

Abstract
Natural genetic variation is a rich resource for identifying novel elements of cellular pathways such as endoplasmic reticulum (ER) stress. ER stress occurs when misfolded proteins accumulate in the ER and cells respond with the conserved unfolded protein response (UPR), which includes large-scale gene expression changes. Although ER stress can be a cause or a modifying factor of human disease, little is known of the amount of variation in the response to ER stress and the genes contributing to such variation. To study natural variation in ER stress response in a model system, we measured the survival time in response to tunicamycin-induced ER stress in flies from 114 lines from the sequenced Drosophila Genetic Reference Panel of wild-derived inbred strains. These lines showed high heterogeneity in survival time under ER stress conditions. To identify the genes that may be driving this phenotypic variation, we profiled ER stress-induced gene expression and performed an association study. Microarray analysis identified variation in transcript levels of numerous known and previously unknown ER stress-responsive genes. Survival time was significantly associated with polymorphisms in candidate genes with known (i.e., Xbp1) and unknown roles in ER stress. Functional testing found that 17 of 25 tested candidate genes from the association study have putative roles in ER stress. In both approaches, one-third of ER stress genes had human orthologs that contribute to human disease. This study establishes Drosophila as a useful model for studying variation in ER stress and identifying ER stress genes that may contribute to human disease.
Natural populations of most organisms harbor extensive variation at the phenotypic and genotypic levels. Studying natural variation is an unbiased way to identify novel elements of a biological pathway. Although variation is often exploited to study adaptive traits, it remains underused in the study of cellular pathways. Complex diseases commonly arise from dysfunction of basic pathways (1). By identifying the variation in these pathways, we may move toward predictions of disease susceptibility among individuals.
Drosophila provides exceptionally powerful tools and approaches for exploring natural variation and underlying mechanisms (2). Differences among inbred lines of flies reflect the array of allelic effects that are extant in a population and provide a means for quantifying aspects of the genetic architecture of the variation (3). For well-conserved, basic, cellular traits, variation among inbred lines can reflect how and which genes within a pathway impact the final organismal phenotype. For instance, we can determine whether branch points in pathways are particularly likely to harbor functional variation.
The endoplasmic reticulum (ER) is a large organelle that is responsible for synthesis, maturation, and delivery of a variety of proteins essential for cellular function (4). The ER is constantly producing and trafficking proteins. ER dysfunction can have devastating consequences for a cell. Such dysfunction occurs when misfolded proteins accumulate in the ER lumen, causing ER stress. The cell responds to ER stress with the evolutionarily conserved unfolded protein response (UPR) (4). The UPR comprises three signaling branches: Inositol-requiring enzyme-1 (Ire1), activating transcription factor 6 (Atf6), and pancreatic eIF-2α kinase (Perk). The UPR induces expression of chaperones and other proteins involved in refolding misfolded proteins. In addition to inducing gene expression (5), the Perk pathway is responsible for attenuating translation under ER stress. The UPR can return the ER to homeostasis by attenuating protein synthesis, activating transcriptional signaling cascades, and refolding or degrading misfolded proteins in the ER (4).
ER stress can be a primary cause and a modifier of many important human diseases (6). For example, diabetes is a common metabolic disease, caused by misregulation of blood glucose levels. Mutations in both PERK and XBP1 cause diabetes-like symptoms in mouse and humans (7–11). Similarly, adipose tissue from obese human individuals shows up-regulation of several key proteins involved in the UPR relative to levels of these proteins in adipose tissue of lean individuals (12). ER stress has also been implicated in the pathogenesis of several human neurological diseases, including Parkinson disease, polyglutamine diseases, amyotrophic lateral sclerosis (ALS), and Alzheimer’s disease (6). Alteration of the UPR may worsen the disease. For example, in animal models of ALS, second-site mutations that increase ER stress result in earlier onset of disease and more severe symptoms (13, 14).
An individual’s ER stress response can be an important factor in determining disease severity. To understand the extent to which ER stress responses can act as a modifier of disease, it is critical to understand the extent and nature of the variation in ER stress responses. Although many aspects of the ER stress response are being studied in depth, little is known of how the pathway might vary in healthy individuals. Only one published study has addressed ER stress-response variation (15). That study demonstrated that there was extensive variation in ER stress-induced transcription among cultured cells derived from different human individuals and that some genes underlying the variation had no previous known function in ER stress response (15). However, no studies have examined the variation in ER stress response in model organisms like Drosophila, where follow-up functional studies can be performed. Furthermore, no studies have performed an unbiased association analysis to identify genetic variation that might drive response to misfolded proteins.
To understand how ER stress may impact human disease, it is important to identify the nature of segregating genetic and phenotypic variation that modifies ER stress. We took advantage of the Drosophila Genetic Reference Panel (DGRP) (2) to measure natural variation in tunicamycin (TM)-induced ER stress response. We first show that among the DGRP lines, there is extensive variation in survival under TM-induced ER stress conditions. We next show that there is transcriptional variation in both known and previously unknown ER stress-responsive genes. Finally, we show that TM-induced ER stress survival differences are associated with both known and unknown ER stress genes. Functional testing indicates that more than half of the candidate genes identified by association analysis play a potential role in ER stress. Of the Drosophila ER stress genes we identified that have human orthologs, one-third of those orthologs have been reported to be involved in human disease. Our data demonstrate that Drosophila genetic variation is a rich resource for identifying novel genes related to ER stress response and have direct implications for the response in humans.
Results
Variation in Survival Under ER Stress.
To evaluate variation in survival time, we tested 114 DGRP lines for their survival under ER stress conditions. We adapted a reported assay (16) and fed each line a diet supplemented with TM and contrasted survival to a TM-free control diet. Flies exposed for as few as 4 h to a diet supplemented by TM displayed systemic ER stress as indicated by an increase in Xbp1 splicing—a standard measure of ER stress—in the head, thorax, and abdomen (Fig. S1). The 114 DGRP lines showed extensive variation in survival time. Hazard Ratios (HR) for the DGRP lines varied by >100-fold (HR range: 5.49–655) (Fig. 1 and Dataset S1; see Dataset S1 for t50 values—time at which 50% of flies have died; see Fig. S2 for representative Kaplan–Meier survival curves). This wide range reflects an exceptional level of variation in survival time mediated by segregating genetic polymorphism that may affect TM-induced ER stress response in the DGRP. Although variation in TM toxicity would also result in changes in survival time, several aspects of the data (cited below) indicate little activation of response to toxicity and instead point to a primary role of ER stress tolerance mediating most of the variation.
Fig. 1.
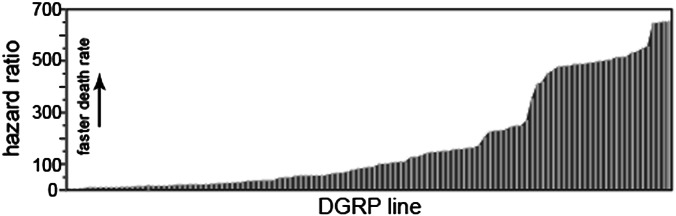
Distribution of ratios of death rates (HR) of 114 DGRP lines on ER stress-inducing TM compared with drug-free control food.
ER Stress-Responsive RNA Levels.
To assess whether variation in survival time under TM-induced ER stress might arise from differences in ER stress-responsive gene expression, we conducted a microarray study on a subset of DGRP lines exposed to TM. Expression was measured in whole TM-treated and control flies at an early exposure time (8 h; 20 DGRP lines) and a late exposure time (20 h; 8 DGRP lines). TM-treated gene expression was compared with control gene expression. A gene was defined as an ER stress-response gene (ERSRG) if it was up-regulated >1.5-fold in at least 25% of the lines (5 of 20 lines) at the early time point and up-regulated >1.5-fold in at least 50% of the lines at the late time point (4 of 8 lines).
At early exposure, we identified 487 ERSRGs (Dataset S2). Gene Ontology (GO) analysis identified enrichment in biological processes expected to be related to ER stress (Dataset S3), including amino acid activation (GO 0043038; false discovery rate, q < 8.6 × 10−4) and other protein translation categories, cellular amino acid metabolic process (GO 0006520; q < 0.014), Golgi vesicle transport (GO 0048193; q < 0.001027), and many stress-related categories, including response to temperature stimulus (GO 0009266; q < 0.017) and response to stress (GO 0006950; q < 0.081). The ER was the only enriched cellular compartment (GO 0005783; q < 1.36 × 10−5). The enrichment of these functional and cellular categories reflects an active ER stress response. Pathway analysis also confirmed this finding, showing enrichment of protein processing in the ER (Kyoto Encyclopedia of Genes and Genomes; q < 0.0001), UPR (REACTOME; q < 0.006), and Activation of Chaperones by IRE1alpha (REACTOME; q < 0.021). Molecular function enrichment included structural constituent of cuticle (GO 0042302; q < 1.87 × 10−7), a category with no known function in ER stress. Not surprisingly, enrichment in drug-response genes was also observed.
At late exposure, we identified 486 ERSRGs (Dataset S2). At this later time point, all functional categories related to ER stress responses showed greater enrichment (Dataset S3). We also found enrichment of retrograde vesicle-mediated transport, Golgi to ER (GO 0006890; q < 0.011), Golgi apparatus (GO 0005794; q < 4.76 × 10−5), and response to unfolded protein (GO 0006986; q < 0.048) (and many others), that were not enriched at the early time point. Genes encoding ER resident proteins were more enriched than at the early time point, and drug-response genes dropped in enrichment. Importantly, we did not find categories enriched for cell death or apoptosis. Overall, gene expression indicated that, at this late time point, the ER stress response is fully induced, but flies are not dying. Indeed, we observed that flies exposed to TM for 20 h and placed back on standard food recovered, and had normal fertility (Fig. S3).
Two hundred ERSRGs were found in both the early and late exposure time points. GO analysis indicated that the overlapping group of genes and genes unique to the late exposure are both enriched for ER stress-related functional groups listed above. Among the overlapping genes is PEK/PERK, which lies at the head of one of the three major UPR pathways. We did not find Ire1 or Atf6, key components of the other two UPR pathways, to be up-regulated at either time point in any of the DGRP lines we measured. A previous study in whole flies also did not detect TM-induced up-regulation of Ire1 or Atf6 (16). Moreover, because we measured whole-fly gene expression, it is likely that differential expression of Ire1 and Atf6 in different tissues could mask up-regulation. Genes unique to the early time point showed no enrichment for functional categories. There were also numerous genes down-regulated in response to ER stress. At both time points, there was GO enrichment for genes whose products function in the extracellular region (GO 0005576; early, q < 3.68 × 10−21; late, q < 1.69 × 10−9) and the lysosome (GO 0005764; early, q < 0.045; late, q < 0.015). This result agrees with previous studies demonstrating that, under ER stress, Ire1 degrades RNA of secreted and lysosomal proteins (17–19). Because down-regulation of these genes appears to relieve ER associated translational load (17–19), these genes were not studied further.
Variation in ER Stress-Responsive RNA Levels.
Profiling TM-induced ER stress gene expression across different DGRP lines created the opportunity to identify variation in the expression of ERSRGs. We found that many canonical ER stress genes showed extensive expression variation at the early and late time points (Fig. 2A). This observation indicates that the transcriptional response to ER stress varies widely, even for so-called essential ER-stress response genes. To identify genes that covary in the ER stress response, correlation analysis was performed on ERSRGs at the early time point (20, 21). Genes that covary in expression might represent expression modules that are regulated by a common mechanism or have common function (21). We identified 45 modules with correlated expression (Fig. 3 and Dataset S4). There are modules that contained mostly genes involved in amino acid metabolism (module 20), cuticle metabolism (module 22), or immunity (module 28). Other modules (e.g., modules 27 and 44) contained many genes, but included only one previously known ER stress gene each (module 27, PEK/PERK; module 44, Hsc70-3/Bip) and showed no functional enrichment. The largest module (module 43) contained 77 genes. Within this module, expression levels of previously known ER stress genes showed correlations with expression levels of genes with unknown ER stress function (average absolute Pearson r = 0.48). Some of the known ER stress genes contained within this module are Gp93, Gadd45, Ero1L, ergic53, and P58IPK (Fig. S4A). The correlation of known ER stress genes with unknown ERSRGs suggests that these candidates might have critical functions in the response. For example, the protein encoded by CG33514 has a potential role in vitamin E metabolism, and the protein encoded by CG11893 has potential protein kinase activity. Both genes have no known function in ER stress but are highly correlated with ER stress genes in this cluster (Fig. S4B).
Fig. 2.

Examples of expression variation in ER stress genes. Expression changes for canonical (A) and previously unknown (B) ER stress genes displayed for early (n = 20) and late (n = 8) TM exposure time points are shown. The line indicates ∼1.5× fold change. Human orthologs are listed below Drosophila gene names. E, early time point; L, late time point; N/A, no human ortholog.
Fig. 3.

Correlation analysis of ERSRGs. Correlation matrix heatmap shows the correlation modules on the diagonal. Red represents the most positive Pearson correlation, and blue represents the most negative correlation. The asterisk marks the largest module (module 43) discussed in the text.
Previously unknown ERSRGs.
We found numerous previously unknown TM-induced ERSRGs at both time points. Respectively, 21.8% and 19.4% of ERSRGs at the early and late time points had no known or predicted function based on GO analysis. For example, CG14545 was one of the most highly induced genes at the late time point, suggesting that it has an important role in ER stress response; however, its predicted protein has no known functions or recognizable domains, and thus there has been no previous basis to associate it with the ER stress response. Similar to the genes shown in Fig. S4B, there are many other genes not previously implicated in ER stress (Fig. 2B). Some of these ER stress genes have putative functions that are relevant to the ER stress response. Some examples include genes that encode ER resident proteins, proteins involved in lipid metabolism, proteins involved in the response to other stresses such as cold shock, and calcium binding proteins. The CHK kinase-like domain (zinc finger C4 and HLH domain-containing kinases domain subfamily of choline kinases: IPR015897; early, q = 8.86 × 10−6; late, q = 5.49 × 10−6) was the most enriched protein domain. Proteins containing the CHK kinase-like domain have no previously known ER stress functions.
Association Study.
Numerous genes show TM-induced ER stress transcriptional variation, but these genes may not be the primary cause of variation. Genetic polymorphisms that associate with TM-induced survival time may identify a more proximate cause of variation in ER stress response. To identify candidate genes that harbor genetic variation contributing to TM-induced ER stress resistance, we performed an association study with the HR derived from the TM exposure with 1,975,718 previously identified SNPs (2). We found 106 SNPs associated with survival time on TM at a nominal P < 10−6. We focused our analysis on the 66 SNPs that fell within 1 kb of a gene (Dataset S5). Forty-six candidate genes are represented by these 66 SNPs (Dataset S5). As reported in other association studies involving the DGRP (22, 23), the minor allele frequency of a SNP was inversely proportional to the effect size of that SNP (Fig. S5). We also asked whether the rarer allele (across the DGRP lines in this study) was associated with increased or decreased susceptibility to ER stress. In all 66 of these SNPs, the rarer allele resulted in a faster death rate (increased sensitivity to ER stress) (Dataset S5). This observation implies that natural selection is a likely factor driving the distribution of allelic effects (24, 25). Seven of the 66 SNPs are nonsynonymous coding changes in Xbp1 (1 SNP), Megator (1 SNP), CG33339 (4 SNPs), and CG11873 (1 SNP) (Dataset S5). These amino acid changes may affect protein function and make these genes particularly good targets for future studies. The 59 remaining SNPs are synonymous, intronic, or intergenic, indicating a potential role in regulating expression.
GO analysis did not reveal overrepresentation of functional classes among the 46 candidate genes. Lack of GO enrichment is not surprising, given that many possible mechanisms may contribute to survival time upon TM treatment. Survival might reflect, but is not limited to, susceptibility to glycosylation defects caused by TM, apoptosis differences, or primary ER stress tolerance. Because TM is a drug, survival might also reflect drug detoxification. Although we did not find any associations with SNPs in canonical drug metabolism genes, we did find an association with a SNP in CG13423. CG13423 has no known function in Drosophila, but the product of its human ortholog, bleomycin hydrolase (BLMH), has been shown to inactivate certain chemotherapy agents (26).
SNP in Xbp1 Is Associated with Survival on TM.
Identifying associations in genes with known ER stress functions could verify that the survival assay is an appropriate proxy for ER stress susceptibility/resistance. In fact, one of the TM survival-associated SNPs was a nonsynonymous SNP in Xbp1 (Dataset S5), a central player in one of the UPR pathways (27). Xbp1 encodes a transcription factor, and under ER stress, Xbp1 mRNA is spliced by Ire1 and is translated (27). Xbp1 protein translocates to the nucleus where it induces expression of genes targeted at returning the ER to homeostasis. The nonsynonymous SNP that we found to associate with survival on TM changes a threonine to an alanine (T40A) in the Xbp1 DNA binding domain and thus may affect Xbp1 transcriptional activity.
Candidate Genes with Likely Roles in ER Stress.
In addition to the classic UPR gene Xbp1, we also found associations in genes with likely roles in ER stress (Dataset S5). CG7140 (human ortholog, H6PD) (28), Calx (NCX) (29), Mgstl (MGST1) (30), Cbs (CBS) (31), and KrT95D (PACS2) (32) are all genes that, when disrupted (in human disease or a model organism), induce or affect the ER stress response. For example, in the mouse, null mutations of H6PD, a gene encoding an ER resident protein, result in up-regulation of UPR associated genes in skeletal muscle (33). Additionally, five genes with TM survival-associated SNPs have orthologs induced by TM in human cells (15): Cbs, Xbp1, CG9518 (CHDH), shep (RBMS3), and pyd (TJP2).
Candidate Genes Not Previously Implicated in ER Stress.
Our association analysis identified many candidates with known direct or indirect roles in ER stress. This finding supports the hypothesis that our screen detects genes relevant to ER stress response. Thus the 34 candidate genes not previously known to play a role in ER stress (Dataset S5) might represent important ER stress genes. Some of these candidates have functions not previously associated with ER stress, but others function in processes known to be important to the ER stress response. Examples of the latter include Golgi function (CG33298 and CG15611) (34, 35), lipid metabolism (CG12428, bgm, Egm, CG9518, RdgA, and CG5162) (36–41), and oxidative stress (CG11594, CG9003, RunxA, and CG11873) (23, 42). In particular, an associated SNP 65 bp downstream of eIF-2β, the beta subunit of the eIF2 translation initiation factor (43), is particularly interesting. In ER stress conditions, the alpha subunit of eIF2 is phosphorylated by PERK, a classic UPR gene, attenuating global translation (44). Although the beta subunit of eIF2 has not been implicated in the UPR, this noncoding SNP downstream of eIF-2β may affect expression and the available amount of eIF2 and the phosphorylation of the alpha subunit.
Functional Testing of Candidate ER Stress Genes.
To validate candidate genes nominated from the SNP associations for a potential function in ER stress, we tested a subset of the candidate genes in a survival assay as performed for the original DGRP lines. The 25 candidates tested were chosen based on availability of P-element insertion strains (Dataset S5). Because most of the P-element insertions were homozygous lethal, we tested flies heterozygous for insertions. We found that 17 of 25 candidates tested showed a significant difference in survival under ER stress as P-element heterozygotes, in comparison with the control w1118 strain (at P < 0.01; Dataset S5 and Fig. 4). With the exception of two lines with slower death rates, the P-element insertion reduced tolerance to ER stress and resulted in a faster death rate upon TM exposure. These results are consistent with the notion that the respective genes play a role in mediating TM-induced ER stress tolerance. Strikingly, CG9005 harbored the most significant SNP and also showed the largest difference in survival relative to w1118. We found that P-element insertion in Xbp1 showed a significant effect on survival. Thirteen of 17 candidates that showed a survival difference had no previously known role in ER stress.
Fig. 4.

Survival times of P-element heterozygotes in several association candidate genes. Flies heterozygous for CG9005, CG15611, and CG11594 P elements died faster than control w1118 flies. Flies heterozygous for CG9518 P element died more slowly than controls. Black curve, w1118; red curve, P element.
Discussion
We report a screen of genetic variation in ER stress response in Drosophila. We find that survival under TM-induced ER stress conditions is highly variable across the DGRP lines. We took two complementary approaches to identify the genetic elements contributing to this variation in TM-induced ER stress response. By profiling TM-induced ER stress gene expression changes, we sought to catalog, across genotypes, the genes that responded the most variably to TM-induced ER stress. However, it is impossible to know from expression data alone whether these variable responsive genes are a cause or effect of the survival differences observed. Thus, we also took a genetic-association approach to identify polymorphisms that may contribute to the variability in survival on TM-induced ER stress conditions. This combined approach of quantifying gene expression and genetic variation identified numerous putative ER stress genes that may be particularly important in understanding inter-individual variation in ER stress response.
Although the canonical ER stress pathway is well studied, there are likely numerous aspects of this process that remain undiscovered. Both methods used in this study identified known ER stress genes, indicating that at least some genes previously unknown to be involved in ER stress have also been identified here. In addition to the many individual ERSRGs identified in the microarray analysis, we found enrichment for genes encoding CHK kinase-like proteins, a subfamily of choline kinases (16 genes; Zinc finger C4, and HLH domain containing kinases domain). The function of these CHK kinase-like proteins is unknown in any organism. A study in Caenorhabditis elegans demonstrated that two genes with choline kinase activity are up-regulated by ER stress (45), but the exact role of choline kinase genes in ER stress is unknown. Choline kinases catalyze the first reaction in phosphatidylcholine (PC) biosynthesis (46). PC is a major component of cellular membranes, and levels of PC can trigger growth arrest and apoptosis (46). Abnormal PC/phosphatidylethanolamine (PE) ratios contribute to impaired ER function and chronic ER stress (47). Regulation of genes encoding CHK kinase-like proteins by ER stress might be a way to normalize PC/PE ratios to relieve ER stress. These genes also contain a zinc finger domain, suggesting a possible role in transcription.
Although survival on a drug might reflect a number of causes, we believe that our assay was actually reflective of ER stress. For example, our association analysis identified a SNP in the classic UPR gene Xbp1. Furthermore, some of the candidate genes identified induce or disrupt the ER stress response when gene function is perturbed. These observations suggest that the 78% of the association candidate genes not previously known to contribute to the ER stress response might be important to this response. Some of these ER stress candidates are within broad functional groups with previously known roles in ER stress. Of note, we identified four genes (CG11594, CG9003, RunxA, and CG11873) that had no prior known function in ER stress, but had been previously identified as candidate genes in oxidative stress response in the same DGRP lines (23). There are functional links between oxidative and ER stress (42), and the fact that these genes appeared as candidates in both processes suggests that there may be a genetic convergence of these two pathways in the DGRP. We also identified six association candidates (CG12428, bgm, Egm, CG9518, RdgA, and CG5162) with known or putative roles in lipid metabolism. Aberrant lipid metabolism is increasingly recognized as a critical cause and effect of ER stress (39, 47, 48). This observation, combined with the overrepresentation of CHK-kinase-like genes in the expression analysis (see above), suggests that lipid metabolism may play a particularly important role in variation in TM-induced ER stress response.
Functional testing demonstrated that many of the candidates identified in our association study have potential roles in TM-induced ER stress response. We found that 76% of the tested candidates with an effect on survival have no known role in ER stress. For example, CG15611 encodes a putative Rho guanyl-nucleotide exchange factor (RhoGEF) that is thought to regulate vesicle budding from the Golgi (34). CG15611 may regulate protein trafficking between the ER and Golgi and function in eliminating misfolded proteins. Megator encodes a nuclear pore protein (49). Megator has no known function in ER stress, but Ire1 has been shown to interact with some components of the nuclear pore complex in yeast (50). Although extensive functional studies are needed to identify specific functions in ER stress response, these verified candidates provide a promising pool of unstudied ER stress genes.
Nominating genes for future study can be challenging. Genes identified by both approaches in this study might be especially important. There were four genes in common between the candidate genes in the association study and genes whose expression was induced during the early time point (CG10962, CG11594, KrT95D, and tkv). There were two genes in common between the association study and the late time point (CG10962 and CG34228). Strikingly, CG10962 was found in the association study and both time points. CG10962 is orthologous to the human gene DHRS11 and is a member of the short-chain dehydrogenases/reductases (SDR) family. Proteins in the SDR family may have a previously unappreciated role in ER stress.
The ultimate goal of this study was to use Drosophila natural variation as a model to identify ER stress genes that may contribute to variation in the human ER stress response. Because the basic UPR pathways are well conserved between Drosophila and human, Drosophila is a good model for studying ER stress. We quantified the potential relevance of our study to humans by identifying the genes with human orthologs. We found that 52%, 68%, and 63% of ER stress genes in this study have human orthologs (early response, late response, and association study, respectively) (Datasets S2 and S5). The majority of genes that contribute to variation in ER stress response in Drosophila are conserved and are very likely to have similar roles in the human ER stress response. To test the parallels between our in vivo Drosophila results and those in mammals, we compared our results to in vivo TM-induced responses in the mouse. Of the genes in our study with clear mouse orthologs, 47%, 48%, and 21% (early response, late response, and association study, respectively) overlap with the in vivo TM-induced liver transcriptional response in mouse (Datasets S2 and S5) (51).
Only one other published study investigated variation in ER stress response in any organism (15). That study measured the variation in ER stress transcriptional response in a collection of human cell lines. A comparison of the transcriptional response of whole flies to that of immortalized cell lines might nominate candidates for important conserved genes that show variation across species. To determine the overlap in variable ER stress genes in Drosophila and human, we compared our results with those reported by Dombroski et al. for human cells (15). Of the conserved genes from our microarray study, 29% and 27% (early and late response, respectively) also showed expression variation in ER-stressed human cells (Dataset S2), as did, strikingly, 17% of the candidate ER stress genes from our association study (Dataset S5). The association candidates found in our Drosophila study, which also showed variation in human cells, might be particularly important candidates. We also performed a PubMed literature search and found that 10%, 12%, and 21% of genes with human orthologs (early response, late response, and association study, respectively), not listed in Dombroski et al. (15), have been implicated in ER stress (Datasets S2 and S5). This high level of conservation between Drosophila and human suggests that results on ER stress variation in Drosophila have promise to be translatable to human biology.
Because ER stress is a major contributor to disease, we also identified the genes detected in our study whose human orthologs have been implicated in human disease. Genes that cause Mendelian-inherited disease, or are associated with disease, have entries in the Online Mendelian Inheritance in Man database (OMIM). We found that 32%, 23%, and 28% of conserved ER stress genes in our study (early response, late response, and association, respectively) have orthologs with entries in OMIM and contribute to human disease (Datasets S2 and S5). Many diseases are affected by these ER stress genes. For example, orthologs of genes associated with neurological disease were identified in the microarray and association study, including RhoGEF3 (human gene: ARHGEF9; disease: epileptic encephalopathy), CG7804 (TARDBP; ALS), CG10420 (SIL1; Marinesco–Sjogren syndrome), Tsp29Fb (TSPAN7; X-linked mental retardation), CG13423 (BLMH; Alzheimer’s disease), and Cbs (CBS; homocystinuria). Orthologs of genes associated with cancer were also identified, including l(2)37Cc (PHB; breast cancer), and RunxA (RUNX1; leukemia). We also identified orthologs involved in diabetes, metabolic disorders, and developmental disorders. Knowledge of the role of these genes in ER stress may lead to a better understanding of how ER stress contributes to the pathophysiology of their associated diseases.
Our results highlight the value of using genetic variation in ER stress response as an avenue to identify genes and understand mechanisms mediating this cellular response. By taking advantage of the rich DGRP resource, we identified numerous genes shared with the human ER stress response as well as many candidate putative ER stress genes. A subset of genes involved in TM-induced ER stress variation in Drosophila also contributes to variation in human cultured cells. Further study of the role of genes identified here in mediating differences among genetic lines in TM-induced ER stress response could proceed rapidly in the Drosophila model. Understanding the genes contributing to variation in ER stress response will not only further our understanding of this basic, conserved pathway, but will also contribute to the understanding of the mechanistic link between ER stress and different diseases.
Materials and Methods
We used 114 lines from the DGRP in this study (Dataset S1). To measure survival under constant ER stress, 3-d-old male flies were fed a diet with or without the ER stress-inducing drug TM (Sigma). The drug exposure protocol was similar to that used by Girardot et al. (16). Details of fly maintenance, drug exposure, and statistical analysis of survival can be found in SI Materials and Methods. Expression was measured on Agilent 4 × 44K Drosophila Gene Expression Microarrays at two time points: 8 h (early time point) and 20 h (late time point) of drug exposure. Details of microarray analysis can be found in SI Materials and Methods. To associate TM-induced ER stress survival time with genome-wide SNPs, we used a web tool developed for the DGRP, which applies a simple linear model to test the null hypothesis that the means of the genotypes at each SNP, tested one at a time, were homogeneous (http://dgrp.gnets.ncsu.edu/). To validate association candidates for a potential role in ER stress, P-element insertion lines for 25 candidate genes were tested for survival on TM. To standardize genetic background, all P-element insertion lines that were not isogenic with the laboratory strain w1118 were backcrossed to the w1118 background for more than five generations. Because many of the P elements were homozygous lethal, we assessed effect of each P element in heterozygotes (over the w1118 background), comparing them to control w1118 flies. Survival analysis was performed as described above. Details of association analysis and functional analysis can be found in SI Materials and Methods.
Supplementary Material
Acknowledgments
We thank Geoff Findlay, Rob Unckless, and Ling Qi for helpful advice and comments; Amanda Manfredo for assistance with microarray preparation; Julien Ayroles for statistical advice; and Dr. Trudy Mackay for providing the DGRP fly lines and sequence data. This work was supported by National Institutes of Health (NIH)/National Institute of Child Health and Human Development (NICHD) Grant R01-HD059060 (to M.F.W. and A.G.C.) and a priming grant from the Cornell Center for Comparative and Population Genomics (to M.F.W. and A.G.C.). For part of this research, C.Y.C. was supported by a traineeship under NIH/NICHD Training Grant in Reproductive Genomics T32-HD052471. Subsequently, C.Y.C. was supported by NIH/National Research Service Award Fellowship F32-GM093663.
Footnotes
The authors declare no conflict of interest.
Data deposition: The sequences reported in this paper have been deposited in the Gene Expression Omnibus database (accession no. GSE46425).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1307125110/-/DCSupplemental.
References
- 1.Dermitzakis ET. Cellular genomics for complex traits. Nat Rev Genet. 2012;13(3):215–220. doi: 10.1038/nrg3115. [DOI] [PubMed] [Google Scholar]
- 2.Mackay TF, et al. The Drosophila melanogaster Genetic Reference Panel. Nature. 2012;482(7384):173–178. doi: 10.1038/nature10811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mackay TF, Stone EA, Ayroles JF. The genetics of quantitative traits: Challenges and prospects. Nat Rev Genet. 2009;10(8):565–577. doi: 10.1038/nrg2612. [DOI] [PubMed] [Google Scholar]
- 4.Bernales S, Papa FR, Walter P. Intracellular signaling by the unfolded protein response. Annu Rev Cell Dev Biol. 2006;22:487–508. doi: 10.1146/annurev.cellbio.21.122303.120200. [DOI] [PubMed] [Google Scholar]
- 5.Teske BF, et al. The eIF2 kinase PERK and the integrated stress response facilitate activation of ATF6 during endoplasmic reticulum stress. Mol Biol Cell. 2011;22(22):4390–4405. doi: 10.1091/mbc.E11-06-0510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhao L, Ackerman SL. Endoplasmic reticulum stress in health and disease. Curr Opin Cell Biol. 2006;18(4):444–452. doi: 10.1016/j.ceb.2006.06.005. [DOI] [PubMed] [Google Scholar]
- 7.Delépine M, et al. EIF2AK3, encoding translation initiation factor 2-alpha kinase 3, is mutated in patients with Wolcott-Rallison syndrome. Nat Genet. 2000;25(4):406–409. doi: 10.1038/78085. [DOI] [PubMed] [Google Scholar]
- 8.Harding HP, et al. Diabetes mellitus and exocrine pancreatic dysfunction in perk-/- mice reveals a role for translational control in secretory cell survival. Mol Cell. 2001;7(6):1153–1163. doi: 10.1016/s1097-2765(01)00264-7. [DOI] [PubMed] [Google Scholar]
- 9.Ladiges WC, et al. Pancreatic beta-cell failure and diabetes in mice with a deletion mutation of the endoplasmic reticulum molecular chaperone gene P58IPK. Diabetes. 2005;54(4):1074–1081. doi: 10.2337/diabetes.54.4.1074. [DOI] [PubMed] [Google Scholar]
- 10.Ozcan U, et al. Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes. Science. 2004;306(5695):457–461. doi: 10.1126/science.1103160. [DOI] [PubMed] [Google Scholar]
- 11.Scheuner D, et al. Control of mRNA translation preserves endoplasmic reticulum function in beta cells and maintains glucose homeostasis. Nat Med. 2005;11(7):757–764. doi: 10.1038/nm1259. [DOI] [PubMed] [Google Scholar]
- 12.Boden G, et al. Increase in endoplasmic reticulum stress-related proteins and genes in adipose tissue of obese, insulin-resistant individuals. Diabetes. 2008;57(9):2438–2444. doi: 10.2337/db08-0604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shimazawa M, et al. An inducer of VGF protects cells against ER stress-induced cell death and prolongs survival in the mutant SOD1 animal models of familial ALS. PLoS ONE. 2010;5(12):e15307. doi: 10.1371/journal.pone.0015307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang L, Popko B, Roos RP. The unfolded protein response in familial amyotrophic lateral sclerosis. Hum Mol Genet. 2011;20(5):1008–1015. doi: 10.1093/hmg/ddq546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dombroski BA, et al. Gene expression and genetic variation in response to endoplasmic reticulum stress in human cells. Am J Hum Genet. 2010;86(5):719–729. doi: 10.1016/j.ajhg.2010.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Girardot F, Monnier V, Tricoire H. Genome wide analysis of common and specific stress responses in adult Drosophila melanogaster. BMC Genomics. 2004;5(1):74. doi: 10.1186/1471-2164-5-74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hollien J, et al. Regulated Ire1-dependent decay of messenger RNAs in mammalian cells. J Cell Biol. 2009;186(3):323–331. doi: 10.1083/jcb.200903014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hollien J, Weissman JS. Decay of endoplasmic reticulum-localized mRNAs during the unfolded protein response. Science. 2006;313(5783):104–107. doi: 10.1126/science.1129631. [DOI] [PubMed] [Google Scholar]
- 19.Gaddam D, Stevens N, Hollien J. Comparison of mRNA localization and regulation during endoplasmic reticulum stress in Drosophila cells. Mol Biol Cell. 2013;24(1):14–20. doi: 10.1091/mbc.E12-06-0491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Stone EA, Ayroles JF. Modulated modularity clustering as an exploratory tool for functional genomic inference. PLoS Genet. 2009;5(5):e1000479. doi: 10.1371/journal.pgen.1000479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ayroles JF, et al. Systems genetics of complex traits in Drosophila melanogaster. Nat Genet. 2009;41(3):299–307. doi: 10.1038/ng.332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jordan KW, et al. Genome-wide association for sensitivity to chronic oxidative stress in Drosophila melanogaster. PLoS ONE. 2012;7(6):e38722. doi: 10.1371/journal.pone.0038722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Weber AL, et al. Genome-wide association analysis of oxidative stress resistance in Drosophila melanogaster. PLoS ONE. 2012;7(4):e34745. doi: 10.1371/journal.pone.0034745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fraser HB, Moses AM, Schadt EE. Evidence for widespread adaptive evolution of gene expression in budding yeast. Proc Natl Acad Sci USA. 2010;107(7):2977–2982. doi: 10.1073/pnas.0912245107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Orr HA. Testing natural selection vs. genetic drift in phenotypic evolution using quantitative trait locus data. Genetics. 1998;149(4):2099–2104. doi: 10.1093/genetics/149.4.2099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schwartz DR, et al. The neutral cysteine protease bleomycin hydrolase is essential for epidermal integrity and bleomycin resistance. Proc Natl Acad Sci USA. 1999;96(8):4680–4685. doi: 10.1073/pnas.96.8.4680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.He Y, et al. Emerging roles for XBP1, a sUPeR transcription factor. Gene Expr. 2010;15(1):13–25. doi: 10.3727/105221610x12819686555051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Senesi S, et al. Hexose-6-phosphate dehydrogenase in the endoplasmic reticulum. Biol Chem. 2010;391(1):1–8. doi: 10.1515/BC.2009.146. [DOI] [PubMed] [Google Scholar]
- 29.Chen X, et al. Endoplasmic reticulum Ca2+ dysregulation and endoplasmic reticulum stress following in vitro neuronal ischemia: role of Na+-K+-Cl- cotransporter. J Neurochem. 2008;106(4):1563–1576. doi: 10.1111/j.1471-4159.2008.05501.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Morgenstern R, Zhang J, Johansson K. Microsomal glutathione transferase 1: Mechanism and functional roles. Drug Metab Rev. 2011;43(2):300–306. doi: 10.3109/03602532.2011.558511. [DOI] [PubMed] [Google Scholar]
- 31.Maclean KN, et al. Cystathionine protects against endoplasmic reticulum stress-induced lipid accumulation, tissue injury, and apoptotic cell death. J Biol Chem. 2012;287(38):31994–32005. doi: 10.1074/jbc.M112.355172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Simmen T, et al. PACS-2 controls endoplasmic reticulum-mitochondria communication and Bid-mediated apoptosis. EMBO J. 2005;24(4):717–729. doi: 10.1038/sj.emboj.7600559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lavery GG, et al. Deletion of hexose-6-phosphate dehydrogenase activates the unfolded protein response pathway and induces skeletal myopathy. J Biol Chem. 2008;283(13):8453–8461. doi: 10.1074/jbc.M710067200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ma Z, Liu Z, Huang X. Membrane phospholipid asymmetry counters the adverse effects of sterol overloading in the Golgi membrane of Drosophila. Genetics. 2012;190(4):1299–1308. doi: 10.1534/genetics.111.137687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schröder M, Sutcliffe L. Consequences of stress in the secretory pathway: The ER stress response and its role in the metabolic syndrome. Methods Mol Biol. 2010;648:43–62. doi: 10.1007/978-1-60761-756-3_3. [DOI] [PubMed] [Google Scholar]
- 36.Jogl G, Hsiao YS, Tong L. Structure and function of carnitine acyltransferases. Ann N Y Acad Sci. 2004;1033:17–29. doi: 10.1196/annals.1320.002. [DOI] [PubMed] [Google Scholar]
- 37.Min KT, Benzer S. Preventing neurodegeneration in the Drosophila mutant bubblegum. Science. 1999;284(5422):1985–1988. doi: 10.1126/science.284.5422.1985. [DOI] [PubMed] [Google Scholar]
- 38.Steinberg SJ, Morgenthaler J, Heinzer AK, Smith KD, Watkins PA. Very long-chain acyl-CoA synthetases. Human “bubblegum” represents a new family of proteins capable of activating very long-chain fatty acids. J Biol Chem. 2000;275(45):35162–35169. doi: 10.1074/jbc.M006403200. [DOI] [PubMed] [Google Scholar]
- 39.Basseri S, Austin RC. Endoplasmic reticulum stress and lipid metabolism: Mechanisms and therapeutic potential. Biochem Res Int. 2012;2012:841362. doi: 10.1155/2012/841362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mourikis P, Hurlbut GD, Artavanis-Tsakonas S. Enigma, a mitochondrial protein affecting lifespan and oxidative stress response in Drosophila. Proc Natl Acad Sci USA. 2006;103(5):1307–1312. doi: 10.1073/pnas.0510564103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Colgan SM, Hashimi AA, Austin RC. Endoplasmic reticulum stress and lipid dysregulation. Expert Rev Mol Med. 2011;13:e4. doi: 10.1017/S1462399410001742. [DOI] [PubMed] [Google Scholar]
- 42.Malhotra JD, Kaufman RJ. Endoplasmic reticulum stress and oxidative stress: A vicious cycle or a double-edged sword? Antioxid Redox Signal. 2007;9(12):2277–2293. doi: 10.1089/ars.2007.1782. [DOI] [PubMed] [Google Scholar]
- 43.Kimball SR. Eukaryotic initiation factor eIF2. Int J Biochem Cell Biol. 1999;31(1):25–29. doi: 10.1016/s1357-2725(98)00128-9. [DOI] [PubMed] [Google Scholar]
- 44.Prostko CR, Brostrom MA, Brostrom CO. Reversible phosphorylation of eukaryotic initiation factor 2 alpha in response to endoplasmic reticular signaling. Mol Cell Biochem. 1993;127-128:255–265. doi: 10.1007/BF01076776. [DOI] [PubMed] [Google Scholar]
- 45.Shen X, Ellis RE, Sakaki K, Kaufman RJ. Genetic interactions due to constitutive and inducible gene regulation mediated by the unfolded protein response in C. elegans. PLoS Genet. 2005;1(3):e37. doi: 10.1371/journal.pgen.0010037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cui Z, Houweling M. Phosphatidylcholine and cell death. Biochim Biophys Acta. 2002;1585(2-3):87–96. doi: 10.1016/s1388-1981(02)00328-1. [DOI] [PubMed] [Google Scholar]
- 47.Fu S, et al. Aberrant lipid metabolism disrupts calcium homeostasis causing liver endoplasmic reticulum stress in obesity. Nature. 2011;473(7348):528–531. doi: 10.1038/nature09968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Fu S, Watkins SM, Hotamisligil GS. The role of endoplasmic reticulum in hepatic lipid homeostasis and stress signaling. Cell Metab. 2012;15(5):623–634. doi: 10.1016/j.cmet.2012.03.007. [DOI] [PubMed] [Google Scholar]
- 49.Qi H, et al. Megator, an essential coiled-coil protein that localizes to the putative spindle matrix during mitosis in Drosophila. Mol Biol Cell. 2004;15(11):4854–4865. doi: 10.1091/mbc.E04-07-0579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Goffin L, et al. The unfolded protein response transducer Ire1p contains a nuclear localization sequence recognized by multiple beta importins. Mol Biol Cell. 2006;17(12):5309–5323. doi: 10.1091/mbc.E06-04-0292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rutkowski DT, et al. UPR pathways combine to prevent hepatic steatosis caused by ER stress-mediated suppression of transcriptional master regulators. Dev Cell. 2008;15(6):829–840. doi: 10.1016/j.devcel.2008.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.