

Abstract
Seventy-five percent of patients with epithelial ovarian cancer present with advanced-stage disease that is extensively disseminated intraperitoneally and prognosticates the poorest outcomes. Primarily metastatic within the abdominal cavity, ovarian carcinomas initially spread to adjacent organs by direct extension and then disseminate via the transcoelomic route to distant sites. Natural fluidic streams of malignant ascites triggered by physiological factors, including gravity and negative subdiaphragmatic pressure, carry metastatic cells throughout the peritoneum. We investigated the role of fluidic forces as modulators of metastatic cancer biology in a customizable microfluidic platform using 3D ovarian cancer nodules. Changes in the morphological, genetic, and protein profiles of biomarkers associated with aggressive disease were evaluated in the 3D cultures grown under controlled and continuous laminar flow. A modulation of biomarker expression and tumor morphology consistent with increased epithelial–mesenchymal transition, a critical step in metastatic progression and an indicator of aggressive disease, is observed because of hydrodynamic forces. The increase in epithelial–mesenchymal transition is driven in part by a posttranslational up-regulation of epidermal growth factor receptor (EGFR) expression and activation, which is associated with the worst prognosis in ovarian cancer. A flow-induced, transcriptionally regulated decrease in E-cadherin protein expression and a simultaneous increase in vimentin is observed, indicating increased metastatic potential. These findings demonstrate that fluidic streams induce a motile and aggressive tumor phenotype. The microfluidic platform developed here potentially provides a flow-informed framework complementary to conventional mechanism-based therapeutic strategies, with broad applicability to other lethal malignancies.
Keywords: tumor microenvironment, stress response, molecular targets, combination therapies, photodynamic therapy
Cancer metastases are responsible for 90% of cancer-related deaths, but the biological and physical factors that determine the fate and heterogeneity of metastatic tumors remain poorly understood (1–6). Ovarian cancer is the leading cause of deaths related to gynecologic malignancies, and is frequently diagnosed at an advanced stage. Initially, ovarian cancer metastasizes by direct extension to sites that are proximal to the primary tumor through a complex series of events including migration, assembly, and proliferation (7–10). Dissemination to distant sites prognosticates the poorest outcomes for ovarian cancer patients and occurs via transcoelomic, lymphatic, or hematogenous routes (7, 9, 10). Among these routes, transcoelomic metastases are the most frequent and are responsible for the highest morbidity and mortality rates, which in turn are associated with the frequent production of malignant ascites (7, 9, 10). Under normal physiologic conditions, the great majority of peritoneal fluid is resorbed by the vasculature and the lymphatics and is removed from the body (11). The buildup of ascites occurs when the rate of fluid production exceeds the rate of clearance, typically because of an underlying pathology such as cancer. Multiple factors drive the accumulation of this transudate, including the overproduction of peritoneal fluid, increased leakiness of tumor microvasculature, and obstruction of lymphatic vessels (11–15). The abnormal buildup of protein- and cell-rich ascitic fluid in the peritoneal cavity occurs frequently in ovarian cancer and is associated with a dismal quality of life and a grim median survival (7, 11). A major route of dissemination for ovarian tumors is along natural ascitic currents in the peritoneal cavity that are established by the physiologic movement of peritoneal fluid (Fig. 1A, blue arrows) (7, 16–18). Among the physiological factors that drive the formation of ascitic currents are gravity, negative subdiaphragmatic pressure, and organ mobility as well as recesses formed by key anatomical structures (7, 16–18). Detached ovarian cancer cells primarily colonize distant sites under the influence of ascitic flow; however, little is known about the impact of dynamic physical forces on the molecular and architectural heterogeneity of ovarian cancer metastases.
Fig. 1.

Modeling fluidic determinants of ovarian cancer dissemination and growth. Tumor cell dissemination and colonization of distant sites is influenced by a complex array of factors including the physical stresses that tumor cells encounter as they interact with stromal beds. (A) Ovarian cancer disseminates predominantly via movement of intraperitoneal fluid leading to a distinctive distribution pattern of tumor nodules (orange) involving four common abdomino-pelvic sites: (1) cul-de-sac (peritoneal fold between the rectum and the posterior wall of the uterus); (2) right infracolic space (the apex formed by the termination of the small intestine of the small bowel mesentery at ileocecal junction); (3) left infracolic space (superior site of sigmoid colon); (4) Right paracolic gutter (communication between the upper and lower abdomen defined by the ascending colon and peritoneal wall). This characteristic distribution is influenced by preferential pathways of ascitic flow (blue arrows) that are established by the hydrodynamics of intraperitoneal fluid motion in the abdomino-pelvic cavity. The direction and strength of these fluidic pathways are determined by physical influences including negative subdiaphragmatic pressure, gravity, and organ mobility as well as by recesses formed by key anatomical structures: (i) cul-de-sac, (ii) termination of small intestine mesentery, (s) sigmoid colon, (iv) falciform ligament, and (v) phrenicocolic ligament. In contrast to this flow-based dissemination, the absence of ascites leads to metastatic spread that is largely proximal to the primary tumor. (B) Schematic of a microfluidic chip used to study the effect of sustained flow on the growth and molecular features of 3D ovarian cancer nodules. (C) Photograph of a microfluidic chip used in the experiments. (D) A side view of a microfluidic chip designed to study the impact of flow on the attachment and growth of ovarian cancer cells to a stromal bed. (E) Ovarian cancer cells were cultured successfully under continuous flow for 7 d in the microfluidic chip and formed 3D micronodules as shown in a 3D rendering (Left) and an optical slice (Right) of a two-photon autofluorescence image.
A key molecular marker relevant to ovarian cancer biology, particularly advanced-stage disease, is the epidermal growth factor receptor (EGFR), a cell-surface receptor that regulates proliferation, growth, and survival (19–21). In ovarian cancer, high EGFR expression is associated with increased proliferation and a more aggressive and invasive phenotype and is observed more frequently in metastases than in samples of primary tumors (22–24). The EGFR may also be an important negative prognostic indicator of disease-free survival and overall survival, depending in part on the stage of the disease and the technique used to evaluate expression of this biomarker (20, 25). EGFR activation has been shown to down-regulate E-cadherin (26–28), a transmembrane glycoprotein involved in intercellular adhesions mediated by a protein complex that includes β-catenin (29). Loss of E-cadherin is associated with a poor clinical outcome and increased metastatic potential of ovarian cancer cells mediated in part by the cell ECM receptor α5β1 integrin, which binds fibronectin (26, 28). Decreased E-cadherin expression also is a hallmark of epithelial-to-mesenchymal transition (EMT) (26–30), a critical step in metastatic progression and a prognostic indicator of aggressive and refractory disease (31–35). Along with loss of E-cadherin, EMT is characterized by a range of molecular and morphological features, including the acquisition of a motile and mesenchymal phenotype with increased spindle-like morphology as well as increased expression of vimentin, an intermediate filament that maintains cell and tissue integrity and helps confer resistance to mechanical stress (30, 32, 35–37). The cell-cycle regulator p27Kip1 modulates proliferation, motility, and apoptosis by inhibiting an array of cyclin-dependent kinases including cell-division control protein kinase 2 (CDC2) and also plays a role in cytoskeletal remodeling (38, 39). Transcriptional and posttranslational regulation of molecular markers provides a cell with the plasticity to respond to environmental changes, particularly in response to stress. However, changes in gene expression do not always correlate with altered protein levels (40–47). Modulation of molecular pathways and architectural features relevant to ovarian cancer progression by dynamic physical forces may elucidate critical metastatic mechanisms.
Microfluidic systems provide unique opportunities to investigate the influence of physical and biological parameters on the growth of metastatic tumors (48–50). The current study integrates these observations with 3D tumor growth under sustained flow to study the effects of dynamic forces on molecular alterations critical to the metastatic process. 3D cancer models restore important architectural and stromal cues (51–65) but fail to evaluate the impact of fluid hydrodynamics on tumor adhesion and micronodule growth. A system that integrates both physical and stromal modulators of metastatic biology could provide unique insights into the events that drive cancer progression and tumor heterogeneity. The microfluidic platform described here produces 3D tumor micronodules that exhibit morphological features and express a panel of molecular markers associated with an aggressive and highly metastatic phenotype. The system reveals a flow-induced posttranslational up-regulation of EGFR expression and activation, a transcriptionally regulated decrease in E-cadherin expression, increased vimentin expression, and increased EMT-like characteristics. Ovarian micronodules grown under flow also show decreased volume and viability compared with nonflow 3D cultures. The data collectively highlight the importance of investigating dynamic physical influences to reflect the complex determinants of metastatic fate that cancer cells encounter under physiological conditions.
Results
Modeling Fluidic Determinants of Ovarian Cancer Dissemination and Growth.
A microfluidic platform was developed to evaluate the effects of flow (Fig. 1A) on the growth of 3D ovarian micronodules as shown in the schematic in Fig. 1B and the corresponding image in Fig. 1C. Additional details of the chip dimensions are provided in Fig. S1 A and B. Tumor cells entered the channels through gas-permeable silicone tubing and flowed over stromal beds of growth-factor reduced (GFR) Matrigel. A portion of the cells was effluxed from the chip via the outlet tubing; those that adhered to the Matrigel beds (Fig. 1D, Fig. S2, and Movie S1) were cultured under continuous flow for 7 d. Fig. 1E shows a representative 3D ovarian micronodule grown under continuous flow (Fig. 1E, Left, a 3D rendering of two-photon autofluorescence high-resolution z-stacks; Fig. 1E, Right, a corresponding optical slice from the 3D rendering showing cellularity throughout the nodule).
Computational Modeling of Flow in the Microfluidic Platform.
To characterize the cross-sectional profile of the stromal bed inside the microchannel (Fig. 2A), 1.02-μm yellow-green fluorescent–labeled microscopheres were incorporated into cooled Matrigel, and the chips were fabricated as described in Microfluidic Chip Design and Fabrication. High-resolution z-stacks were obtained by confocal microscopy, which demonstrated uniform thickness and spreading of the Matrigel in the middle of the channels. To demonstrate that flow velocity, shear rate, and vorticity are uniform in the midchannel region, where biological characterization of the ovarian cancer cells was performed, we numerically simulated the flow conditions in microchannels based on the cross-sectional profile of the Matrigel coating from Fig. 2A. Results showed that the capillary curvatures of the Matrigel bed at the two edges of the channel did not have a significant effect on the vorticity and shear-rate profiles in the midchannel region, where shear stress reached up to 0.3 Pa at the walls. To validate temperature and gaseous (CO2) equilibration of culture medium under constant flow, we performed computational modeling of culture medium flow in the porous tubing used in this study (Fig. 2B). Results showed that at the flow rates used the CO2 level (Fig. 2C) and temperature (Fig. 2D) of the culture medium reached equilibrium with the cell-culture conditions in the incubator [5% (vol/vol) CO2, 37 °C] before reaching the microchannel (Fig. S1 C and D).
Fig. 2.

Computational modeling of flow in the microfluidic platform. (A) Schematics of the cross-section of microchannels with a stromal bed (Matrigel coating). (Top to Bottom) Velocity field (mm/s), shear rate (1/s), and y- and z- components of vorticity distribution (1/s) are plotted across the cross-section above the Matrigel. (B) Schematic for gaseous and thermal equilibration across porous tubing. The length of the tube inside the incubator was designed to be sufficiently long to allow gaseous and thermal equilibration before entry of the medium into microchannels. (C) CO2 concentration (moles/m3) and (D) Temperature (°C) contours on the axisymmetric layer of medium and porous tubing that feeds the microchannel with culture medium that is sufficiently equilibrated to the appropriate temperature and CO2 levels before entering the channel.
Tumor Volume and Viability Are Significantly Lower in 3D Ovarian Nodules Grown Under Continuous Flow than in Equivalent Nonflow Cultures.
Intrachannel tumor cell adherence densities were quantified immediately postflow in three regions of the channels: proximal to the inlet (1.6–6.5 mm), middle (11.4–14.7 mm), and proximal to the outlet (19.4–24.3 mm) (Fig. 3 A and B). Malignant ascites contains a mix of tumor cells and other cell types as well proteinaceous secretions (11–15). The concentration of ovarian cancer cells in this milieu likely changes as the disease progresses and the volume of ascites increases. For the present study (Fig. 3B), we chose cell concentrations that span three orders of magnitude (104, 105, and 106 cells/mL) to account for a broad range of possibly relevant seeding densities (12, 66). Quantitative analysis (Fig. S3A) of intrachannel tumor cell adherence densities for the three initial cell concentrations shows a concentration-dependent increase in the number of adherent cells (Fig. 3B). A significant increase in intrachannel tumor cell adherence density is observed at an initial cell concentration of 106 cells/mL compared with the other cell concentrations (P < 0.05). There was a slight trend toward decreasing tumor cell adherence density along the channel, particularly in the section distal to the inlet, but these differences were not statistically significant. A more detailed characterization of the number of adherent cells per linear mm [λ (x)] immediately postflow (Fig. 3C) shows a similar trend as a function of distance along the channel at an initial cell concentration of 106 cells/mL. The number of cells adhered per linear mm across the channel [λ (y)] (Fig. 3D) indicated higher concentrations toward the channel center, possibly because of increased vorticity at the edges of the channel (Fig. 2A). After 7 d of growth under continuous flow, 3D micronodules were distributed along the full length of the channel (x axis) (Fig. 3E) and across the width of the channel (y axis) (Fig. 3F). The middle region (5–15 mm along the channel) produced the highest number of viable 3D tumors, possibly because of the stabilization of flow in this section of the channel. To evaluate the impact of flow on 3D nodule volume and viability (Fig. 3 G–I and Fig. S3B), nonflow 3D cultures were plated at densities equivalent to the intrachannel tumor cell adherence densities quantified in Fig. 3B (120, 40, and 8 cells/mm2) and were grown for 7 d (Fig. S4). Growth under continuous flow resulted in a significant decrease in mean tumor volume relative to nonflow cultures, at equivalent plating densities (Fig. 3G). At an equivalent plating density of 120 cells/mm2, mean tumor volume after 7 d of growth in nonflow cultures was 5.2 × 108 µm3 (± 4.6 × 107 µm3) (n = 6) compared with 2.7 × 104 ± 8.2 × 103 µm3 (n = 5) under continuous flow (P < 0.05). Sporadic 3D tumor formation under flow was observed at an equivalent plating density of 40 cells/mm2 [mean tumor volume: 1.1 × 104 µm3 (± 5.2 × 103 µm3)] (n = 3) compared with nonflow conditions and resulted in a significantly higher mean tumor volume (3.3 × 108 µm3 ± 1.9 × 107 µm3) (n = 6) (P < 0.05). Tumor formation at the lowest equivalent plating density (8 cells/mm2) was negligible under flow [mean tumor volume: 3.7 × 103 µm3 (± 2.5 × 103)] (n = 3) but was higher in nonflow cultures [8.6 × 107 µm3 (± 1.9 × 107 µm3)] (n = 6) (P < 0.05). Compared with nonflow 3D cultures, the distribution of nodule volumes following culture under continuous flow produced a shift toward smaller tumors (Fig. 3H). A significant decrease in fractional viability was observed at all equivalent plating densities (Fig. 3I). Nodules grown under continuous flow had a significantly lower fractional viability (0.44 ± 0.08; n = 5) than did nonflow 3D cultures (0.99 ± 0.0014; n = 6) (P < 0.05) at an equivalent density of 120 cells/mm2. These results for cell adherence and 3D tumor growth are dependent on the flow rates, microfluidic chip design, and stromal matrices as well as on the cells being evaluated. If these parameters are modified, similar characterization studies would have to be performed to account for variability in the biological characteristics and fluid dynamics of alternative systems.
Fig. 3.
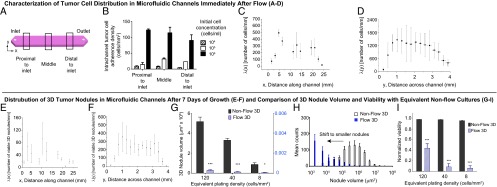
Characterization of tumor cell distribution in microfluidic channels immediately after flow (A–D), distribution of 3D tumor nodules in microfluidic channels after 7 d of growth (E and F), and comparison of 3D nodule volume and viability with equivalent nonflow cultures (G–I). (A) Intrachannel distribution of adherent ovarian cancer cells immediately after the introduction of three initial cell concentrations (104, 105, and 106 cells/mL) into the channels was quantified in three regions: proximal to the inlet (1.6–6.5 mm), middle (11.4–14.7 mm), and proximal to the outlet (19.4–24.3). (B) A concentration-dependent increase in the intrachannel tumor cell adherence density was observed in all three regions (P < 0.05). Within each initial cell concentration, results indicated no statistically significant difference in adherent cell densities across the channel in the three regions analyzed (P > 0.05). There was a trend toward decreased cell adherence density distal to the outlet at the initial cell concentration of 106 cells/mL. (C) Characterization of the number of adherent cells per linear mm [λ (x)] showed similar trends as a function of distance along the channel at the initial concentration of 106 cells/mL. (D) The number of cells adhered per linear distance across the channel [λ (y)] indicated higher concentration toward the channel center. (E and F) At an initial concentration of 106 cells/mL, adherent ovarian cancer cells that grew into 3D micronodules under the influence of continuous flow for 7 d were distributed along the full length of the channel (x axis) and across the width of the channel (y axis). (G) Compared with nonflow 3D cultures at all equivalent plating densities (black bars), growth under continuous flow (blue striped bars) resulted in a significant decrease in mean tumor volume. ***P < 0.001, *P = 0.01 to < 0.05. (H) A shift toward smaller tumors was observed in 3D nodules cultured under continuous flow (blue bars) compared with equivalent nonflow 3D cultures (white bars). (I) At all equivalent plating densities, 3D nodules grown under continuous flow (blue striped bars) had a significantly lower fractional viability than corresponding nonflow cultures (black bars). ***P < 0.001.
Flow Induces EMT and an Aggressive Phenotype in 3D Ovarian Micronodules.
To characterize the molecular and biological features of 3D nodules cultured under flow and nonflow conditions, we performed on-chip immunofluorescence staining of the EGFR (Fig. 4A), on-chip mRNA isolation and quantitative real-time PCR analysis (Fig. 4C), and on-chip protein isolation and quantification of select biomarkers relevant to ovarian cancer progression and resistance (Fig. 4D). Compared with nonflow cultures, 3D ovarian cancer nodules grown under flow exhibited morphological features indicative of increased EMT and a more motile phenotype (Fig. 4A). To quantify this contrast in morphology (Fig. 4B), the fractal dimension, df, was calculated by box counting in sets of brightfield images of cultures grown under flow and nonflow conditions. The df was lower under flow (1.78 ± 0.01; n = 8 fields) than in nonflow cultures (1.87 ± 0.01; n = 6 fields) (P < 0.05). These results were consistent with the expectation that fields containing cells with pronounced linear extensions characteristic of mesenchymal morphology have a lower df than the nonflow fields, which contain relatively densely packed nodules that fill the 2D image space more completely. A significant down-regulation of E-cadherin and CDC2 gene expression (Fig. 4C) was induced by flow (1.0 ± 0.10 and 1.0 ± 0.14, respectively) compared with nonflow cultures (1.5 ± 0.16 and 2.0 ± 0.25, respectively) (n = 6) (P < 0.05). EGFR gene expression levels were not significantly different between nonflow (1.1 ± 0.17) and flow (1.2 ± 0.12) (n = 6), whereas a significant increase in p27Kip1 gene levels was observed under flow (5.3 ± 0.71) relative to nonflow cultures (1.0 ± 0.17) (n = 6) (P < 0.05). In the presence of flow, a significant increase in EGFR protein expression and phosphorylation was observed (Fig. 4D) (0.70 ± 0.09 and 0.86 ± 0.10, n = 6 and 4, respectively) compared with nonflow cultures (0.28 ± 0.06, and 0.51 ± 0.09, n = 6 and 4, respectively) (P < 0.05). E-cadherin protein expression decreased significantly as a result of flow (0.19 ± 0.04) compared with nonflow 3D cultures (0.45 ± 0.10) (n = 9) (P < 0.05), and vimentin expression increased significantly under flow (1.14 ± 0.19, n = 9) compared with nonflow (0.11 ± 0.04, n = 8) (P < 0.05) (Fig. 4D). A flow-induced down-regulation of β-catenin expression was observed (mean normalized protein expression under flow was 0.67 ± 0.10 compared with 1.3 ± 0.10 in nonflow cultures) (n = 10) (P < 0.05). There was no change in the protein levels of integrin α5 between flow (0.57 ± 0.10, n = 8) and nonflow (0.72 ± 0.09, n = 8) cultures.
Fig. 4.

Biological characterization of 3D nodules cultured under flow vs. nonflow. (A) Compared with nonflow, 3D ovarian cancer nodules grown under flow exhibited morphological features indicative of increased EMT and a more motile phenotype, as observed by DIC and two-photon autofluorescence microscopy and on-chip immunofluorescence staining of a clinically relevant biomarker, EGFR. (B) Fractal dimension (a ratio describing how an object fills space out to its maximum radius with a value of 1 describing a line and a value of 2 a circle) was used to quantify the morphological features of 3D tumors grown under flow versus nonflow conditions. Fractal dimension was significantly lower under flow (1.78 ± 0.01, n = 8 fields) than in nonflow (1.87 ± 0.01, n = 6 fields) conditions (P < 0.05), supporting the observation that flow induces morphological changes consistent with increased EMT in 3D tumors. *** P < 0.001. (C) A significant down-regulation of E-cadherin and CDC2 gene expression was induced by flow (1.0 ± 0.10 and 1.0 ± 0.14, respectively) compared with nonflow cultures (1.5 ± 0.16 and 2.0 ± 0.25, respectively) (n = 6) (P < 0.05). EGFR gene expression levels were not significantly different between nonflow (1.1 ± 0.17) and flow (1.2 ± 0.12) conditions (n = 6), whereas a significant increase in p27Kip1 gene levels was observed under flow (5.3 ± 0.71) relative to nonflow cultures (1.0 ± 0.17) (n = 6) (P < 0.05). *** P < 0.001. (D) In the presence of flow, a significant increase in EGFR expression and phosphorylation was observed (0.70 ± 0.09 and 0.86 ± 0.10, n = 6 and 4, respectively) as compared with nonflow cultures (0.28 ± 0.06, and 0.51 ± 0.09, n = 6 and 4, respectively) (P < 0.05). E-cadherin protein expression decreased significantly in flow (0.19 ± 0.04) as compared with nonflow 3D cultures (0.45 ± 0.10) (n = 9) (P < 0.05), and vimentin expression increased significantly under flow (1.14 ± 0.19, n = 9) compared with nonflow (0.11 ± 0.04, n = 8) conditions (P < 0.05). A flow-induced down-regulation of β-catenin expression was observed (0.67 ± 0.10 under flow compared with 1.3 ± 0.10 in nonflow cultures) (n = 10) (P < 0.05). There was a trend toward a reversal of the protein expression of p27Kip1 and CDC2 (and activation for CDC2), relative to the gene expression for these markers, as a consequence of flow. ***P < 0.001, **P = 0.001 to <0.01, *P = 0.01 to <0.05.
Relative to the gene expression profiles for CDC2 and p27Kip1, there was a trend toward a reversal of the protein expression (and activation for CDC2), as a consequence of flow. Recent studies have provided increasing evidence that changes in gene expression do not always correlate with protein levels, highlighting the complexities of analyzing genetic and molecular responses in biological systems (40–47). Posttranscriptional regulation allows a more rapid response to stress and may play a more important role than previously assumed. These observations may help explain the discrepancy between RNA and protein expression of CDC2 and p27Kip1.
Discussion
The metastatic cascade is characterized by a high attrition rate and significant modulation of molecular biology caused in part by the dynamic physical forces encountered by tumor cells as they interact with distant stromal microenvironments and develop into micronodules (1–6). The present study describes a unique microfluidic platform that addresses the significant challenge of 3D cell growth under sustained flow to explore the impact of shear stress on tumor cell attachment and micronodule formation on a stromal matrix. Our findings indicate that fluidic streams significantly modulate the architectural and biological features of ovarian micronodule growth and induce physiologically relevant characteristics that cannot be revealed with existing systems. The present study describes a flow-induced modulation of tumor morphology, biomarker expression, and proliferative rate indicative of EMT. This transition is driven in part by a posttranslational up-regulation of EGFR expression and activation, which induces a transcriptionally regulated decrease in E-cadherin expression and a concomitant increase in vimentin protein expression with no change in the expression of integrin α5 (Fig. 5). These morphological features and molecular characteristics are associated with the worst prognosis for ovarian cancer patients (19, 20, 26, 31–33, 35) and collectively suggest the induction of a highly aggressive tumor phenotype.
Fig. 5.
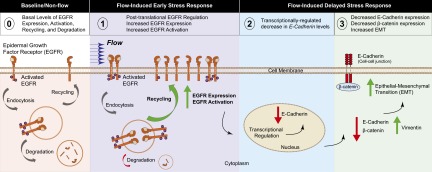
Proposed model for hydrodynamic stress-induced modulation of ovarian cancer biology. (0) Baseline levels of EGFR activity and protein expression are maintained by a complex array of factors including recycling and degradation of the activated receptor complex. (1) Flow-induced stress causes a posttranslational up-regulation of EGFR expression and activation, which may result from decreased EGFR degradation and increased receptor recycling. (2) The resultant increase in EGFR signaling modulates molecular pathways (26, 27) that induce a transcriptionally regulated decrease in (3) E-cadherin protein expression. A concomitant reduction in β-catenin and an increase in vimentin protein expression are observed, indicative of EMT resulting from stress induced by hydrodynamic physical forces.
Colonization of distant sites by metastatic nodules is a highly inefficient process because of the numerous physiological and biological hurdles as well as the physical stresses encountered by cancer cells throughout the metastatic cascade. The great majority of cancer cells that manage to colonize distant sites either persists as small colonies or suffers attrition over time (1, 6, 67, 68). Recent elegant studies have evaluated the influence of chemo-mechanical gradients on tumor migration and the impact of flow on cell proliferation using fluidic platforms (48–50). However, these studies have not investigated the impact of flow on the growth and molecular expression profiles of biologically relevant 3D tumor nodules. Shields et al. (48) have shown that interstitial flow enhances the migratory potential of metastatic cancer cells. Subsequently, Polacheck et al. (49) demonstrated that the directionality of tumor cell migration depends on the flow strength and cell density in collagen I scaffolds. Chang et al. (50) have shown that flow-induced shear stress causes cell-cycle arrest in monolayer cultures of multiple cancer cell lines. Microfluidic systems also have been used to mimic intravasation and extravasation to study cancer metastasis (69, 70). Cells were embedded in Matrigel; however, no flow was present during culture. In addition, gas equilibrium and nutrient transport were not considered during long-term culture in the absence of flow. These studies have provided valuable insights into early steps in the metastatic cascade but have limitations in terms of investigating the effect of flow on cell adhesion and 3D microtumor growth. The present findings provide an understanding of how fluidic streams significantly modulate the morphologic features, biomarker expression, and the volume and viability of 3D tumor nodules. Compared with nonflow cultures plated at equivalent densities for the same growth period, 3D tumors cultured under sustained flow showed increased EMT and expressed biomarkers associated with an aggressive and metastatic phenotype (19, 20, 26, 31–33, 35). A corresponding significant decrease in 3D tumor volume and viability was observed in 3D nodules grown under flow. These findings are consistent with the decreased proliferation that is a characteristic of EMT (30, 32, 35–37) and the high attrition rates that metastatic nodules experience as they cope with the physical and biological stresses associated with colonizing distant sites (1, 6, 67, 68). The results reported here highlight the value of the microfluidic platform as a tool to gain mechanistic insights into the influence of stromal and hydrodynamic cues on metastatic progression.
This versatile platform for sterile culture of 3D micrometastatic nodules under biologically relevant conditions of continuous flow is broadly applicable to other tumors and mechanisms of metastatic progression to provide improved biological insights and therapeutic strategies for an array of lethal malignancies. This platform integrates customized image analysis routines (Fig. S3) and provides an opportunity for on-chip immunofluorescence-based evaluation of clinically relevant biomarker expression. Additional capabilities relevant to the biological characterization capabilities of the microfluidic system include on-chip mRNA isolation for genomic analysis and on-chip protein isolation to evaluate biomarker expression and potential molecular targets during tumor growth and in response to applied therapies.
Our findings highlight the importance of the effect of flow on the growth, morphology, and biomarker expression of ovarian tumor micronodules. The fluidic streams depicted in Fig. 1A are established as part of a dynamic process that evolves as the disease progresses and ascites accumulate in the peritoneum (7). With increased volume of ascitic fluid, there is increased abdominal distention and intraabdominal pressure, which may impact the fluid dynamics of ascitic currents. However, the flow rates and fluid dynamics of these currents and the effects of disease progression and changes in ascites volume on these fluidic streams remain poorly characterized. The hydrodynamic characteristics of ascitic currents need to be investigated further to refine the flow conditions studied here. Guided by insights from the improved in vivo and clinical characterization of fluid dynamics in the peritoneal cavity, the microfluidic system can be customized to reflect the spatial and hydrodynamic complexities that arise from disease progression, ascites accumulation, and key anatomical features within the abdomen.
The attachment and growth of ovarian cancer cells in the peritoneal cavity involves interaction with a complex mixture of stromal molecules. We and others have shown that collagen IV, laminin, and fibronectin are among the components of the extracellular matrix that are critical to the adhesion and growth of tumor metastases, including ovarian micronodules (8, 71–74). Laminin and collagen IV are major components in Matrigel, and fibronectin is also present. This commercially available reconstituted basement membrane has been used by us and others to study the growth of 3D tumor nodules (51, 53, 55, 57, 59, 60, 75, 76), including ovarian cancer, and was a suitable stromal bed for the microfluidic chip used in the present study. The specific interaction between ovarian cancer cells and components of the stromal matrix is an important topic that may be the focus of future investigations with this platform.
A comprehensive treatment plan for a disease as complex as metastatic ovarian cancer likely will involve a multifaceted understanding of the factors that drive tumor heterogeneity and contribute to differential treatment efficacy. The present studies, performed with the OVCAR5 cell line derived from a patient with metastatic ovarian cancer that had not been previously treated, demonstrate the induction of an aggressive and metastatic tumor phenotype under the influence of flow in these cells that are naive to treatment. Future studies will test whether these changes that are indicative of aggressive disease also demonstrate increased resistance to treatment and how these outcomes are influenced by ovarian tumors with varying histories (e.g., adherent vs. nonadherent cells or treatment-naive vs. resistant cells). An increase in EMT by hydrodynamic forces described here provides preliminary insights into the factors that may influence differential sensitivity to chemotherapeutics and targeted inhibitors. Haslehurst et al. (33) showed that modulation of key regulators of EMT contributed directly to cisplatin resistance in ovarian cancer cells and primary tumors. In a study by Thomson et al. (34), non–small-cell lung carcinoma cells were differentially sensitive to the EGFR inhibitor erlotinib, and EMT status was a key determinant of sensitivity to treatment. Overexpression of the EGFR occurs in many tumors, including ovarian cancer, and is associated with increased aggressiveness and invasiveness, as well as the induction of EMT (22, 77–79). Several studies have identified the EGFR as an independent prognostic indicator for poor outcomes (20, 80), although the clinical consequences of EGFR expression remain controversial (23, 24, 81). Nonetheless, the EGFR remains an important and relevant focus for molecularly targeted therapies, as evidenced by recent clinical trials (23, 80, 82, 83). The broad impact of EGFR-targeted therapies alone remains to be borne out, but combination regimens with anti-EGFR agents have shown increased overall survival in patients with recurrent and refractory ovarian cancer (83).
The flow-induced decrease in E-cadherin expression concomitant with no change in integrin α5, observed in this study, is consistent with an expression profile that promotes metastases in ovarian cancer (8, 84, 85). Integrin antagonists, including monoclonal antibodies and peptides against integrin α5, reduced ovarian tumor burden and controlled metastases in preclinical models and have shown modest efficacy in clinical studies (85–87). Negative selection for E-cadherin was one of the factors described by Meirelles et al. (88) to predict increased colony formation, shorter tumor-free intervals in vivo, and not only resistance but stimulation by doxorubicin. However, these cells were sensitive to Mullerian inhibiting substance (MIS), a fetal testicular protein that targets the MIS receptor type II, which is expressed by a majority of ovarian carcinomas (88). The microfluidic platform provides a potentially valuable system to test chemotherapeutic agents and targeted biologics in unseparated and stem cell-enriched tumors. This system could be leveraged to gain insights into the evolution and reversibility of the morphological and molecular changes that occur under flow and to investigate the role of flow in the selection vs. induction of particular malignant characteristics such as invasiveness and genetic instability. The effectiveness of targeted agents and chemotherapeutics can be enhanced by the rational deployment of mechanistically distinct treatment modalities such as photodynamic therapy (PDT) (89). A biophysical cytotoxic therapy that involves the light-based activation of a photosensitive molecule, PDT has demonstrated clinical promise for the treatment of peritoneal cancers, particularly ovarian carcinoma (90, 91). PDT synergizes with chemotherapeutics and biological agents (19, 60, 89, 92), and these combinations are in early clinical trials. This study attempts to address the significant challenge of studying metastatic tumor cell growth in physiologically relevant conditions and suggests the need to develop complementary interventional strategies that account for the impact of dynamic physical forces. Microfluidic systems, such as the one described here, may enable the development of therapeutic strategies informed by locoregional differences in tumor characteristics caused in part by the influence of hydrodynamic stress. This advance will provide improved biological insights and therapeutic strategies for an array of lethal malignancies and may enable the study of a variety of physiologic and pathological conditions and drug combinations.
Materials and Methods
Microfluidic Chip Design and Fabrication.
The microfluidic chip (Fig. S1 A and B) was fabricated using 3.175-mm-thick polymethyl methacrylate (PMMA) (McMaster Carr). Microfluidic chip components were cut to 24 × 40 mm dimensions (VersaLASER; Universal Laser Systems Inc.). Three channels with a width of 4 mm were cut into a 254-μm-thick medical-grade double-sided adhesive (DSA) film (ARcare 90485; Adhesives Research) to provide the channel height with a 3-mm space in between. The channel inlet and outlet regions were fabricated with a 127° angle to facilitate fluid entrance to and exit from the channels (93, 94). The inlet and outlet ports of microchannels were 2.2 mm in diameter and were positioned 5 mm from the edge of the chip. The channel length is defined as the distance between inlet and outlet. All components were sterilized using isopropyl alcohol followed by air drying under sterile conditions. Microfluidic chip fabrication was performed in a laminar flow hood for long-term culture studies. Glass coverslips (Corning) with DSA films attached were placed on an ice pack (to prevent Matrigel polymerization), and the Matrigel was spread evenly on the channel surface (Figs. 1 B and C and 2A). A PMMA layer was used to cover the Matrigel-coated glass coverslip and DSA films, forming microfluidic channels. The microfluidic chips were incubated at room temperature, allowing gelation of Matrigel within channels. Next, gas-permeable silicon tubing (TYGON 3350 silicone; Cole-Parmer) was attached to the inlet and outlet ports of microfluidic chips. Epoxy glue (5 Minute Epoxy; Devcon) was used to seal the external tubing edges at the inlet and outlet. We have designed and fabricated microfluidic chips with easily accessible, inexpensive components and adhesives, allowing widespread use of this method by other researchers around the world.
Cell Line and Culture.
Epithelial ovarian cancer NIH:OVCAR5 cells were obtained from Thomas Hamilton (Fox Chase Cancer Institute, Philadelphia, PA) (95). The cells were grown under standard conditions (19, 59, 60, 95) in recommended culture medium: RPMI 1640 (Mediatech) plus 10% heat-inactivated FBS (GIBCO) plus 100 U/mL penicillin and 100 µg/mL streptomycin (Mediatech). The basement membrane used for 3D cell culture was GFR Matrigel (354230; BD Biosciences).
Thermal and Gas Equilibration Across Gas-Permeable Tubing During Long-Term Culture.
We developed a computational model to analyze the effects of thermal and gaseous gradients across the gas-permeable silicone tube (TYGON 3350 silicone; Cole-Parmer). For 3D growth, culture medium with 2% GFR Matrigel was supplied through 100-cm-long tubing (of which 40 cm ran from the incubator lid to the microchannel inlet). The outer and inner diameters of the gas-permeable tube were 2.4 mm and 0.76 mm, respectively. The tube inside the incubator was designed to be long enough so that gaseous and thermal equilibrations were reached before the medium entered the microchannels. The incubator was maintained at standard tissue-culture conditions (5% CO2 and 37 °C).
In our computational model, we assumed that the medium flowing in the gas-permeable tube was at ambient temperature (25 °C) and CO2 concentration (0.03% CO2). Therefore, there was a gradient of CO2 from 5% CO2 (incubator) to 0.03% CO2 (medium flowing inside the tube) across the gas-permeable silicone tube. Similarly, there was a thermal gradient from 37 °C (incubator) to 25 °C (medium flowing inside the tube).
Fig. 2B demonstrates the schematic for the computational model. Axisymmetric Navier–Stokes equations were solved in the fluidic domain to evaluate velocity distribution. Convection–diffusion transport equations of temperature and concentration were solved in both fluidic and permeable tube domains to assess the equilibration of CO2 and distribution of temperature. We assumed that the diffusion coefficient of CO2 in medium can be approximated as the diffusion coefficient of CO2 in water, which was reported as 2 × 10−9 m2/s (96). The diffusion coefficient of CO2 across the gas-permeable tube was evaluated by incorporating the permeability coefficient (processsystems.saint-gobain.com) and pressure drop across the tubing thickness. Pressure distribution along the tubing (ΔP) was determined based on the following equation: P = 8µLQ/πR4, where µ = 0.0006965 Pa/s (at 37 °C), L = 100 cm, Q = 100 µL/min, and R = 0.38 mm. Note that here ΔP is the difference in pressure between the inlet and the outlet of the tube. The outlet of tube was set to atmospheric pressure. There was a linear pressure gradient along the tube. The axisymmetric Navier–Stokes equations were solved for modeling fluidic flow inside the gas-permeable silicone tube. To model thermal and gas equilibration, axisymmetric convection and diffusion equations were solved.
Flow Modeling.
Flow inside the microfluidic channel with Matrigel coating (Fig. 2A) is modeled by Cartesian Navier–Stokes equations. The cells moving closer to the edges of channel were constantly exposed to a small rotational field (see vorticity in z-direction; Fig. 2A). This rotational field might force cells to move back and forth toward the channel edges, creating an additional shear imbalance on the cells. The larger number of cells adhering in the middle section of the microchannels can be attributed to this phenomenon.
Characterization of Intrachannel Tumor Cell Adherence Density Immediately Post-Flow.
The microfluidic chips were fabricated as described above. One-milliliter syringes (309628; BD) fitted with 18-G blunt needles (B18-100; SAI Infusion Technologies Inc.) were loaded with OVCAR5 cells at three stock concentrations (104, 105, and 106 cells/mL) in complete culture medium. The syringes were attached to gas-permeable silicone tubing (20-cm inlet, 20-cm outlet) and were loaded onto a vertically placed syringe pump (Multi-Phaser, NE-1600; New Era Pump Systems) set appropriately for the syringe being used in these experiments. The cell suspensions were pumped into the microchannels at a flow rate of 100 µL/min for 5 min. During flow, cells adhered on the channel surface; those cells that did not adhere flowed out of the channels and were collected and counted in the outlet reservoir. The cell concentrations in the inlet and outlet reservoirs were quantified using a hemocytometer before and after flow. The adhered cells in the channels were imaged and the intrachannel distribution was quantified as described in Microscopy and Quantitative Image Analysis.
Growth of 3D Tumor Nodules in Microchannels Under Continuous Flow.
All procedures used to prepare the chips and to introduce OVCAR5 cells into the microfluidic channels for 3D cultures were conducted under sterile conditions in a laminar-flow tissue-culture hood. A detailed description of microfluidic chip fabrication is provided in Microfluidic Chip Design and Fabrication. The gas-permeable silicone tubing for 3D culture experiments was 100 cm long from the pump to the inlet and 20 cm long at the outlet. Suspensions of OVCAR5 cells were prepared at three concentrations (104, 105, 106 cells/mL) in complete culture medium and were loaded into 1-mL syringes fitted with 18-G blunt needles. The cell-loaded syringes were attached to the inlet tubing and were loaded onto a vertically placed syringe pump with appropriate settings for the 1-mL BD syringe used during this phase of the experiment. The outlet tubes were inserted through holes made in the caps of 50-mL conical tubes (430291; Corning) to collect the effluent. The cell suspensions were pumped into the silicone tubing at a flow rate of 100 µL/min for 5 min. Upon completion, the tubing was clipped at the inlets and outlets, and the syringes were removed from the pump. Twenty-milliliter syringes (309661; BD) filled with 3D culture medium were loaded onto the pump, and the settings were adjusted for the larger syringes. The pump was run until the air was removed and the volumes were equalized. The needles were detached from the 1-mL cell-loaded syringes, and the residual medium in the hub was aspirated using a Gilson pipette. The hubs were filled with fresh culture medium to prevent air bubbles from entering the tubing, and then were attached to the 20-mL syringes. The cells were pumped from the tubing into the microfluidic channels at a flow rate of 100 µL/min for 5 min. Upon completion, the entire setup (pump, tubing, chips, and effluent reservoirs) was moved carefully from the tissue-culture hood to the incubator. The cultures were maintained under continuous flow for 7 d at a flow rate of 2.0 µL/min. Additional details relevant to the temperature and concentration of CO2 in the medium that entered the channels are provided in Thermal and Gas Equilibration Across Gas-Permeable Tubing During Long-Term Culture. The intrachannel distribution, volume, and viability of the resultant 3D nodules were quantified as described below in Microscopy and Quantitative Image Analysis.
Nonflow 3D Tumor Culture.
The protocols for nonflow 3D ovarian cancer cultures were based on previously published methods developed by us and others (51, 53, 55, 57, 59, 60, 75, 76). Briefly, stromal beds were prepared by pipetting 250 µL of chilled GFR Matrigel into each well of a chilled 24-well plate (662892; Greiner Bio-One). After a brief incubation at 37 °C, OVCAR5 cells were added to the polymerized beds at plating densities ranging from 4 cells/mm2 to 200 cells/mm2 (Fig. S4) in 1 mL of 2% GFR Matrigel complete growth medium per well. The 3D cultures were maintained at 37 °C at 5% CO2 for 7 d with medium changes every 3 d. Based on the range of intrachannel tumor cell adherence densities observed in microfluidic channels (Fig. 3B), the most relevant densities for flow and nonflow comparison studies were 4, 8, and 120 mm2.
Microscopy and Quantitative Image Analysis.
Intrachannel distribution of adherent tumor cells immediately post-flow.
To characterize the distribution of adherent cells to Matrigel beds immediately after flowing through channels, differential interference contrast (DIC) image mosaics of microfluidic channels (Fig. S3A) were acquired for all three stock concentrations (104, 105, and 106 cells/mL). Multiarea mosaics were obtained with a 10× 0.4 NA objective on an Olympus FV-1000 confocal microscope (Olympus), equipped with a Prior Proscan XY positioning stage (Prior Scientific, Rockland). Stitching of image mosaics and all subsequent image processing described herein was conducted using custom routines developed in the MATLAB software package (MathWorks) adapted from a set of analysis protocols previously described for image-based quantification of in vitro 3D tumor growth and treatment response (59). The routine used here first applies a wavelet transform to remove gradient background and then converts images to binary using a threshold operation based on Otsu’s method to identify high-contrast objects as cells. The routine then tabulates segmented features to obtain lists of size (based on an ellipsoid approximation of 3D volume), eccentricity, and position, identifying cells as features with a minimum estimated 3D volume of 5,400 μm3. To exclude the sharply sloped regions at the edge of channels (Fig. 2A) containing objects at multiple focal depths, edges are identified automatically in the analysis by applying a wavelet transform with a narrow bandpass to each image and then performing two consecutive rounds of thresholding as described above. Edge positions are determined by identifying objects in the “edge” masks, tabulating the y-positions of objects with eccentricities greater than 0.93, and taking the crop parameters to be the maximum and minimum y-positions in the object list for the top and bottom edges, respectively. Stitched images then are cropped according to these parameters. Finally the spatial seeding density for each flow condition is obtained by dividing the cell count over the area of the matrix bed imaged. Sensitivity and specificity of the object identification algorithm were validated for all initial cell concentrations by manual counting of objects in representative DIC image fields compared with the same fields with arrows overlaid at each computer-identified object position (Fig. S3A).
Intrachannel distribution, volume, and viability of 3D tumor nodules after 7 d growth under flow.
To characterize nodule formation and viability over extended periods under flow, cultures were stained on day 7 by introducing calcein AM and ethidium bromide (Invitrogen) at a flow rate of 10 μL/min for 40 min while the chips remained in an incubator at 37 °C. The fluorescent signal of cleaved calcein is proportional to esterase activity as a reporter of cell viability, whereas ethidium bromide becomes brightly fluorescent upon passing through the cell membrane of dead or dying cells and intercalating into DNA. Mosaics of fluorescence image data (Fig. S3B) were obtained and stitched as described above. Calcein and intercalated ethidium bromide were excited with 488-nm and 559-nm laser sources, respectively. For quantification of overall viability, each channel (calcein, and intercalated ethidium bromide) was binarized as described above, and a mask of nodule regions in the 2D projection was computed as the summation of binary pixel values from each channel. In cases where “live” and “dead” pixels were colocalized, a logical operator was applied to assign the value to 1. Viability was reported as the ratio of total live pixels to total nodule area. This protocol is an adaptation of a methodology previously described to assess cytotoxic response (59, 60), modified here to draw comparisons among multiple untreated arms with differing growth conditions. Using the segmentation methodology above, object size analysis was performed on viable (calcein-positive) cells, and lists of estimated volumes from segmented images were binned into logarithmically spaced bins to create size-distribution histograms as previously described (59). The sensitivity and specificity of the object-identification algorithm in fluorescence images were validated for all initial cell concentrations by manual cell counting of representative fields compared with the same fields with arrows overlaid at each computer-identified object position (Fig. S3B). Data represent averages of multiarea mosaics from 3–6 channels per cell density from three independent experiments.
Volume and viability of 3D tumor nodules in nonflow 3D cultures after 7 d growth.
To image nodules under nonflow conditions, cultures were incubated with calcein and ethidium bromide reagents for 40 min. Fluorescence images were obtained with the Olympus FV-1000 confocal microscope as 2 × 2 image mosaics from the central region of each well using a 4× 0.16 NA objective. Analysis of nodule size and viability was performed in the same manner as for flow-based cultures. Data represent six wells per cell density from two independent experiments.
Measurement of the Matrigel bed profile.
To measure the profile of the Matrigel bed in microfluidic channels, 1.02 μm yellow-green fluorescent carboxylate-modified polystyrene microscopheres (Polysciences Inc) were incorporated into the cold GFR Matrigel before gelation to provide fluorescence contrast into beds (otherwise prepared as described above in Microfluidic Chip Design and Fabrication. Confocal fluorescence z-stacks were obtained along the length of the channel, merged, and rotated 90° to visualize the profile.
Quantification of morphology by fractal dimension calculation.
To quantify the contrasting morphology of tumor nodules formed under flow and nonflow conditions, the fractal dimension, df, (which describes the scaling relationship of an object of mass, m, with a radius, r, as m~rdf) was calculated from sets of DIC images obtained by random sampling throughout respective channels imaged after 7 d of growth in the specified conditions. The applicability of this approach is suggested by previous literature in which df is used to differentiate structural and morphological properties of different tissues (97). Based on the qualitative appearance of nearly circular multicellular objects forming in the absence of flow, we anticipate <df> approaching 2, in contrast to the more fibroblast-like morphology with less complete filling of 2D space within the spatial extent of objects formed under flow, corresponding to a lower value of <df> between 1 and 2. Calculation of df from image data was based on an open-source box-counting script for MATLAB (mathworks.com). Measurements over six to eight fields from two separate experiments were obtained to report <df>in each condition. Data represent a minimum of eight randomly selected fields pooled from two experiments for flow and non-flow conditions.
Quantitative Real-Time PCR Analysis.
The on-chip total RNA isolation was performed using the Qiagen RNeasy Mini Kit (Qiagen, Inc.). On day 7 of 3D culture under flow, the microfluidic chips were placed immediately on ice, and the inlet and outlet tubes were cut to 2 cm. The shortened inlet tube was connected to the tip of an 18-G 1-inch blunt needle (B18-100; SAI Inc.) on a 1-mL syringe (309268; BD Biosciences). Each channel was filled with 0.5 mL of RLT lysis buffer (79216; Qiagen Inc.), and the lysate was pushed out of the outlet tube and collected in a 1.5-mL Eppendorf tube. This step was repeated with lysate; then each channel was flushed with an additional 0.5 mL of fresh RLT buffer, which was combined with the lysate to obtain a total 1 mL of lysate per channel. First-strand cDNA synthesis used 1 mmol/L of oligo dT of 18-mer (Invitrogen) and 1.0 μg of total RNA per 100-μL reaction volume with Ready-To-Go You-Prime First-Strand Beads (Amersham Biosciences). To validate the efficiency of the real-time PCR assay, a fourfold dilution series was generated with the cDNA. The equivalent of 0.83 μL of the cDNA generated in its synthesis step was used per reaction. PCR reactions were performed in the final volume of 25 μL containing 20 mM Tris⋅HCl (pH 8.4), 50 mM KCl, 200 μM of each of four dNTPs (dATP, dCTP, dGTP, dTTP), 0.5 μM of each primer, 3.5 mM MgCl2, and 1 × SYBR Green (Bio-Rad). Six samples for each treatment group and PCR assays in duplicate were used. PCR was carried out with an initial 5-min denaturing step at 95 °C followed by 40 cycles of an annealing step at 58 °C for 30 s, an extension step at 72 °C for 20 s, and a denaturing step at 95 °C for 20 s. After completion of the PCR amplification reaction, a melting curve analysis was performed by heating the samples to 95 °C and immediately cooling them to 50 °C for 1 min. The samples were then incrementally heated to 95 °C in steps of 1 °C for 10 s per step with the fluorescence measured continuously. GAPDH was used as a reference housekeeping gene for internal control. All real-time PCR reactions were run on the iCycler iQ5 (Bio-Rad). The relative gene expression was quantified by using the ΔΔcycle threshold method. The data represent pooled samples from six channels (flow) or 12 wells (non-flow) from two independent experiments for flow and non-flow conditions, evaluated in triplicate and repeated twice.
Western Blot Analysis.
Flow 3D samples cultured for 7 d were placed on ice, and the inlet and outlet tubes were cut to 2 cm and clipped to avoid efflux of samples and medium from the channels. A 1-mL syringe fitted with an 18-G blunt needle was used to flow 70 µL of chilled RIPA lysis buffer (89900; Thermo Scientific) containing a mixture of phosphatase inhibitors (P5726; Sigma) and protease inhibitors (P8340; Sigma) into the channels. Any efflux was collected into Eppendorf tubes and placed on ice. The end of each tube was clipped, and the chip was incubated on ice for 30 min. An additional 30 µL of RIPA lysis buffer was used to flush the lysed cells and Matrigel beds out of channels into Eppendorf tubes at the end of the 30-min incubation period. The blunt needle-fitted syringe was used to push air through the tubing of each channel three times to improve removal of the lysate from chip. The samples then were incubated for another 30 min on ice with mixing by pipetting every 10 min during the incubation period. The lysed samples were centrifuged at 16,000 × g at 4 °C for 15 min (Model 5415 with 18-place fixed angle rotor for 1.5–2 mL tubes; Eppendorf); then the supernatant containing the protein lysate was removed and aliquoted. Each sample was a pool of three channels.
To prepare protein lysates from 7-d nonflow cultures in 24-well plates, the samples were placed on ice. The culture medium was aspirated carefully, and 200 µL of chilled RIPA buffer containing a mixture of phosphatase and protease inhibitor was added to each well and incubated for 30 min to ensure complete lysis of the cells and Matrigel beds. The lysed samples were centrifuged at 16,000 × g at 4 °C for 15 min; then the supernatant containing the protein lysate was removed and aliquoted. Each sample was a pool of 12 wells.
The Pierce Bicinchoninic acid (BCA) protein assay kit (23225; Thermo Scientific) was used to quantify the protein lysate. Protein lysates were separated on either 4–15% or 10% (wt/vol) SDS/PAGE gel (Bio-Rad) at 70 V for ∼4 h or 1.5 h, respectively. The proteins were transferred onto an immunoblot polyvinylidene difluoride (PVDF) membrane (Bio-Rad) at 120 V for 1 h on ice in the cold room. The membranes were probed with the following primary antibodies (obtained from Cell Signaling Technology unless otherwise stated): anti–β-catenin (5982), anti-p27Kip1 (3686), anti–E-cadherin (5296), anti-cdc2 (9112), anti–phospho-cdc2 (9111), anti-integrin alpha 5 (4705), anti-EGFR (4405), and anti–phospho-EGFR Tyr1173 (4407). Rabbit polyclonal vimentin antibody (sc-5565) was obtained from Santa Cruz Biotechnology. All primary antibody dilutions were at 1:1,000 except for vimentin, which was used at 1:200 (final antibody concentration was 0.2 µg/µL). Anti-rabbit IgG HRP-conjugated secondary antibody (7074; Cell Signaling Technology) was used to blot for all primary antibodies except for anti–E-cadherin, for which anti-mouse IgG HRP-linked secondary antibody was used. β-Actin rabbit HRP-conjugated antibody (5125; CST) was used for actin-loading control. All proteins were detected under nonreducing conditions, except for anti-p27Kip1, which was performed under reducing conditions. The HRP signal from protein bands was detected using the Immun-Star WesternC substrate ECL kit (Bio-Rad) on a Kodak image station 4000R. Protein-of-interest signal intensities were normalized to their respective β-actin loading control. Densitometry was performed using Image J analysis software (National Institutes of Health). Data represent at least two independent experiments each for flow and non-flow conditions, repeated a minimum of five times using two to four replicates of randomly selected pooled samples.
Statistical Analysis.
The experimental results were analyzed using ANOVA with Tukey’s post hoc test for multiple comparisons and Student’s two-tailed t test for single comparisons with statistical significance threshold set at 0.05 (P < 0.05). Unless otherwise stated, mean values represent three experiments with two or three channels per experiment, and error bars represent standard error of the mean. Statistical analyses were performed with Minitab (Minitab) and GraphPad Prism (GraphPad Software).
Supplementary Material
Acknowledgments
We thank Drs. Patricia Donahoe and Marcela del Carmen for valuable guidance, feedback, and clinical perspective; Dr. Bryan Spring for assistance with the immunofluorescence protocol and fluorescence imaging; Michael Glidden for assistance with image processing; and Drs. Maxim Olchanyi and Bala Sundaram for useful conversations regarding interpretation of fractal geometries in image data. This work was supported by National Institutes of Health Grants K99CA155045 (to J.P.C.); R21-HL112114, R21-AI087107, R01AI081534, and R01EB015776 (to U.D.); and R01CA158415, R01CA160998, and 5PO1CA084203 (to T.H.); and by the Eleanor and Miles Shore Scholars in Medicine Program (J.P.C.). U.D. is the recipient of National Science Foundation CAREER Award 1150733.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission. D.K. is a guest editor invited by the Editorial Board.
See Author Summary on page 8773 (volume 110, number 22).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1216989110/-/DCSupplemental.
References
- 1.Gupta GP, Massagué J. Cancer metastasis: Building a framework. Cell. 2006;127(4):679–695. doi: 10.1016/j.cell.2006.11.001. [DOI] [PubMed] [Google Scholar]
- 2.Nguyen DX, Bos PD, Massagué J. Metastasis: From dissemination to organ-specific colonization. Nat Rev Cancer. 2009;9(4):274–284. doi: 10.1038/nrc2622. [DOI] [PubMed] [Google Scholar]
- 3.Joyce JA, Pollard JW. Microenvironmental regulation of metastasis. Nat Rev Cancer. 2009;9(4):239–252. doi: 10.1038/nrc2618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wirtz D, Konstantopoulos K, Searson PC. The physics of cancer: The role of physical interactions and mechanical forces in metastasis. Nat Rev Cancer. 2011;11(7):512–522. doi: 10.1038/nrc3080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chaffer CL, Weinberg RA. A perspective on cancer cell metastasis. Science. 2011;331(6024):1559–1564. doi: 10.1126/science.1203543. [DOI] [PubMed] [Google Scholar]
- 6.Valastyan S, Weinberg RA. Tumor metastasis: Molecular insights and evolving paradigms. Cell. 2011;147(2):275–292. doi: 10.1016/j.cell.2011.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tan DS, Agarwal R, Kaye SB. Mechanisms of transcoelomic metastasis in ovarian cancer. Lancet Oncol. 2006;7(11):925–934. doi: 10.1016/S1470-2045(06)70939-1. [DOI] [PubMed] [Google Scholar]
- 8.Lengyel E. Ovarian cancer development and metastasis. Am J Pathol. 2010;177(3):1053–1064. doi: 10.2353/ajpath.2010.100105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Naora H, Montell DJ. Ovarian cancer metastasis: Integrating insights from disparate model organisms. Nat Rev Cancer. 2005;5(5):355–366. doi: 10.1038/nrc1611. [DOI] [PubMed] [Google Scholar]
- 10.Bast RC, Jr, Hennessy B, Mills GB. The biology of ovarian cancer: New opportunities for translation. Nat Rev Cancer. 2009;9(6):415–428. doi: 10.1038/nrc2644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Adam RA, Adam YG. Malignant ascites: Past, present, and future. J Am Coll Surg. 2004;198(6):999–1011. doi: 10.1016/j.jamcollsurg.2004.01.035. [DOI] [PubMed] [Google Scholar]
- 12.Scarberry KE, Dickerson EB, Zhang ZJ, Benigno BB, McDonald JF. Selective removal of ovarian cancer cells from human ascites fluid using magnetic nanoparticles. Nanomedicine. 2010;6(3):399–408. doi: 10.1016/j.nano.2009.11.003. [DOI] [PubMed] [Google Scholar]
- 13.Ho C-M, Chang S-F, Hsiao C-C, Chien T-Y, Shih DT. Isolation and characterization of stromal progenitor cells from ascites of patients with epithelial ovarian adenocarcinoma. J Biomed Sci. 2012;19:23. doi: 10.1186/1423-0127-19-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Feldman GB, Knapp RC, Order SE, Hellman S. The role of lymphatic obstruction in the formation of ascites in a murine ovarian carcinoma. Cancer Res. 1972;32(8):1663–1666. [PubMed] [Google Scholar]
- 15.Holm-Nielsen P. Pathogenesis of ascites in peritoneal carcinomatosis. Acta Pathol Microbiol Scand. 1953;33(1):10–21. doi: 10.1111/j.1699-0463.1953.tb04805.x. [DOI] [PubMed] [Google Scholar]
- 16.Sugarbaker PH. Observations concerning cancer spread within the peritoneal cavity and concepts supporting an ordered pathophysiology. Cancer Treat Res. 1996;82:79–100. doi: 10.1007/978-1-4613-1247-5_6. [DOI] [PubMed] [Google Scholar]
- 17.Feki A, et al. Dissemination of intraperitoneal ovarian cancer: Discussion of mechanisms and demonstration of lymphatic spreading in ovarian cancer model. Crit Rev Oncol Hematol. 2009;72(1):1–9. doi: 10.1016/j.critrevonc.2008.12.003. [DOI] [PubMed] [Google Scholar]
- 18.Carmignani CP, Sugarbaker TA, Bromley CM, Sugarbaker PH. Intraperitoneal cancer dissemination: Mechanisms of the patterns of spread. Cancer Metastasis Rev. 2003;22(4):465–472. doi: 10.1023/a:1023791229361. [DOI] [PubMed] [Google Scholar]
- 19.del Carmen MG, et al. Synergism of epidermal growth factor receptor-targeted immunotherapy with photodynamic treatment of ovarian cancer in vivo. J Natl Cancer Inst. 2005;97(20):1516–1524. doi: 10.1093/jnci/dji314. [DOI] [PubMed] [Google Scholar]
- 20.Psyrri A, et al. Effect of epidermal growth factor receptor expression level on survival in patients with epithelial ovarian cancer. Clin Cancer Res. 2005;11(24 Pt 1):8637–8643. doi: 10.1158/1078-0432.CCR-05-1436. [DOI] [PubMed] [Google Scholar]
- 21.Avraham R, Yarden Y. Feedback regulation of EGFR signalling: Decision making by early and delayed loops. Nat Rev Mol Cell Biol. 2011;12(2):104–117. doi: 10.1038/nrm3048. [DOI] [PubMed] [Google Scholar]
- 22.Alper O, et al. Epidermal growth factor receptor signaling and the invasive phenotype of ovarian carcinoma cells. J Natl Cancer Inst. 2001;93(18):1375–1384. doi: 10.1093/jnci/93.18.1375. [DOI] [PubMed] [Google Scholar]
- 23.Zeineldin R, Muller CY, Stack MS, Hudson LG. Targeting the EGF receptor for ovarian cancer therapy. J Oncol. 2010;2010:414676. doi: 10.1155/2010/414676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Posadas EM, et al. A phase II and pharmacodynamic study of gefitinib in patients with refractory or recurrent epithelial ovarian cancer. Cancer. 2007;109(7):1323–1330. doi: 10.1002/cncr.22545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dimou A, et al. Standardization of epidermal growth factor receptor (EGFR) measurement by quantitative immunofluorescence and impact on antibody-based mutation detection in non-small cell lung cancer. Am J Pathol. 2011;179(2):580–589. doi: 10.1016/j.ajpath.2011.04.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sawada K, et al. Loss of E-cadherin promotes ovarian cancer metastasis via alpha 5-integrin, which is a therapeutic target. Cancer Res. 2008;68(7):2329–2339. doi: 10.1158/0008-5472.CAN-07-5167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cowden Dahl KD, et al. Matrix metalloproteinase 9 is a mediator of epidermal growth factor-dependent e-cadherin loss in ovarian carcinoma cells. Cancer Res. 2008;68(12):4606–4613. doi: 10.1158/0008-5472.CAN-07-5046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Caswell PT, et al. Rab-coupling protein coordinates recycling of alpha5beta1 integrin and EGFR1 to promote cell migration in 3D microenvironments. J Cell Biol. 2008;183(1):143–155. doi: 10.1083/jcb.200804140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cavallaro U, Dejana E. Adhesion molecule signalling: Not always a sticky business. Nat Rev Mol Cell Biol. 2011;12(3):189–197. doi: 10.1038/nrm3068. [DOI] [PubMed] [Google Scholar]
- 30.Moreno-Bueno G, et al. The morphological and molecular features of the epithelial-to-mesenchymal transition. Nat Protoc. 2009;4(11):1591–1613. doi: 10.1038/nprot.2009.152. [DOI] [PubMed] [Google Scholar]
- 31.Ahmed N, Abubaker K, Findlay J, Quinn M. Epithelial mesenchymal transition and cancer stem cell-like phenotypes facilitate chemoresistance in recurrent ovarian cancer. Curr Cancer Drug Targets. 2010;10(3):268–278. doi: 10.2174/156800910791190175. [DOI] [PubMed] [Google Scholar]
- 32.Ahmed N, Thompson EW, Quinn MA. Epithelial-mesenchymal interconversions in normal ovarian surface epithelium and ovarian carcinomas: An exception to the norm. J Cell Physiol. 2007;213(3):581–588. doi: 10.1002/jcp.21240. [DOI] [PubMed] [Google Scholar]
- 33.Haslehurst AM, et al. EMT transcription factors snail and slug directly contribute to cisplatin resistance in ovarian cancer. BMC Cancer. 2012;12:91. doi: 10.1186/1471-2407-12-91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Thomson S, et al. Epithelial to mesenchymal transition is a determinant of sensitivity of non-small-cell lung carcinoma cell lines and xenografts to epidermal growth factor receptor inhibition. Cancer Res. 2005;65(20):9455–9462. doi: 10.1158/0008-5472.CAN-05-1058. [DOI] [PubMed] [Google Scholar]
- 35.Iwatsuki M, et al. Epithelial-mesenchymal transition in cancer development and its clinical significance. Cancer Sci. 2010;101(2):293–299. doi: 10.1111/j.1349-7006.2009.01419.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jungmann O, et al. The dermatan sulfate proteoglycan decorin modulates α2β1 integrin and the vimentin intermediate filament system during collagen synthesis. PLoS ONE. 2012;7(12):e50809. doi: 10.1371/journal.pone.0050809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gilles C, et al. Vimentin contributes to human mammary epithelial cell migration. J Cell Sci. 1999;112(Pt 24):4615–4625. doi: 10.1242/jcs.112.24.4615. [DOI] [PubMed] [Google Scholar]
- 38.Besson A, Assoian RK, Roberts JM. Regulation of the cytoskeleton: An oncogenic function for CDK inhibitors? Nat Rev Cancer. 2004;4(12):948–955. doi: 10.1038/nrc1501. [DOI] [PubMed] [Google Scholar]
- 39.Chu IM, Hengst L, Slingerland JM. The Cdk inhibitor p27 in human cancer: Prognostic potential and relevance to anticancer therapy. Nat Rev Cancer. 2008;8(4):253–267. doi: 10.1038/nrc2347. [DOI] [PubMed] [Google Scholar]
- 40.Holcik M, Sonenberg N. Translational control in stress and apoptosis. Nat Rev Mol Cell Biol. 2005;6(4):318–327. doi: 10.1038/nrm1618. [DOI] [PubMed] [Google Scholar]
- 41.Nishizuka S, et al. Proteomic profiling of the NCI-60 cancer cell lines using new high-density reverse-phase lysate microarrays. Proc Natl Acad Sci USA. 2003;100(24):14229–14234. doi: 10.1073/pnas.2331323100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gygi SP, et al. Quantitative analysis of complex protein mixtures using isotope-coded affinity tags. Nat Biotechnol. 1999;17(10):994–999. doi: 10.1038/13690. [DOI] [PubMed] [Google Scholar]
- 43.Rajasekhar VK, Holland EC. Postgenomic global analysis of translational control induced by oncogenic signaling. Oncogene. 2004;23(18):3248–3264. doi: 10.1038/sj.onc.1207546. [DOI] [PubMed] [Google Scholar]
- 44.Rajasekhar VK, et al. Oncogenic Ras and Akt signaling contribute to glioblastoma formation by differential recruitment of existing mRNAs to polysomes. Mol Cell. 2003;12(4):889–901. doi: 10.1016/s1097-2765(03)00395-2. [DOI] [PubMed] [Google Scholar]
- 45.Celis JE, et al. Gene expression profiling: Monitoring transcription and translation products using DNA microarrays and proteomics. FEBS Lett. 2000;480(1):2–16. doi: 10.1016/s0014-5793(00)01771-3. [DOI] [PubMed] [Google Scholar]
- 46.Hanash SM, et al. Integrating cancer genomics and proteomics in the post-genome era. Proteomics. 2002;2(1):69–75. [PubMed] [Google Scholar]
- 47.Ideker T, et al. Integrated genomic and proteomic analyses of a systematically perturbed metabolic network. Science. 2001;292(5518):929–934. doi: 10.1126/science.292.5518.929. [DOI] [PubMed] [Google Scholar]
- 48.Shields JD, et al. Autologous chemotaxis as a mechanism of tumor cell homing to lymphatics via interstitial flow and autocrine CCR7 signaling. Cancer Cell. 2007;11(6):526–538. doi: 10.1016/j.ccr.2007.04.020. [DOI] [PubMed] [Google Scholar]
- 49.Polacheck WJ, Charest JL, Kamm RD. Interstitial flow influences direction of tumor cell migration through competing mechanisms. Proc Natl Acad Sci USA. 2011;108(27):11115–11120. doi: 10.1073/pnas.1103581108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chang SF, et al. Tumor cell cycle arrest induced by shear stress: Roles of integrins and Smad. Proc Natl Acad Sci USA. 2008;105(10):3927–3932. doi: 10.1073/pnas.0712353105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Petersen OW, Rønnov-Jessen L, Howlett AR, Bissell MJ. Interaction with basement membrane serves to rapidly distinguish growth and differentiation pattern of normal and malignant human breast epithelial cells. Proc Natl Acad Sci USA. 1992;89(19):9064–9068. doi: 10.1073/pnas.89.19.9064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bissell MJ, et al. 1999. Tissue structure, nuclear organization, and gene expression in normal and malignant breast. Cancer Res 59(7 Suppl):1757s–1763s; discussion 1763s–1764s.
- 53.Muthuswamy SK, Li D, Lelievre S, Bissell MJ, Brugge JS. ErbB2, but not ErbB1, reinitiates proliferation and induces luminal repopulation in epithelial acini. Nat Cell Biol. 2001;3(9):785–792. doi: 10.1038/ncb0901-785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kenny PA, et al. The morphologies of breast cancer cell lines in three-dimensional assays correlate with their profiles of gene expression. Mol Oncol. 2007;1(1):84–96. doi: 10.1016/j.molonc.2007.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lee GY, Kenny PA, Lee EH, Bissell MJ. Three-dimensional culture models of normal and malignant breast epithelial cells. Nat Methods. 2007;4(4):359–365. doi: 10.1038/nmeth1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Debnath J, et al. The role of apoptosis in creating and maintaining luminal space within normal and oncogene-expressing mammary acini. Cell. 2002;111(1):29–40. doi: 10.1016/s0092-8674(02)01001-2. [DOI] [PubMed] [Google Scholar]
- 57.Debnath J, Muthuswamy SK, Brugge JS. Morphogenesis and oncogenesis of MCF-10A mammary epithelial acini grown in three-dimensional basement membrane cultures. Methods. 2003;30(3):256–268. doi: 10.1016/s1046-2023(03)00032-x. [DOI] [PubMed] [Google Scholar]
- 58.Xu F, et al. A three-dimensional in vitro ovarian cancer coculture model using a high-throughput cell patterning platform. Biotechnol J. 2011;6(2):204–212. doi: 10.1002/biot.201000340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Celli JP, Rizvi I, Evans CL, Abu-Yousif AO, Hasan T. Quantitative imaging reveals heterogeneous growth dynamics and treatment-dependent residual tumor distributions in a three-dimensional ovarian cancer model. J Biomed Opt. 2010;15(5):051603. doi: 10.1117/1.3483903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Rizvi I, et al. Synergistic enhancement of carboplatin efficacy with photodynamic therapy in a three-dimensional model for micrometastatic ovarian cancer. Cancer Res. 2010;70(22):9319–9328. doi: 10.1158/0008-5472.CAN-10-1783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Rahmanzadeh R, et al. Ki-67 as a molecular target for therapy in an in vitro three-dimensional model for ovarian cancer. Cancer Res. 2010;70(22):9234–9242. doi: 10.1158/0008-5472.CAN-10-1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Abu-Yousif AO, Rizvi I, Evans CL, Celli JP, Hasan T. PuraMatrix encapsulation of cancer cells. J Vis Exp. 2009;(34):e1692. doi: 10.3791/1692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kenny HA, Lengyel E. MMP-2 functions as an early response protein in ovarian cancer metastasis. Cell Cycle. 2009;8(5):683–688. doi: 10.4161/cc.8.5.7703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kenny HA, Krausz T, Yamada SD, Lengyel E. Use of a novel 3D culture model to elucidate the role of mesothelial cells, fibroblasts and extra-cellular matrices on adhesion and invasion of ovarian cancer cells to the omentum. Int J Cancer. 2007;121(7):1463–1472. doi: 10.1002/ijc.22874. [DOI] [PubMed] [Google Scholar]
- 65.Ohmori T, Yang JL, Price JO, Arteaga CL. Blockade of tumor cell transforming growth factor-betas enhances cell cycle progression and sensitizes human breast carcinoma cells to cytotoxic chemotherapy. Exp Cell Res. 1998;245(2):350–359. doi: 10.1006/excr.1998.4261. [DOI] [PubMed] [Google Scholar]
- 66.Croonen A, van der Valk P, Herman C, Lindeman J. 1988. Cytology, immunopathology and flow cytometry in the diagnosis of pleural and peritoneal effusions. Lab Invest 58(6):725–732. [PubMed]
- 67.Chambers AF, Groom AC, MacDonald IC. Dissemination and growth of cancer cells in metastatic sites. Nat Rev Cancer. 2002;2(8):563–572. doi: 10.1038/nrc865. [DOI] [PubMed] [Google Scholar]
- 68.Hanahan D, Weinberg RA. Hallmarks of cancer: The next generation. Cell. 2011;144(5):646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 69.Shin MK, Kim SK, Jung H. Integration of intra- and extravasation in one cell-based microfluidic chip for the study of cancer metastasis. Lab Chip. 2011;11(22):3880–3887. doi: 10.1039/c1lc20671k. [DOI] [PubMed] [Google Scholar]
- 70.Chaw KC, Manimaran M, Tay EH, Swaminathan S. Multi-step microfluidic device for studying cancer metastasis. Lab Chip. 2007;7(8):1041–1047. doi: 10.1039/b707399m. [DOI] [PubMed] [Google Scholar]
- 71.Runnels JM, Chen N, Ortel B, Kato D, Hasan T. BPD-MA-mediated photosensitization in vitro and in vivo: Cellular adhesion and beta1 integrin expression in ovarian cancer cells. Br J Cancer. 1999;80(7):946–953. doi: 10.1038/sj.bjc.6690448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Buczek-Thomas JA, Chen N, Hasan T. Integrin-mediated adhesion and signalling in ovarian cancer cells. Cell Signal. 1998;10(1):55–63. doi: 10.1016/s0898-6568(97)00074-0. [DOI] [PubMed] [Google Scholar]
- 73.Burleson KM, et al. Ovarian carcinoma ascites spheroids adhere to extracellular matrix components and mesothelial cell monolayers. Gynecol Oncol. 2004;93(1):170–181. doi: 10.1016/j.ygyno.2003.12.034. [DOI] [PubMed] [Google Scholar]
- 74.Burleson KM, Boente MP, Pambuccian SE, Skubitz AP. Disaggregation and invasion of ovarian carcinoma ascites spheroids. J Transl Med. 2006;4:6. doi: 10.1186/1479-5876-4-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Schmeichel KL, Bissell MJ. Modeling tissue-specific signaling and organ function in three dimensions. J Cell Sci. 2003;116(Pt 12):2377–2388. doi: 10.1242/jcs.00503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Nelson CM, Bissell MJ. Of extracellular matrix, scaffolds, and signaling: Tissue architecture regulates development, homeostasis, and cancer. Annu Rev Cell Dev Biol. 2006;22:287–309. doi: 10.1146/annurev.cellbio.22.010305.104315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Ahmed N, et al. Molecular pathways regulating EGF-induced epithelio-mesenchymal transition in human ovarian surface epithelium. Am J Physiol Cell Physiol. 2006;290(6):C1532–C1542. doi: 10.1152/ajpcell.00478.2005. [DOI] [PubMed] [Google Scholar]
- 78.Cheng J-C, Auersperg N, Leung PC. EGF-induced EMT and invasiveness in serous borderline ovarian tumor cells: A possible step in the transition to low-grade serous carcinoma cells? PLoS ONE. 2012;7(3):e34071. doi: 10.1371/journal.pone.0034071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Holz C, et al. 2011. Epithelial-mesenchymal-transition induced by EGFR activation interferes with cell migration and response to irradiation and cetuximab in head and neck cancer cells. Radiother Oncol 101(1):158–164.
- 80.Seiden MV, et al. A phase II trial of EMD72000 (matuzumab), a humanized anti-EGFR monoclonal antibody, in patients with platinum-resistant ovarian and primary peritoneal malignancies. Gynecol Oncol. 2007;104(3):727–731. doi: 10.1016/j.ygyno.2006.10.019. [DOI] [PubMed] [Google Scholar]
- 81.Elie C, et al. GINECO Group Lack of relationship between EGFR-1 immunohistochemical expression and prognosis in a multicentre clinical trial of 93 patients with advanced primary ovarian epithelial cancer (GINECO group) Br J Cancer. 2004;91(3):470–475. doi: 10.1038/sj.bjc.6601961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Posadas EM, et al. A phase II study of ixabepilone (BMS-247550) in metastatic renal-cell carcinoma. Cancer Biol Ther. 2007;6(4):490–493. doi: 10.4161/cbt.6.4.3831. [DOI] [PubMed] [Google Scholar]
- 83.Gui T, Shen K. 2012. The epidermal growth factor receptor as a therapeutic target in epithelial ovarian cancer. Cancer Epidemiol 36(5):490–496.
- 84.Galaz S, et al. Loss of E-cadherin mediated cell-cell adhesion as an early trigger of apoptosis induced by photodynamic treatment. J Cell Physiol. 2005;205(1):86–96. doi: 10.1002/jcp.20374. [DOI] [PubMed] [Google Scholar]
- 85.Sawada K, Ohyagi-Hara C, Kimura T, Morishige K. Integrin inhibitors as a therapeutic agent for ovarian cancer. J Oncol. 2012;2012:915140. doi: 10.1155/2012/915140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Desgrosellier JS, Cheresh DA. Integrins in cancer: Biological implications and therapeutic opportunities. Nat Rev Cancer. 2010;10(1):9–22. doi: 10.1038/nrc2748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Cox D, Brennan M, Moran N. Integrins as therapeutic targets: lessons and opportunities. Nat Rev Drug Discov. 2010;9(10):804–820. doi: 10.1038/nrd3266. [DOI] [PubMed] [Google Scholar]
- 88.Meirelles K, et al. Human ovarian cancer stem/progenitor cells are stimulated by doxorubicin but inhibited by Mullerian inhibiting substance. Proc Natl Acad Sci USA. 2012;109(7):2358–2363. doi: 10.1073/pnas.1120733109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Celli JP, et al. Imaging and photodynamic therapy: Mechanisms, monitoring, and optimization. Chem Rev. 2010;110(5):2795–2838. doi: 10.1021/cr900300p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Hendren SK, et al. 2001. Phase II trial of debulking surgery and photodynamic therapy for disseminated intraperitoneal tumors. Ann Surg Oncol 8(1English):65–71.
- 91.Cengel KA, Glatstein E, Hahn SM. Intraperitoneal photodynamic therapy. Cancer Treat Res. 2007;134:493–514. doi: 10.1007/978-0-387-48993-3_34. [DOI] [PubMed] [Google Scholar]
- 92.Zuluaga MF, Lange N. Combination of photodynamic therapy with anti-cancer agents. Curr Med Chem. 2008;15(17):1655–1673. doi: 10.2174/092986708784872401. [DOI] [PubMed] [Google Scholar]
- 93.Tasoglu S, Gurkan UA, Wang S, Demirci U. Manipulating biological agents and cells in micro-scale volumes for applications in medicine. Chem Soc Rev. 2013 doi: 10.1039/c3cs60042d. 10.1039/C3CS60042D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Moon S, et al. Enumeration of CD4+ T-cells using a portable microchip count platform in Tanzanian HIV-infected patients. PLoS One. 2011;6(7):e21409. doi: 10.1371/journal.pone.0021409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Louie KG, et al. Adriamycin accumulation and metabolism in adriamycin-sensitive and -resistant human ovarian cancer cell lines. Biochem Pharmacol. 1986;35(3):467–472. doi: 10.1016/0006-2952(86)90221-2. [DOI] [PubMed] [Google Scholar]
- 96.Tamimi A, Rinker EB, Sandall OC. Diffusion-coefficients for hydrogen-sulfide, carbon-dioxide, and nitrous-oxide in water over the temperature-range 293-368-K. J Chem Eng Data. 1994;39(2):330–332. [Google Scholar]
- 97.Cross SS. Fractals in pathology. J Pathol. 1997;182(1):1–8. doi: 10.1002/(SICI)1096-9896(199705)182:1<1::AID-PATH808>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
