Res Cardiol. 2007;102:393–411. doi: 10.1007/s00395-007-0674-z. [DOI] [PubMed] [Google Scholar]
- 13.Haghikia A, Stapel B, Hoch M, Hilfiker-Kleiner D. STAT3 and cardiac remodeling. Heart Fail Rev. 2011;16:35–47. doi: 10.1007/s10741-010-9170-x. [DOI] [PubMed] [Google Scholar]
- 14.Higuchi T, Yamauchi-Takihara K, Matsumiya G, Fukushima N, Ichikawa H, Kuratani T, et al. Granulocyte colony-stimulating factor prevents reperfusion injury after heart preservation. Ann Thorac Surg. 2008;85:1367–73. doi: 10.1016/j.athoracsur.2007.12.053. [DOI] [PubMed] [Google Scholar]
- 15.Izumi M, Masaki M, Hiramoto Y, Sugiyama S, Kuroda T, Terai K, et al. Cross-talk between bone morphogenetic protein 2 and leukemia inhibitory factor through ERK 1/2 and Smad1 in protection against doxorubicin-induced injury of cardiomyocytes. J Mol Cell Cardiol. 2006;40:224–33. doi: 10.1016/j.yjmcc.2005.11.007. [DOI] [PubMed] [Google Scholar]
- 16.Negoro S, Kunisada K, Fujio Y, Funamoto M, Darville MI, Eizirik DL, et al. Activation of signal transducer and activator of transcription 3 protects cardiomyocytes from hypoxia/reoxygenation-induced oxidative stress through the upregulation of manganese superoxide dismutase. Circulation. 2001;104:979–81. doi: 10.1161/hc3401.095947. [DOI] [PubMed] [Google Scholar]
- 17.Oshima Y, Fujio Y, Nakanishi T, Itoh N, Yamamoto Y, Negoro S, et al. STAT3 mediates cardioprotection against ischemia/reperfusion injury through metallothionein induction in the heart. Cardiovasc Res. 2005;65:428–35. doi: 10.1016/j.cardiores.2004.10.021. [DOI] [PubMed] [Google Scholar]
- 18.Osugi T, Oshima Y, Fujio Y, Funamoto M, Yamashita A, Negoro S, et al. Cardiac-specific activation of signal transducer and activator of transcription 3 promotes vascular formation in the heart. J Biol Chem. 2002;277:6676–81. doi: 10.1074/jbc.M108246200. [DOI] [PubMed] [Google Scholar]
- 19.Wang M, Zhang W, Crisostomo P, Markel T, Meldrum KK, Fu XY, et al. Endothelial STAT3 plays a critical role in generalized myocardial proinflammatory and proapoptotic signaling. Am J Physiol Heart Circ Physiol. 2007;293:H2101–8. doi: 10.1152/ajpheart.00125.2007. [DOI] [PubMed] [Google Scholar]
- 20.Manukyan MC, Alvernaz CH, Poynter JA, Wang Y, Brewster BD, Weil BR, et al. Interleukin-10 protects the ischemic heart from reperfusion injury via the STAT3 pathway. Surgery. 2011;150:231–9. doi: 10.1016/j.surg.2011.05.017. [DOI] [PubMed] [Google Scholar]
- 21.Obana M, Maeda M, Takeda K, Hayama A, Mohri T, Yamashita T, et al. Therapeutic activation of signal transducer and activator of transcription 3 by interleukin-11 ameliorates cardiac fibrosis after myocardial infarction. Circulation. 2010;121:684–91. doi: 10.1161/CIRCULATIONAHA.109.893677. [DOI] [PubMed] [Google Scholar]
- 22.Haghikia A, Missol-Kolka E, Tsikas D, Venturini L, Brundiers S, Castoldi M, et al. Signal transducer and activator of transcription 3-mediated regulation of miR-199a-5p links cardiomyocyte and endothelial cell function in the heart: a key role for ubiquitin-conjugating enzymes. Eur Heart J. 2011;32:1287–97. doi: 10.1093/eurheartj/ehq369. [DOI] [PubMed] [Google Scholar]
- 23.Szczepanek K, Chen Q, Larner AC, Lesnefsky EJ. Cytoprotection by the modulation of mitochondrial electron transport chain: The emerging role of mitochondrial STAT3. Mitochondrion. 2012;12:180–9. doi: 10.1016/j.mito.2011.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pfeffer LM, Mullersman JE, Pfeffer SR, Murti A, Shi W, Yang CH. STAT3 as an adapter to couple phosphatidylinositol 3-kinase to the IFNAR1 chain of the type I interferon receptor. Science. 1997;276:1418–20. doi: 10.1126/science.276.5317.1418. [DOI] [PubMed] [Google Scholar]
- 25.Li L, Cheung SH, Evans EL, Shaw PE. Modulation of gene expression and tumor cell growth by redox modification of STAT3. Cancer Res. 2010;70:8222–32. doi: 10.1158/0008-5472.CAN-10-0894. [DOI] [PubMed] [Google Scholar]
- 26.Kojima H, Sasaki T, Ishitani T, Iemura S, Zhao H, Kaneko S, et al. STAT3 regulates Nemo-like kinase by mediating its interaction with IL-6-stimulated TGFbeta-activated kinase 1 for STAT3 Ser-727 phosphorylation. Proc Natl Acad Sci U S A. 2005;102:4524–9. doi: 10.1073/pnas.0500679102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Uozumi H, Hiroi Y, Zou Y, Takimoto E, Toko H, Niu P, et al. gp130 plays a critical role in pressure overload-induced cardiac hypertrophy. J Biol Chem. 2001;276:23115–9. doi: 10.1074/jbc.M100814200. [DOI] [PubMed] [Google Scholar]
- 28.Li Y, Zhang H, Liao W, Song Y, Ma X, Chen C, et al. Transactivated EGFR mediates α₁-AR-induced STAT3 activation and cardiac hypertrophy. Am J Physiol Heart Circ Physiol. 2011;301:H1941–51. doi: 10.1152/ajpheart.00338.2011. [DOI] [PubMed] [Google Scholar]
- 29.Yue H, Li W, Desnoyer R, Karnik SS. Role of nuclear unphosphorylated STAT3 in angiotensin II type 1 receptor-induced cardiac hypertrophy. Cardiovasc Res. 2010;85:90–9. doi: 10.1093/cvr/cvp285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hilfiker-Kleiner D, Shukla P, Klein G, Schaefer A, Stapel B, Hoch M, et al. Continuous glycoprotein-130-mediated signal transducer and activator of transcription-3 activation promotes inflammation, left ventricular rupture, and adverse outcome in subacute myocardial infarction. Circulation. 2010;122:145–55. doi: 10.1161/CIRCULATIONAHA.109.933127. [DOI] [PubMed] [Google Scholar]
- 31.Yajima T, Murofushi Y, Zhou H, Park S, Housman J, Zhong ZH, et al. Absence of SOCS3 in the cardiomyocyte increases mortality in a gp130-dependent manner accompanied by contractile dysfunction and ventricular arrhythmias. Circulation. 2011;124:2690–701. doi: 10.1161/CIRCULATIONAHA.111.028498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shen Y, Schlessinger K, Zhu X, Meffre E, Quimby F, Levy DE, et al. Essential role of STAT3 in postnatal survival and growth revealed by mice lacking STAT3 serine 727 phosphorylation. Mol Cell Biol. 2004;24:407–19. doi: 10.1128/MCB.24.1.407-419.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Qin HR, Kim HJ, Kim JY, Hurt EM, Klarmann GJ, Kawasaki BT, et al. Activation of signal transducer and activator of transcription 3 through a phosphomimetic serine 727 promotes prostate tumorigenesis independent of tyrosine 705 phosphorylation. Cancer Res. 2008;68:7736–41. doi: 10.1158/0008-5472.CAN-08-1125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hazan-Halevy I, Harris D, Liu Z, Liu J, Li P, Chen X, et al. STAT3 is constitutively phosphorylated on serine 727 residues, binds DNA, and activates transcription in CLL cells. Blood. 2010;115:2852–63. doi: 10.1182/blood-2009-10-230060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kim JH, Yoon MS, Chen J. Signal transducer and activator of transcription 3 (STAT3) mediates amino acid inhibition of insulin signaling through serine 727 phosphorylation. J Biol Chem. 2009;284:35425–32. doi: 10.1074/jbc.M109.051516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schaefer LK, Wang S, Schaefer TS. c-Src activates the DNA binding and transcriptional activity of Stat3 molecules: serine 727 is not required for transcriptional activation under certain circumstances. Biochem Biophys Res Commun. 1999;266:481–7. doi: 10.1006/bbrc.1999.1853. [DOI] [PubMed] [Google Scholar]
- 37.Kurdi M, Booz GW. Can the protective actions of JAK-STAT in the heart be exploited therapeutically? Parsing the regulation of interleukin-6-type cytokine signaling. J Cardiovasc Pharmacol. 2007;50:126–41. doi: 10.1097/FJC.0b013e318068dd49. [DOI] [PubMed] [Google Scholar]
- 38.Cheon H, Yang J, Stark GR. The functions of signal transducers and activators of transcriptions 1 and 3 as cytokine-inducible proteins. J Interferon Cytokine Res. 2011;31:33–40. doi: 10.1089/jir.2010.0100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hou T, Ray S, Lee C, Brasier AR. The STAT3 NH2-terminal domain stabilizes enhanceosome assembly by interacting with the p300 bromodomain. J Biol Chem. 2008;283:30725–34. doi: 10.1074/jbc.M805941200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ohbayashi N, Ikeda O, Taira N, Yamamoto Y, Muromoto R, Sekine Y, et al. LIF- and IL-6-induced acetylation of STAT3 at Lys-685 through PI3K/Akt activation. Biol Pharm Bull. 2007;30:1860–4. doi: 10.1248/bpb.30.1860. [DOI] [PubMed] [Google Scholar]
- 41.Booz GW, Day JN, Baker KM. Interplay between the cardiac renin angiotensin system and JAK-STAT signaling: role in cardiac hypertrophy, ischemia/reperfusion dysfunction, and heart failure. J Mol Cell Cardiol. 2002;34:1443–53. doi: 10.1006/jmcc.2002.2076. [DOI] [PubMed] [Google Scholar]
- 42.Wegrzyn J, Potla R, Chwae YJ, Sepuri NB, Zhang Q, Koeck T, et al. Function of mitochondrial Stat3 in cellular respiration. Science. 2009;323:793–7. doi: 10.1126/science.1164551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Harada M, Qin Y, Takano H, Minamino T, Zou Y, Toko H, et al. G-CSF prevents cardiac remodeling after myocardial infarction by activating the Jak-Stat pathway in cardiomyocytes. Nat Med. 2005;11:305–11. doi: 10.1038/nm1199. [DOI] [PubMed] [Google Scholar]
- 44.Lecour S, James RW. When are pro-inflammatory cytokines SAFE in heart failure? Eur Heart J. 2011;32:680–5. doi: 10.1093/eurheartj/ehq484. [DOI] [PubMed] [Google Scholar]
- 45.Gross ER, Hsu AK, Gross GJ. The JAK/STAT pathway is essential for opioid-induced cardioprotection: JAK2 as a mediator of STAT3, Akt, and GSK-3 β. Am J Physiol Heart Circ Physiol. 2006;291:H827–34. doi: 10.1152/ajpheart.00003.2006. [DOI] [PubMed] [Google Scholar]
- 46.Wang W, Kagaya Y, Asaumi Y, Fukui S, Takeda M, Shimokawa H. Protective effects of recombinant human erythropoietin against pressure overload-induced left ventricular remodeling and premature death in mice. Tohoku J Exp Med. 2011;225:131–43. doi: 10.1620/tjem.225.131. [DOI] [PubMed] [Google Scholar]
- 47.Fuglesteg BN, Suleman N, Tiron C, Kanhema T, Lacerda L, Andreasen TV, et al. Signal transducer and activator of transcription 3 is involved in the cardioprotective signalling pathway activated by insulin therapy at reperfusion. Basic Res Cardiol. 2008;103:444–53. doi: 10.1007/s00395-008-0728-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kleinbongard P, Schulz R, Heusch G. TNFα in myocardial ischemia/reperfusion, remodeling and heart failure. Heart Fail Rev. 2011;16:49–69. doi: 10.1007/s10741-010-9180-8. [DOI] [PubMed] [Google Scholar]
- 49.Hausenloy DJ, Lecour S, Yellon DM. Reperfusion injury salvage kinase and survivor activating factor enhancement prosurvival signaling pathways in ischemic postconditioning: two sides of the same coin. Antioxid Redox Signal. 2011;14:893–907. doi: 10.1089/ars.2010.3360. [DOI] [PubMed] [Google Scholar]
- 50.Huffman LC, Koch SE, Butler KL. Coronary effluent from a preconditioned heart activates the JAK-STAT pathway and induces cardioprotection in a donor heart. Am J Physiol Heart Circ Physiol. 2008;294:H257–62. doi: 10.1152/ajpheart.00769.2007. [DOI] [PubMed] [Google Scholar]
- 51.McCormick J, Barry SP, Sivarajah A, Stefanutti G, Townsend PA, Lawrence KM, et al. Free radical scavenging inhibits STAT phosphorylation following in vivo ischemia/reperfusion injury. FASEB J. 2006;20:2115–7. doi: 10.1096/fj.06-6188fje. [DOI] [PubMed] [Google Scholar]
- 52.Kurdi M, Booz GW. JAK redux: a second look at the regulation and role of JAKs in the heart. Am J Physiol Heart Circ Physiol. 2009;297:H1545–56. doi: 10.1152/ajpheart.00032.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Giese B, Roderburg C, Sommerauer M, Wortmann SB, Metz S, Heinrich PC, et al. Dimerization of the cytokine receptors gp130 and LIFR analysed in single cells. J Cell Sci. 2005;118:5129–40. doi: 10.1242/jcs.02628. [DOI] [PubMed] [Google Scholar]
- 54.Zolk O, Ng LL, O’Brien RJ, Weyand M, Eschenhagen T. Augmented expression of cardiotrophin-1 in failing human hearts is accompanied by diminished glycoprotein 130 receptor protein abundance. Circulation. 2002;106:1442–6. doi: 10.1161/01.CIR.0000033117.39335.DF. [DOI] [PubMed] [Google Scholar]
- 55.Eiken HG, Øie E, Damås JK, Yndestad A, Bjerkeli V, Aass H, et al. Myocardial gene expression of leukaemia inhibitory factor, interleukin-6 and glycoprotein 130 in end-stage human heart failure. Eur J Clin Invest. 2001;31:389–97. doi: 10.1046/j.1365-2362.2001.00795.x. [DOI] [PubMed] [Google Scholar]
- 56.González A, Ravassa S, Loperena I, López B, Beaumont J, Querejeta R, et al. Association of depressed cardiac gp130-mediated antiapoptotic pathways with stimulated cardiomyocyte apoptosis in hypertensive patients with heart failure. J Hypertens. 2007;25:2148–57. doi: 10.1097/HJH.0b013e32828626e2. [DOI] [PubMed] [Google Scholar]
- 57.Podewski EK, Hilfiker-Kleiner D, Hilfiker A, Morawietz H, Lichtenberg A, Wollert KC, et al. Alterations in Janus kinase (JAK)-signal transducers and activators of transcription (STAT) signaling in patients with end-stage dilated cardiomyopathy. Circulation. 2003;107:798–802. doi: 10.1161/01.CIR.0000057545.82749.FF. [DOI] [PubMed] [Google Scholar]
- 58.Jougasaki M, Leskinen H, Larsen AM, Cataliotti A, Chen HH, Burnett JC., Jr. Leukemia inhibitory factor is augmented in the heart in experimental heart failure. Eur J Heart Fail. 2003;5:137–45. doi: 10.1016/S1388-9842(02)00236-2. [DOI] [PubMed] [Google Scholar]
- 59.Kanazawa H, Ieda M, Kimura K, Arai T, Kawaguchi-Manabe H, Matsuhashi T, et al. Heart failure causes cholinergic transdifferentiation of cardiac sympathetic nerves via gp130-signaling cytokines in rodents. J Clin Invest. 2010;120:408–21. doi: 10.1172/JCI39778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Baumann H, Marinkovic-Pajovic S, Won KA, Jones VE, Campos SP, Jahreis GP, et al. The action of interleukin 6 and leukaemia inhibitory factor on liver cells. Ciba Found Symp. 1992;167:100–14, discussion 114-24. doi: 10.1002/9780470514269.ch7. [DOI] [PubMed] [Google Scholar]
- 61.Metcalfe SM. LIF in the regulation of T-cell fate and as a potential therapeutic. Genes Immun. 2011;12:157–68. doi: 10.1038/gene.2011.9. [DOI] [PubMed] [Google Scholar]
- 62.Gearing DP, Thut CJ, VandeBos T, Gimpel SD, Delaney PB, King J, et al. Leukemia inhibitory factor receptor is structurally related to the IL-6 signal transducer, gp130. EMBO J. 1991;10:2839–48. doi: 10.1002/j.1460-2075.1991.tb07833.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ernst M, Novak U, Nicholson SE, Layton JE, Dunn AR. The carboxyl-terminal domains of gp130-related cytokine receptors are necessary for suppressing embryonic stem cell differentiation. Involvement of STAT3. J Biol Chem. 1999;274:9729–37. doi: 10.1074/jbc.274.14.9729. [DOI] [PubMed] [Google Scholar]
- 64.Schaeffer M, Schneiderbauer M, Weidler S, Tavares R, Warmuth M, de Vos G, et al. Signaling through a novel domain of gp130 mediates cell proliferation and activation of Hck and Erk kinases. Mol Cell Biol. 2001;21:8068–81. doi: 10.1128/MCB.21.23.8068-8081.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Clahsen T, Lehmann U, Stross C, Hermanns HM, Volkmer-Engert R, Schneider-Mergener J, et al. The tyrosine 974 within the LIF-R-chain of the gp130/LIF-R heteromeric receptor complex mediates negative regulation of LIF signalling. Cell Signal. 2005;17:559–69. doi: 10.1016/j.cellsig.2004.09.016. [DOI] [PubMed] [Google Scholar]
- 66.Blanchard F, Duplomb L, Wang Y, Robledo O, Kinzie E, Pitard V, et al. Stimulation of leukemia inhibitory factor receptor degradation by extracellular signal-regulated kinase. J Biol Chem. 2000;275:28793–801. doi: 10.1074/jbc.M003986200. [DOI] [PubMed] [Google Scholar]
- 67.Gibson RM, Schiemann WP, Prichard LB, Reno JM, Ericsson LH, Nathanson NM. Phosphorylation of human gp130 at Ser-782 adjacent to the Di-leucine internalization motif. Effects on expression and signaling. J Biol Chem. 2000;275:22574–82. doi: 10.1074/jbc.M907658199. [DOI] [PubMed] [Google Scholar]
- 68.Novotny-Diermayr V, Zhang T, Gu L, Cao X. Protein kinase C δ associates with the interleukin-6 receptor subunit glycoprotein (gp) 130 via Stat3 and enhances Stat3-gp130 interaction. J Biol Chem. 2002;277:49134–42. doi: 10.1074/jbc.M206727200. [DOI] [PubMed] [Google Scholar]
- 69.Hirai H, Karian P, Kikyo N. Regulation of embryonic stem cell self-renewal and pluripotency by leukaemia inhibitory factor. Biochem J. 2011;438:11–23. doi: 10.1042/BJ20102152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Cohen MV, Downey JM. Ischemic postconditioning: from receptor to end-effector. Antioxid Redox Signal. 2011;14:821–31. doi: 10.1089/ars.2010.3318. [DOI] [PubMed] [Google Scholar]
- 71.Ovize M, Baxter GF, Di Lisa F, Ferdinandy P, Garcia-Dorado D, Hausenloy DJ, et al. Working Group of Cellular Biology of Heart of European Society of Cardiology Postconditioning and protection from reperfusion injury: where do we stand? Position paper from the Working Group of Cellular Biology of the Heart of the European Society of Cardiology. Cardiovasc Res. 2010;87:406–23. doi: 10.1093/cvr/cvq129. [DOI] [PubMed] [Google Scholar]
- 72.Miura T, Tanno M, Sato T. Mitochondrial kinase signalling pathways in myocardial protection from ischaemia/reperfusion-induced necrosis. Cardiovasc Res. 2010;88:7–15. doi: 10.1093/cvr/cvq206. [DOI] [PubMed] [Google Scholar]
- 73.Yang X, Cohen MV, Downey JM. Mechanism of cardioprotection by early ischemic preconditioning. Cardiovasc Drugs Ther. 2010;24:225–34. doi: 10.1007/s10557-010-6236-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Nakaoka Y, Nishida K, Fujio Y, Izumi M, Terai K, Oshima Y, et al. Activation of gp130 transduces hypertrophic signal through interaction of scaffolding/docking protein Gab1 with tyrosine phosphatase SHP2 in cardiomyocytes. Circ Res. 2003;93:221–9. doi: 10.1161/01.RES.0000085562.48906.4A. [DOI] [PubMed] [Google Scholar]
- 75.Nicol RL, Frey N, Pearson G, Cobb M, Richardson J, Olson EN. Activated MEK5 induces serial assembly of sarcomeres and eccentric cardiac hypertrophy. EMBO J. 2001;20:2757–67. doi: 10.1093/emboj/20.11.2757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Suleman N, Somers S, Smith R, Opie LH, Lecour SC. Dual activation of STAT-3 and Akt is required during the trigger phase of ischaemic preconditioning. Cardiovasc Res. 2008;79:127–33. doi: 10.1093/cvr/cvn067. [DOI] [PubMed] [Google Scholar]
- 77.Lacerda L, Somers S, Opie LH, Lecour S. Ischaemic postconditioning protects against reperfusion injury via the SAFE pathway. Cardiovasc Res. 2009;84:201–8. doi: 10.1093/cvr/cvp274. [DOI] [PubMed] [Google Scholar]
- 78.Tamareille S, Mateus V, Ghaboura N, Jeanneteau J, Croué A, Henrion D, et al. RISK and SAFE signaling pathway interactions in remote limb ischemic perconditioning in combination with local ischemic postconditioning. Basic Res Cardiol. 2011;106:1329–39. doi: 10.1007/s00395-011-0210-z. [DOI] [PubMed] [Google Scholar]
- 79.Liao Z, Brar BK, Cai Q, Stephanou A, O’Leary RM, Pennica D, et al. Cardiotrophin-1 (CT-1) can protect the adult heart from injury when added both prior to ischaemia and at reperfusion. Cardiovasc Res. 2002;53:902–10. doi: 10.1016/S0008-6363(01)00531-4. [DOI] [PubMed] [Google Scholar]
- 80.Brar BK, Stephanou A, Liao Z, O’Leary RM, Pennica D, Yellon DM, et al. Cardiotrophin-1 can protect cardiac myocytes from injury when added both prior to simulated ischaemia and at reoxygenation. Cardiovasc Res. 2001;51:265–74. doi: 10.1016/S0008-6363(01)00294-2. [DOI] [PubMed] [Google Scholar]
- 81.Ghosh S, Ng LL, Talwar S, Squire IB, Galiñanes M. Cardiotrophin-1 protects the human myocardium from ischemic injury. Comparison with the first and second window of protection by ischemic preconditioning. Cardiovasc Res. 2000;48:440–7. doi: 10.1016/S0008-6363(00)00186-3. [DOI] [PubMed] [Google Scholar]
- 82.Wang F, Seta Y, Baumgarten G, Engel DJ, Sivasubramanian N, Mann DL. Functional significance of hemodynamic overload-induced expression of leukemia-inhibitory factor in the adult mammalian heart. Circulation. 2001;103:1296–302. doi: 10.1161/01.CIR.103.9.1296. [DOI] [PubMed] [Google Scholar]
- 83.Negoro S, Oh H, Tone E, Kunisada K, Fujio Y, Walsh K, et al. Glycoprotein 130 regulates cardiac myocyte survival in doxorubicin-induced apoptosis through phosphatidylinositol 3-kinase/Akt phosphorylation and Bcl-xL/caspase-3 interaction. Circulation. 2001;103:555–61. doi: 10.1161/01.CIR.103.4.555. [DOI] [PubMed] [Google Scholar]
- 84.Nelson SK, Wong GH, McCord JM. Leukemia inhibitory factor and tumor necrosis factor induce manganese superoxide dismutase and protect rabbit hearts from reperfusion injury. J Mol Cell Cardiol. 1995;27:223–9. doi: 10.1016/S0022-2828(08)80021-1. [DOI] [PubMed] [Google Scholar]
- 85.Zou Y, Takano H, Mizukami M, Akazawa H, Qin Y, Toko H, et al. Leukemia inhibitory factor enhances survival of cardiomyocytes and induces regeneration of myocardium after myocardial infarction. Circulation. 2003;108:748–53. doi: 10.1161/01.CIR.0000081773.76337.44. [DOI] [PubMed] [Google Scholar]
- 86.Funamoto M, Fujio Y, Kunisada K, Negoro S, Tone E, Osugi T, et al. Signal transducer and activator of transcription 3 is required for glycoprotein 130-mediated induction of vascular endothelial growth factor in cardiac myocytes. J Biol Chem. 2000;275:10561–6. doi: 10.1074/jbc.275.14.10561. [DOI] [PubMed] [Google Scholar]
- 87.Fujio Y, Kunisada K, Hirota H, Yamauchi-Takihara K, Kishimoto T. Signals through gp130 upregulate bcl-x gene expression via STAT1-binding cis-element in cardiac myocytes. J Clin Invest. 1997;99:2898–905. doi: 10.1172/JCI119484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Nagai T, Tanaka-Ishikawa M, Aikawa R, Ishihara H, Zhu W, Yazaki Y, et al. Cdc42 plays a critical role in assembly of sarcomere units in series of cardiac myocytes. Biochem Biophys Res Commun. 2003;305:806–10. doi: 10.1016/S0006-291X(03)00838-6. [DOI] [PubMed] [Google Scholar]
- 89.Fujio Y, Matsuda T, Oshima Y, Maeda M, Mohri T, Ito T, et al. Signals through gp130 upregulate Wnt5a and contribute to cell adhesion in cardiac myocytes. FEBS Lett. 2004;573:202–6. doi: 10.1016/j.febslet.2004.07.082. [DOI] [PubMed] [Google Scholar]
- 90.Takahashi E, Fukuda K, Miyoshi S, Murata M, Kato T, Ita M, et al. Leukemia inhibitory factor activates cardiac L-Type Ca2+ channels via phosphorylation of serine 1829 in the rabbit Cav1.2 subunit. Circ Res. 2004;94:1242–8. doi: 10.1161/01.RES.0000126405.38858.BC. [DOI] [PubMed] [Google Scholar]
- 91.Florholmen G, Aas V, Rustan AC, Lunde PK, Straumann N, Eid H, et al. Leukemia inhibitory factor reduces contractile function and induces alterations in energy metabolism in isolated cardiomyocytes. J Mol Cell Cardiol. 2004;37:1183–93. doi: 10.1016/j.yjmcc.2004.09.008. [DOI] [PubMed] [Google Scholar]
- 92.Florholmen G, Andersson KB, Yndestad A, Austbø B, Henriksen UL, Christensen G. Leukaemia inhibitory factor alters expression of genes involved in rat cardiomyocyte energy metabolism. Acta Physiol Scand. 2004;180:133–42. doi: 10.1046/j.0001-6772.2003.01245.x. [DOI] [PubMed] [Google Scholar]
- 93.Florholmen G, Thoresen GH, Rustan AC, Jensen J, Christensen G, Aas V. Leukaemia inhibitory factor stimulates glucose transport in isolated cardiomyocytes and induces insulin resistance after chronic exposure. Diabetologia. 2006;49:724–31. doi: 10.1007/s00125-006-0150-6. [DOI] [PubMed] [Google Scholar]
- 94.Hagiwara Y, Miyoshi S, Fukuda K, Nishiyama N, Ikegami Y, Tanimoto K, et al. SHP2-mediated signaling cascade through gp130 is essential for LIF-dependent I CaL, [Ca2+]i transient, and APD increase in cardiomyocytes. J Mol Cell Cardiol. 2007;43:710–6. doi: 10.1016/j.yjmcc.2007.09.004. [DOI] [PubMed] [Google Scholar]
- 95.Hiraoka E, Kawashima S, Takahashi T, Rikitake Y, Hirase T, Yokoyama M. PI 3-kinase-Akt-p70 S6 kinase in hypertrophic responses to leukemia inhibitory factor in cardiac myocytes. Kobe J Med Sci. 2003;49:25–37. [PubMed] [Google Scholar]
- 96.Iwakura T, Mohri T, Hamatani T, Obana M, Yamashita T, Maeda M, et al. STAT3/Pim-1 signaling pathway plays a crucial role in endothelial differentiation of cardiac resident Sca-1+ cells both in vitro and in vivo. J Mol Cell Cardiol. 2011;51:207–14. doi: 10.1016/j.yjmcc.2011.04.013. [DOI] [PubMed] [Google Scholar]
- 97.Kucia M, Dawn B, Hunt G, Guo Y, Wysoczynski M, Majka M, et al. Cells expressing early cardiac markers reside in the bone marrow and are mobilized into the peripheral blood after myocardial infarction. Circ Res. 2004;95:1191–9. doi: 10.1161/01.RES.0000150856.47324.5b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Silver JS, Hunter CA. gp130 at the nexus of inflammation, autoimmunity, and cancer. J Leukoc Biol. 2010;88:1145–56. doi: 10.1189/jlb.0410217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Lang R, Pauleau AL, Parganas E, Takahashi Y, Mages J, Ihle JN, et al. SOCS3 regulates the plasticity of gp130 signaling. Nat Immunol. 2003;4:546–50. doi: 10.1038/ni932. [DOI] [PubMed] [Google Scholar]
- 100.Croker BA, Krebs DL, Zhang JG, Wormald S, Willson TA, Stanley EG, et al. SOCS3 negatively regulates IL-6 signaling in vivo. Nat Immunol. 2003;4:540–5. doi: 10.1038/ni931. [DOI] [PubMed] [Google Scholar]
- 101.Stephanou A. Role of STAT-1 and STAT-3 in ischaemia/reperfusion injury. J Cell Mol Med. 2004;8:519–25. doi: 10.1111/j.1582-4934.2004.tb00476.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Antoniv TT, Ivashkiv LB. Interleukin-10-induced gene expression and suppressive function are selectively modulated by the PI3K-Akt-GSK3 pathway. Immunology. 2011;132:567–77. doi: 10.1111/j.1365-2567.2010.03402.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Kurdi M, Booz GW. Deciphering STAT3 signaling in the heart: plasticity and vascular inflammation. Congest Heart Fail. 2010;16:234–8. doi: 10.1111/j.1751-7133.2010.00175.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Yuan ZL, Guan YJ, Chatterjee D, Chin YE. Stat3 dimerization regulated by reversible acetylation of a single lysine residue. Science. 2005;307:269–73. doi: 10.1126/science.1105166. [DOI] [PubMed] [Google Scholar]
- 105.Wang R, Cherukuri P, Luo J. Activation of Stat3 sequence-specific DNA binding and transcription by p300/CREB-binding protein-mediated acetylation. J Biol Chem. 2005;280:11528–34. doi: 10.1074/jbc.M413930200. [DOI] [PubMed] [Google Scholar]
- 106.Braunstein J, Brutsaert S, Olson R, Schindler C. STATs dimerize in the absence of phosphorylation. J Biol Chem. 2003;278:34133–40. doi: 10.1074/jbc.M304531200. [DOI] [PubMed] [Google Scholar]
- 107.Arany I, Reed DK, Grifoni SC, Chandrashekar K, Booz GW, Juncos LA. A novel U-STAT3-dependent mechanism mediates the deleterious effects of chronic nicotine exposure on renal injury. Am J Physiol Renal Physiol. 2012;302:F722–9. doi: 10.1152/ajprenal.00338.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Tanner JJ, Parsons ZD, Cummings AH, Zhou H, Gates KS. Redox regulation of protein tyrosine phosphatases: structural and chemical aspects. Antioxid Redox Signal. 2011;15:77–97. doi: 10.1089/ars.2010.3611. [DOI] [PubMed] [Google Scholar]
- 109.Kurdi M, Booz GW. Evidence that IL-6-type cytokine signaling in cardiomyocytes is inhibited by oxidative stress: parthenolide targets JAK1 activation by generating ROS. J Cell Physiol. 2007;212:424–31. doi: 10.1002/jcp.21033. [DOI] [PubMed] [Google Scholar]
- 110.Smith JK, Patil CN, Patlolla S, Gunter BW, Booz GW, Duhé RJ. Identification of a redox-sensitive switch within the JAK2 catalytic domain. Free Radic Biol Med. 2012;52:1101–10. doi: 10.1016/j.freeradbiomed.2011.12.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Shaw PE. Could STAT3 provide a link between respiration and cell cycle progression? Cell Cycle. 2010;9:4294–6. doi: 10.4161/cc.9.21.13677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Zgheib C, Zouein FA, Chidiac R, Kurdi M, Booz GW. Calyculin a reveals serine/threonine phosphatase protein phosphatase 1 as a regulatory nodal point in canonical signal transducer and activator of transcription 3 signaling of human microvascular endothelial cells. J Interferon Cytokine Res. 2012;32:87–94. doi: 10.1089/jir.2011.0059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Boengler K, Hilfiker-Kleiner D, Heusch G, Schulz R. Inhibition of permeability transition pore opening by mitochondrial STAT3 and its role in myocardial ischemia/reperfusion. Basic Res Cardiol. 2010;105:771–85. doi: 10.1007/s00395-010-0124-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Phillips D, Reilley MJ, Aponte AM, Wang G, Boja E, Gucek M, et al. Stoichiometry of STAT3 and mitochondrial proteins: Implications for the regulation of oxidative phosphorylation by protein-protein interactions. J Biol Chem. 2010;285:23532–6. doi: 10.1074/jbc.C110.152652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Sarafian TA, Montes C, Imura T, Qi J, Coppola G, Geschwind DH, et al. Disruption of astrocyte STAT3 signaling decreases mitochondrial function and increases oxidative stress in vitro. PLoS One. 2010;5:e9532. doi: 10.1371/journal.pone.0009532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Petrosillo G, Matera M, Moro N, Ruggiero FM, Paradies G. Mitochondrial complex I dysfunction in rat heart with aging: critical role of reactive oxygen species and cardiolipin. Free Radic Biol Med. 2009;46:88–94. doi: 10.1016/j.freeradbiomed.2008.09.031. [DOI] [PubMed] [Google Scholar]
- 117.Paradies G, Petrosillo G, Pistolese M, Di Venosa N, Federici A, Ruggiero FM. Decrease in mitochondrial complex I activity in ischemic/reperfused rat heart: involvement of reactive oxygen species and cardiolipin. Circ Res. 2004;94:53–9. doi: 10.1161/01.RES.0000109416.56608.64. [DOI] [PubMed] [Google Scholar]
- 118.Szczepanek K, Chen Q, Derecka M, Salloum FN, Zhang Q, Szelag M, et al. Mitochondrial-targeted Signal transducer and activator of transcription 3 (STAT3) protects against ischemia-induced changes in the electron transport chain and the generation of reactive oxygen species. J Biol Chem. 2011;286:29610–20. doi: 10.1074/jbc.M111.226209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Zhou L, Too HP. Mitochondrial localized STAT3 is involved in NGF induced neurite outgrowth. PLoS One. 2011;6:e21680. doi: 10.1371/journal.pone.0021680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Bernier M, Paul RK, Martin-Montalvo A, Scheibye-Knudsen M, Song S, He HJ, et al. Negative regulation of STAT3 protein-mediated cellular respiration by SIRT1 protein. J Biol Chem. 2011;286:19270–9. doi: 10.1074/jbc.M110.200311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Park J, Kusminski CM, Chua SC, Scherer PE. Leptin receptor signaling supports cancer cell metabolism through suppression of mitochondrial respiration in vivo. Am J Pathol. 2010;177:3133–44. doi: 10.2353/ajpath.2010.100595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Reich NC. STAT3 revs up the powerhouse. Sci Signal. 2009;2:pe61. doi: 10.1126/scisignal.290pe61. [DOI] [PubMed] [Google Scholar]
- 123.Heusch G, Musiolik J, Gedik N, Skyschally A. Mitochondrial STAT3 activation and cardioprotection by ischemic postconditioning in pigs with regional myocardial ischemia/reperfusion. Circ Res. 2011;109:1302–8. doi: 10.1161/CIRCRESAHA.111.255604. [DOI] [PubMed] [Google Scholar]
- 124.Boengler K, Heusch G, Schulz R. Nuclear-encoded mitochondrial proteins and their role in cardioprotection. Biochim Biophys Acta. 2011;1813:1286–94. doi: 10.1016/j.bbamcr.2011.01.009. [DOI] [PubMed] [Google Scholar]