

Abstract
In many bacteria the flavoenzyme thymidylate synthase ThyX produces the DNA nucleotide deoxythymidine monophosphate from dUMP, using methylenetetrahydrofolate as carbon donor and NADPH as hydride donor. Because all three substrates bind in close proximity to the catalytic flavin adenine dinucleotide group, substantial flexibility of the ThyX active site has been hypothesized. Using femtosecond time-resolved fluorescence spectroscopy, we have studied the conformational heterogeneity and the conformational interconversion dynamics in real time in ThyX from the hyperthermophilic bacterium Thermotoga maritima. The dynamics of electron transfer to excited flavin adenine dinucleotide from a neighboring tyrosine residue are used as a sensitive probe of the functional dynamics of the active site. The fluorescence decay spanned a full three orders of magnitude, demonstrating a very wide range of conformations. In particular, at physiological temperatures, multiple angstrom cofactor-residue displacements occur on the picoseconds timescale. These experimental findings are supported by molecular dynamics simulations. Binding of the dUMP substrate abolishes this flexibility and stabilizes the active site in a configuration where dUMP closely interacts with the flavin cofactor and very efficiently quenches fluorescence itself. Our results indicate a dynamic selected-fit mechanism where binding of the first substrate dUMP at high temperature stabilizes the enzyme in a configuration favorable for interaction with the second substrate NADPH, and more generally have important implications for the role of active site flexibility in enzymes interacting with multiple poly-atom substrates and products. Moreover, our data provide the basis for exploring the effect of inhibitor molecules on the active site dynamics of ThyX and other multisubstrate flavoenzymes.
Keywords: protein dynamics, flavoprotein, ultrafast fluorescence spectroscopy, quenching
Configurational flexibility is essential for enzyme function during catalysis. Binding of one or more substrates, accommodation of the transition state where the actual reaction takes place, relaxation to the product state, and release of the product(s) is possible because different configurations of the enzyme are continuously sampled, by thermal or reaction-driven motions, on timescales ranging from femtoseconds to microseconds. The configurational space sampled in a certain time range will depend on the local protein flexibility/energy landscape and the temperature (1). During these configurational changes, distances between constituents of the enzyme complex change. It has been recognized that the fastest (femtosecond to picosecond) localized motions exist alongside the slower motions that occur on the (typically millisecond) timescale of catalysis (2–4).
Various experimental techniques allow monitoring changes in interactions resulting from configurational changes. Long-range micro/millisecond domain motions in flexible proteins have been studied by NMR (5) and FRET techniques (6, 7). Molecular dynamics simulations and NMR experiments indicate that more localized structural fluctuations in these flexible regions occur on the picoseconds-to-nanoseconds timescale (6, 8). These fluctuations can only be directly investigated using very high time-resolution optical techniques. In particular, the fluorescence properties of intrinsic or external fluorophores can be exquisitely sensitive to the protein environment. In flavoproteins, interaction of the flavin cofactor with the protein environment has been shown to diminish the lifetime of the flavin fluorescence from the intrinsic nanosecond timescale to the femtoseconds-to-picoseconds timescale, due to quenching by photooxidation of nearby aromatic residues (3, 9–15). In the present study, we use ultrafast fluorescence spectroscopy to investigate conformational flexibility of the flavoenzyme thymidylate synthase ThyX.
ThyX is a homotetrameric enzyme discovered a decade ago (16), which is essential for de novo synthesis of the DNA precursor 2′-deoxythymidine-5′-monophosphate (dTMP) in a large number of bacterial systems. ThyX shows no structural homology to thymidylate synthase ThyA, which is used in most eukaryotes (17). Because the ThyX pathway is used by a number of pathogenic bacteria and absent in humans, ThyX is considered a promising antimicrobial target (16, 18); it catalyses carbon transfer from N5,N10-methylene-5,6,7,8-tetrahydrofolate (MTHF or CH2H4folate) to deoxyuridine monophosphate (dUMP) using NADPH as a hydride donor and consequently has three substrates (dUMP, MTHF, NADPH) with the flavin adenine dinucleotide cofactor shuttling between the fully oxidized (FAD) and fully reduced (FADH2) forms. dUMP binds in close interaction with the flavin group, displacing a nearby Tyr residue (19). A very recent study shows that folate derivatives may bind to the opposite side of the flavin cofactor with respect to dU(20). The binding site of NADPH has not been determined by structural studies, but inhibition studies indicate that folate and NADPH binding sites may partially coincide (21). Furthermore, reduction of flavin by NADPH appears gated by the presence of dUMP (21), further pointing at substrate-induced, functional structural rearrangements. Steady-state crystallographic studies of this enzyme also indicate substantial flexibility of the active site: in the substrate-free structure the flavin group and its close environment appear disordered (19); for the folate-bound form, multiple flavin configurations have been suggested (20).
Using a newly developed, ultrafast fluorescence spectrometer with full spectral resolution, we performed studies on the dynamics of FAD fluorescence in wild-type and genetically modified ThyX enzymes from several bacterial species. In the present work we focus on the enzyme from the hyperthermophilic bacterium Thermotoga maritima that allows studies over a wide temperature range. We identified a close-lying tyrosine as well as the substrate dUMP itself as fluorescence quenchers. The observed fluorescence decay revealed the presence of a wide range of conformations, whose interconversion accelerates at the physiological temperature of this hyperthermophilic enzyme. Binding of dUMP was found to stabilize the active site in a configuration allowing close interaction between dUMP and FAD and favorable for interaction with NADPH. Our data have important implications for the role of active site flexibility in multisubstrate enzymes and ultimately permit exploring the effect of inhibitor molecules on the active site dynamics.
Results
The absorption spectrum of ThyX from T. maritima (TmThyX; Fig. S1A) is very similar to that published previously (22), and the fluorescence spectrum (Fig. S1C) typical for oxidized flavins. The enzyme contains four FAD binding sites (closest center-to-center distance 27 Å). Because in principle, resonance energy transfer between identical molecules within the protein (homo-FRET) can take place, potentially complicating the analysis, we measured fluorescence anisotropy. The anisotropy was close to the theoretical maximum of 0.4, implying that no homo-FRET occurs. This finding is consistent with an estimation of energy transfer times of >3 ns using the Förster formalism and the observation (see below) that quenching takes place on faster timescales.
Transient fluorescence spectra and kinetics at the emission maximum are shown in Fig. 1; kinetics at different wavelengths are shown in Fig. S2, and they show a highly multiphasic fluorescence decay, spanning timescales from ∼1 ps to ∼1 ns. However, all decay occurs substantially faster than the intrinsic nanosecond (∼3 ns) decay of FAD (23). The most probable origin of this quenching is electron transfer (ET) from close-by aromatic residues to the excited flavin cofactor. According to the crystal structure of TmThyX (19), Tyr-91, a widely conserved residue (24), is the closest aromatic residue to the FAD cofactor, and therefore likely to act as main fluorescence quencher. To test this hypothesis, we investigated the Y91F mutant protein, designed to prevent electron transfer while maximally preserving the structural environment of the flavin.
Fig. 1.

(A) Transient fluorescence spectra of WT TmThyX measured at 20 °C. The feature at ∼460 nm in the 0-ps spectrum is due to Raman scattering of water. (B) Kinetics at 520 nm of WT and Y91F ThyX. The time axis is linear until 10 ps and logarithmic thereafter. Solid lines are fits with a four-parameter exponential decay; the dashed line with power law.
The overall fluorescence decay kinetics of the mutant ThyX protein are much slower than those of WT (Fig. 1B; note the logarithmic scaling after 10 ps), implying that ET from Tyr-91 is indeed the dominant quenching process. However, in both cases, a fast decay in the order of ∼1 ps is present (see below), which we assign to relaxation processes in the excited state. Furthermore, the decay of the mutant protein still takes place faster than that of FAD in solution, indicating that aromatic residues other than Tyr-91 also contribute to quenching. The shape of the fluorescence spectra was found to be constant after ∼200 ps and to be significantly different from that of FAD in solution (Fig. S3A). This observation further confirms that free FAD does not contribute to the signal.
Analysis of the full data set in terms of multiexponential decay of both the WT and the Y91F mutant protein requires at least four decay rates (Fig. S3 A and B). This analysis indicates that red shifts occur concomitant with fluorescence decay on the timescale <200 ps, and presumably reflect charge relaxation processes of the flavin environment (25). The nonexponentiality of the decay can be assigned to a distribution of conformations of the donor–acceptor pairs that do not or only partly interconvert during the excited state lifetime. The fact that the nonexponentiality is also found in the mutant protein implies that this distribution is not uniquely due to heterogeneity specifically of the Tyr-91 conformation.
Temperature Studies.
To obtain detailed insight into the conformational flexibility of the active site, we measured the fluorescence properties of the enzyme as a function of temperature. Here, we take advantage of the fact that the ThyX enzyme from the hyperthermophile T. maritima allows studies over a wide range of temperatures, up to 70 °C, into the physiological range of the enzyme (at higher temperatures the isolated enzyme starts to precipitate). Fig. S1C shows that the total fluorescence decreases with increasing temperature, and that this decrease is fully reversible upon cooling. The fluorescence decrease rather than increase at higher temperatures indicates that FAD is not released at high temperature (up to 70 °C), as has been observed in flavodoxin (26).
As shown in Fig. 2, the decrease of the total fluorescence is the result of the acceleration of the fluorescence decay kinetics. In a multiexponential global analysis, at all temperatures at least four components (eight fit parameters in total) are required to satisfactorily fit the data. It was possible to describe the data at all temperatures with the same set of four rate constants (Fig. S3 C and D). In this case, the amplitudes of the two longest decay components decreased and those of the shortest decay components increased with temperature. Qualitatively, the result of this analysis implies that with increasing temperature the distribution of fluorescence decays shifts toward the shorter-lived side, without the faster decays themselves becoming faster.
Fig. 2.

Temperature dependence of peak-normalized fluorescence decay of WT TmThyX at 520 nm. The time axis is linear until 10 ps and logarithmic thereafter.
The high number of exponentials (and fit parameters) required to fit the data suggests that a continuous distribution of rates, rather than four distinct rates, may provide a more adequate and simpler description. To explore this possibility, we attempted to fit the integrated fluorescence decay with a power law. For all temperatures, a satisfactory fit was obtained (at t >2 ps) using Eq. S1, which contains only three fit parameters: the amplitude I0, mean lifetime τ0, and distribution width parameters q. The corresponding lifetime distributions are shown in Fig. 3C. The average lifetime τ0 (Fig. 3A) is seen to gradually decrease with temperature. Overall, the relative width of the distribution (Fig. 3B) decreases at higher temperatures and in particular at 70 °C, which is in the physiological temperature range for T. maritima. Through its influence on the electronic coupling, the ET rate k critically depends on the edge-to-edge distance r between the flavin cofactor and the quenching residue as 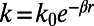 (3, 27). Assuming ET between Tyr and excited FAD is near barrierless (3, 11), and β = 1.36 Å−1 (28), we can convert the lifetime distribution (Fig. 3C) to a distribution of distances from which ET takes place (Fig. 3D). In view of uncertainty in
(3, 27). Assuming ET between Tyr and excited FAD is near barrierless (3, 11), and β = 1.36 Å−1 (28), we can convert the lifetime distribution (Fig. 3C) to a distribution of distances from which ET takes place (Fig. 3D). In view of uncertainty in  , we show the distribution as relative to a mean distance R0. At 20 °C the distribution is very large (FWHM >2 Å), implying strong conformational heterogeneity. We note that this distribution should be considered qualitatively and does not necessarily directly correspond to the static distribution of flavin–quencher pairs, because conformational flexibility within the fluorescence lifetime may bias the distribution toward shorter distances. This point is illustrated in Fig. 3D. At higher temperatures, the distance distribution shifts to shorter distances, and becomes narrower—in particular, close to the physiological temperature range (70 °C). Because warming should enhance the configurational distribution, the picture that emerges is that (i) at all temperatures a very large distribution of configurations is populated, and that (ii) at higher temperatures the interconversion between these is accelerated so that the configurations that give rise to the fastest quenching rates are more easily reached within the timescale of fluorescence decay.
, we show the distribution as relative to a mean distance R0. At 20 °C the distribution is very large (FWHM >2 Å), implying strong conformational heterogeneity. We note that this distribution should be considered qualitatively and does not necessarily directly correspond to the static distribution of flavin–quencher pairs, because conformational flexibility within the fluorescence lifetime may bias the distribution toward shorter distances. This point is illustrated in Fig. 3D. At higher temperatures, the distance distribution shifts to shorter distances, and becomes narrower—in particular, close to the physiological temperature range (70 °C). Because warming should enhance the configurational distribution, the picture that emerges is that (i) at all temperatures a very large distribution of configurations is populated, and that (ii) at higher temperatures the interconversion between these is accelerated so that the configurations that give rise to the fastest quenching rates are more easily reached within the timescale of fluorescence decay.
Fig. 3.

Analysis of the temperature dependence of the fluorescence kinetics of WT TmThyX using Eq. S1. (A) Mean value τ0 of the lifetime distribution. (B) Heterogeneity factor q of the lifetime distribution. The solid lines are guides for the eye. (C) Lifetimes distribution. (D) Distribution of edge-to-edge distances between FAD and electron donors normalized to the total integral.
dUMP Binding.
In the absence of the two other substrates, dUMP binds close to oxidized FAD through aromatic stacking against its isoalloxazine ring system (19). Fig. 4 shows that this binding leads to dramatic quenching of FAD fluorescence: the dominant decay phase occurs with a time constant of ∼200 fs in this case, and the slower decay phases are strongly, although not completely, suppressed (∼90% decay within the first few picoseconds).
Fig. 4.

Effect of dUMP binding on fluorescence decay of WT and Y91F TmThyX at 520 nm. The time axis is linear until 4 ps and logarithmic thereafter. A global analysis in terms of DAS of WT TmThyX fluorescence spectra in the presence of dUMP is shown in Fig. S3F.
The origin of this quenching may be either a direct interaction between dUMP and the FAD cofactor, or a change in the interaction of FAD and the quenching residues, in particular Tyr-91. Indeed, the available TmThyX crystal structures suggest that upon dUMP binding, Tyr-91 moves toward the FAD cofactor (19). To discriminate between these two possibilities, we investigated the Y91F mutant enzyme, which binds dUMP with comparable affinity and shows a similar perturbation of the absorption spectrum (Fig. S1B) as WT (see below). We observed (Fig. 4) that the effect of dUMP binding on the fluorescence decay is very similar for WT and Y91F TmThyX, which led us to conclude that it is dUMP itself that acts as quencher of flavin fluorescence. The minor (∼10%), slower decay phases in the presence of dUMP (Fig. S3E) may reflect partial and/or heterogeneous dUMP binding.
The strong quenching of FAD binding by dUMP can be used as a sensitive probe to determine the affinity of TmThyX for dUMP. In agreement with the time-resolved data of Fig. 4, the overall fluorescence was found to decrease ∼10-fold upon dUMP binding to WT TmThyX. Analysis of the dUMP titration to 5-µM FAD binding sites yielded a Kd of 400 nM at 60 °C and in the presence of 250 mM NaCl (Fig. S4). The apparent binding site concentration deduced from the binding curve was very close to the FAD concentration, implying that flavin-devoid sites, which are also present in ThyX in solution (SI Materials and Methods), do not bind dUMP in the same affinity range. This finding strongly suggests that stacking with FAD is the major binding determinant of dUMP. A similar affinity was found using the change in the flavin absorption spectrum (Fig. S1A) as a measure for dUMP binding, although the high dynamic range of the fluorescence measurements allows a more precise determination. Detailed binding studies will be presented elsewhere.
We also used FAD fluorescence to investigate whether dUMP binding influences the thermal stability of the complex. As mentioned previously, in the absence of dUMP up to 70 °C FAD does not dissociate from the protein. Above 70 °C, the FAD fluorescence rises and redshifts, and at 90 °C an increase corresponding to ∼4% free FAD is observed (Fig. S5). When dUMP is bound, this increase amounts to less than 0.5%, and no spectral shift is observed (Fig. S5). Although protein precipitation above 90 °C prohibited the determination of full dissociation curves of this hyperthermophilic enzyme, these data indicate that dUMP binding shifts denaturation to higher temperatures and thus thermally stabilizes the complex.
Molecular Dynamics Simulations.
To investigate the molecular parameters underlying the experimentally observed conformational flexibility, we performed molecular dynamics (MD) simulations of WT TmThyX in the presence and absence of dUMP and at two temperatures (27 °C and 70 °C). The models that included all four subunits were based on the X-ray structure of dUMP-bound TmThyX that was obtained at −173 °C (19). Fig. S6 shows that after warming and equilibration of the model, the rmsd of the backbone atoms of the model from the X-ray structure remained roughly constant and ∼1 Å for the 2.5-ns free dynamics trajectories. In the absence of dUMP, the deviation is significantly larger for the trajectories at 70 °C than at 27 °C, indicating a larger conformational heterogeneity. Interestingly, such a difference is not observed in the absence of dUMP, indicating that this substrate effectively rigidifies the protein.
We now focus on the vicinity of the FAD. Fig. 5 compares time-averaged simulated structures of the four active sites; a corresponding full tetramer is shown in Fig. S7. Though the structures in the presence of dUMP are similar to the X-ray structure (19), in the absence of dUMP the flavin is rotated in all subunits by ∼50° with respect to the X-ray structure and shows substantial orientational freedom (Fig. 5B; Fig. S8). Similar observations were made with models based directly on the dUMP-devoid structure. Interestingly, these rotated conformations are similar to those observed recently in structural studies in some of the active sites of mutant TmThyX (20). As in the crystal structures of the TmThyX WT proteins (19), in all subunits, the presence of dUMP results in a shorter FAD–Tyr-91 distance.
Fig. 5.

Superimposed structures of elements of the four active sites, time averaged over the last 500 ps of the free trajectories of MD simulations of ThyX in the presence (A) and absence (B) of dUMP. The superposition is on the entire backbone of each subunit. The His53 residues associated with the active sites belong to different subunits as the other elements. The carbon atoms are color-coded with the respective subunits.
Fig. 6 shows the fluctuations of the FAD–Tyr-91 distance during the free dynamics in each of the four subunits. The shortest edge-to-edge distance is thought to be relevant for the electronic coupling determining the electron transfer rate. Therefore, at each instant, this distance is plotted rather than the distance between two fixed atoms. In the crystal structure the shortest distance is between the C6 atom of FAD and the Cε1 atom of Tyr-91. In our simulations, other atom pairs, such as FAD N5-Tyr-91 Cδ1, also occur as shortest distance. The active site structures of Fig. 5 indicate that the distance fluctuations in the dUMP-devoid model are rather the result of fluctuations of the flavin cofactor than of Tyr-91.
Fig. 6.

Dynamics (Left) and distribution histograms (Right) of the distance between FAD and Tyr-91 (shortest distance between an atom on the isoalloxazine ring and the tyrosine aromatic ring) in the four subunits during the 2.5-ns free dynamics.
In the dUMP-devoid model, the FAD–Tyr-91 distance fluctuates substantially in all four subunits, at 27 °C from ∼3 to ∼8 Å. Larger fluctuations of 2 Å or more appear infrequently (intervals of several nanoseconds) on the simulation timescale, and the statistics, especially at 27 °C, are clearly undersampled, indicating that substantial conformational changes also take place on a longer timescale. These observations are in agreement with our analysis of the broad range of fluorescence decay times that indicate that FAD–Tyr-91 distance changes take place on the timescale of ∼1 ns or more. At 70 °C the fluctuations become substantially more frequent, both the smaller, high-frequency and the larger, low-frequency fluctuations.
Interestingly, our simulations also indicate that the presence of dUMP drastically reduces the distance fluctuations for both temperatures. This finding implies that substrate binding leads to a rigidification of the active site, and in particular of the FAD cofactor positioning, and is generally consistent with the better resolution of the active site in the X-ray crystallography studies at cryogenic temperatures (19) and our finding of dUMP-induced thermal stabilization of the FAD–protein complex (Fig. S5). Similar analyses for other residues involved in substrate binding and catalysis also indicate substantial distance variation specifically in the absence of dUMP. For instance Arg-147, Arg-174, and Ser-88, which all H-bond to dUMP, were all found to be able to intermittently H-bond to FAD in the absence of dUMP.
Discussion
In this work, the quenching of flavin fluorescence in an enzyme involved in DNA synthesis, associated with MD simulations, was used to probe the dynamic properties of the active site. Experimentally, we exploited the fact that photoexcited FAD (FAD*) can accept an electron from the aromatic residues Tyr and Trp on the femtosecond-to-nanosecond timescale, and showed that in the case of TmThyX from, Tyr-91 is the principal electron donor. It has been argued that the FAD*Tyr→FAD−Tyr°+ reaction occurs with a driving force in the range of 0.7–1 eV and in a near-activationless regime (3, 11). Indeed, the rates of this electron transfer reaction were found to be temperature independent in flavoproteins with a much less heterogeneous fluorescence decay distribution than TmThyX (11, 13). Moreover, in TmThyX, increase in temperature leads to a shift of slow-decaying populations to fast-decaying populations rather than an increase of the rates itself. For these reasons we interpret the temperature dependence observed in this work as originating from changes in the distance-dependent electronic coupling between the reactants, rather than from activation barriers. We note that temperature-dependent fluorescence decay times have been reported for the flavoprotein lipoamide dehydrogenase (29). The FAD cofactors in this protein have an extremely high fluorescence quantum yield and are not quenched; here, the activation of the fluorescence decay was due to distinct conformational changes that are static on the nanosecond timescale (29). The analysis in the present work by contrast highlights dynamic conformational changes occurring on the picosecond timescale.
Our analysis is based on the widely used (3, 11, 14) notion that excited-state quenching in flavoproteins is due to electron transfer from aromatic residues. The validity of this notion has been demonstrated for tryptophan quenchers (9, 30) and strongly suggested for tyrosine quenchers (31, 32). In both cases, the radical pair can be subsequently stabilized by proton transfer from the residue. Simultaneous proton and electron transfer, avoiding high-energy charge-separation intermediates, has been observed for phenol oxidation reactions in aqueous solutions (33, 34). Though we cannot fully exclude that such simultaneous processes (and hence motions between Tyr-91 and potential proton acceptors) could play a role in FAD* quenching in ThyX, we consider it unlikely in view of the evidence from other flavoproteins and the fact that the high-driving force generated by FAD* formation (see above) energetically allows formation of the FAD−Tyr°+ intermediate by electron transfer only.
We observed that not only aromatic residues, but also the substrate dUMP quenches FAD fluorescence, strongly suggesting that bound dUMP also donates an electron to FAD*. dUMP indeed binds very close to FAD, and the isoalloxazine and uracil rings are actually in, or close to, van der Waals contact (distance ∼3.5 Å in the crystal structure). The relevant redox properties of dUMP have not been determined, but the light-induced oxidation of uracil in the presence of a photoreducing material has been reported (35), suggesting that the reaction is energetically possible. We will further investigate this hypothesis by transient absorption spectroscopy of the photoproducts formed in the ThyX–dUMP complex.
The FAD* decay dynamics of ThyX are strikingly multiphasic, spanning three orders of magnitude in time. We assign the large and continuous lifetime distribution in ThyX fluorescence to conformational heterogeneity in the active site; this assignment is strongly supported by the MD simulation that shows large variations of active site configurations. In itself, nonmonoexponential decay of the excited state of quenched flavin cofactors in flavoproteins is frequent (15). Biexponential decay has been documented spanning shorter time spans (14, 36, 37), and in flavodoxin multiexponentiality has been associated predominantly with partial flavin dissociation from the protein (26). In blue light-sensing using FAD (BLUF) photosensor proteins (13, 31, 32) and in flavin reductase (3), broader and more heterogeneous lifetime distributions have been interpreted in terms of ground-state heterogeneity. The lifetime distribution in TmThyX is considerably larger than in the abovementioned flavoproteins, and the associated donor–acceptor distance distribution is substantially wider (∼2 Å FWHM at 20 °C; Fig. 3D) than the ∼1 Å FWHM distribution in Yang et al. (3).
Our results show that a very broad range of configurations is accessible for TmThyX in the absence of substrate, and that these do not fully interconvert on the fluorescence timescale and at subcatalytic temperatures. These findings can be considered consistent with an early crystallographic study of substrate-free TmThyX, where fractions of the protein, and in particular the flavin ring, appear disordered, in contrast to the dUMP-containing protein (19), and appear general for ThyX enzymes (38, 39). More specifically for TmThyX, our MD simulations indicate the presence of substantial variation in the distance between FAD and the Tyr-91 quencher, with large changes (up to ∼4 Å) occurring infrequently on the timescale of up to several nanoseconds. This flexibility is no longer apparent in the presence of dUMP. Single-molecule studies of flavin reductase have demonstrated that interconversion of the lifetime-distinguishable configurations for a substantial part takes place on the timescale of milliseconds and beyond (3). Our temperature dependence studies of ThyX show that the distribution does not broaden at higher temperature, as could be expected from pure increase of accessible configurational space, but narrows and shifts to shorter effective distances. We assign this finding to acceleration of the interconversion rates of configurations on the same timescale as fluorescence decay. This interpretation is supported by our MD simulations that indicate that substantial fluctuations in donor–acceptor distance do occur on the picosecond-to-nanosecond timescale, and that these, and in particular the less-frequent large amplitude fluctuations, occur substantially more frequent at higher, physiologically relevant, temperatures.
We suggest that the flexibility in the active site of this enzyme, as reflected in the picosecond–nanosecond multiangstrom conformational fluctuations, is related to its capacity of binding multiple different aromatic substrates, of which at least two (NADPH/dUMP and MTHF/dUMP) simultaneously. The picture that emerges is that the extremely rapid conformational sampling of the protein allows dUMP to enter the active site and be accommodated close to the flavin. The binding of dUMP then stabilizes the flavin cofactor in the active site, as indicated by our MD simulations, presumably in a configuration favorable for NADPH binding and subsequent flavin reduction. This mechanism is in agreement with the reported drastic acceleration of flavin reduction by NADPH in the presence of dUMP (21). We stress that our observations have further functional consequences. Indeed, low stereospecificity and a very high Km for NADPH have been observed for the TmThyX protein at 37 °C (40, 41). Moreover, this protein turns over substantially faster at 65 °C (within the physiological range) than at 37 °C (41, 42).
Our results indicate that a selected fit-type mechanism applies, where the substrate binding does not necessarily change the enzyme configuration, but rather stabilizes a favorable configuration among many sampled. The accelerated and more extensive sampling at higher temperature does not substantially influence the equilibrium binding of the substrate, but may allow faster exchange with the solvent of substrate and product.
In conclusion, we exploited ultrafast time-resolved FAD fluorescence quenching by a neighboring aromatic residue in ThyX and show that, in combination with temperature dependence studies, it is a sensitive probe for conformational fluctuations. We have shown that the active site of ThyX, which has to accommodate multiple substrates, is highly flexible and, in particular, samples configurations with very high speed, with large, multiple angstrom displacements occurring on the picoseconds timescale in the physiological temperature range. We suggest that this structural plasticity allows efficient binding of the dUMP substrate. Binding of dUMP, which acts as a very efficient FAD fluorescence quencher itself, arrests these movements and stabilizes the enzyme in a configuration allowing FAD reduction by the NADPH substrate. We conjecture that the rapid large amplitude motions of the active site monitored in ThyX enzymes may be related to their capacity of interacting with multiple different aromatic and relatively rigid substrates and products. The method that we have explored here can be used to study the effect of other substrates and, in particular, inhibitors, including potential antimicrobial agents (18), on the dynamics of the active site of ThyX. More generally, our results emphasize the important role of enzyme dynamics in the interaction with the substrates. Whereas flavoenzymes provide an intrinsically well-suited tool for real-time monitoring of configurational evolution, extensive high-speed sampling of active site configurations on the intrinsic picosecond timescale of global protein motions is likely to be a general feature for flexible protein domains and important for understanding enzyme substrate reactivity (2).
Materials and Methods
Proteins were expressed using a codon-optimized expression vector, and purified following standard procedures. Time-resolved fluorescence experiments were performed using a recently developed spectrally resolved Kerr-gate femtosecond fluorometer (43). The models for the MD simulations (performed with CHARMM) in the presence and absence of dUMP shown in the main text were based on the structure of the dUMP-containing protein (PDB ID code 1O26) (19); a model based on the substrateless structure (PDB ID code 1O2A) gave similar results (Fig. S9). A detailed description of the experimental procedures, data analysis, and simulation protocols is given in SI Materials and Methods.
Supplementary Material
Acknowledgments
This work was supported by the Agence National de la Recherche Grant ANR-09-PIRI-0019.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1218729110/-/DCSupplemental.
References
- 1.Frauenfelder H, Sligar SG, Wolynes PG. The energy landscapes and motions of proteins. Science. 1991;254(5038):1598–1603. doi: 10.1126/science.1749933. [DOI] [PubMed] [Google Scholar]
- 2.Henzler-Wildman KA, et al. A hierarchy of timescales in protein dynamics is linked to enzyme catalysis. Nature. 2007;450(7171):913–916. doi: 10.1038/nature06407. [DOI] [PubMed] [Google Scholar]
- 3.Yang H, et al. Protein conformational dynamics probed by single-molecule electron transfer. Science. 2003;302(5643):262–266. doi: 10.1126/science.1086911. [DOI] [PubMed] [Google Scholar]
- 4.Boekelheide N, Salomón-Ferrer R, Miller TF., III Dynamics and dissipation in enzyme catalysis. Proc Natl Acad Sci USA. 2011;108(39):16159–16163. doi: 10.1073/pnas.1106397108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Williams JC, McDermott AE. Dynamics of the flexible loop of triosephosphate isomerase: The loop motion is not ligand gated. Biochemistry. 1995;34(26):8309–8319. doi: 10.1021/bi00026a012. [DOI] [PubMed] [Google Scholar]
- 6.Henzler-Wildman KA, et al. Intrinsic motions along an enzymatic reaction trajectory. Nature. 2007;450(7171):838–844. doi: 10.1038/nature06410. [DOI] [PubMed] [Google Scholar]
- 7.Sinev MA, Sineva EV, Ittah V, Haas E. Domain closure in adenylate kinase. Biochemistry. 1996;35(20):6425–6437. doi: 10.1021/bi952687j. [DOI] [PubMed] [Google Scholar]
- 8.Pontiggia F, Zen A, Micheletti C. Small- and large-scale conformational changes of adenylate kinase: A molecular dynamics study of the subdomain motion and mechanics. Biophys J. 2008;95(12):5901–5912. doi: 10.1529/biophysj.108.135467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brazard J, et al. Spectro-temporal characterization of the photoactivation mechanism of two new oxidized cryptochrome/photolyase photoreceptors. J Am Chem Soc. 2010;132(13):4935–4945. doi: 10.1021/ja1002372. [DOI] [PubMed] [Google Scholar]
- 10.Chosrowjan H, et al. Ultrafast fluorescence dynamics of FMN-binding protein from Desulfovibrio vulgaris (Miyazaki F) and its site-directed mutated proteins. Chem Phys Lett. 2008;462(1–3):121–124. doi: 10.1021/jp073702k. [DOI] [PubMed] [Google Scholar]
- 11.Mataga N, et al. Dynamics and mechanisms of ultrafast fluorescence quenching reactions of flavin chromophores in protein nanospace. J Phys Chem B. 2000;104(45):10667–10677. [Google Scholar]
- 12.Pan J, et al. Excited-state properties of flavin radicals in flavoproteins: femtosecond spectroscopy of DNA photolyase, glucose oxidase and flavodoxin. J Phys Chem B. 2004;108(28):10160–10167. [Google Scholar]
- 13.Shibata Y, et al. Acceleration of electron-transfer-induced fluorescence quenching upon conversion to the signaling state in the blue-light receptor, TePixD, from Thermosynechococcus elongatus. J Phys Chem B. 2009;113(23):8192–8198. doi: 10.1021/jp901631b. [DOI] [PubMed] [Google Scholar]
- 14.Zhong D, Zewail AH. Femtosecond dynamics of flavoproteins: Charge separation and recombination in riboflavine (vitamin B2)-binding protein and in glucose oxidase enzyme. Proc Natl Acad Sci USA. 2001;98(21):11867–11872. doi: 10.1073/pnas.211440398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Callis PR, Liu TQ. Short range photoinduced electron transfer in proteins: QM-MM simulations of tryptophan and flavin fluorescence quenching in proteins. Chem Phys. 2006;326(1):230–239. [Google Scholar]
- 16.Myllykallio H, et al. An alternative flavin-dependent mechanism for thymidylate synthesis. Science. 2002;297(5578):105–107. doi: 10.1126/science.1072113. [DOI] [PubMed] [Google Scholar]
- 17.Finer-Moore JS, Santi DV, Stroud RM. Lessons and conclusions from dissecting the mechanism of a bisubstrate enzyme: Thymidylate synthase mutagenesis, function, and structure. Biochemistry. 2003;42(2):248–256. doi: 10.1021/bi020599a. [DOI] [PubMed] [Google Scholar]
- 18.Basta T, et al. 2012. Mechanistic and structural basis for inhibition of thymidylate synthase ThyX. Open Biol 2:2046–2441.
- 19.Mathews II, et al. Functional analysis of substrate and cofactor complex structures of a thymidylate synthase-complementing protein. Structure. 2003;11(6):677–690. doi: 10.1016/s0969-2126(03)00097-2. [DOI] [PubMed] [Google Scholar]
- 20.Koehn EM, et al. Folate binding site of flavin-dependent thymidylate synthase. Proc Natl Acad Sci USA. 2012;109(39):15722–15727. doi: 10.1073/pnas.1206077109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Graziani S, et al. Functional analysis of FAD-dependent thymidylate synthase ThyX from Paramecium bursaria Chlorella virus-1. J Biol Chem. 2004;279(52):54340–54347. doi: 10.1074/jbc.M409121200. [DOI] [PubMed] [Google Scholar]
- 22.Gattis SG, Palfey BA. Direct observation of the participation of flavin in product formation by thyX-encoded thymidylate synthase. J Am Chem Soc. 2005;127(3):832–833. doi: 10.1021/ja0432214. [DOI] [PubMed] [Google Scholar]
- 23.Brazard J, et al. New insights into the ultrafast photophysics of oxidized and reduced FAD in solution. J Phys Chem A. 2011;115(15):3251–3262. doi: 10.1021/jp110741y. [DOI] [PubMed] [Google Scholar]
- 24.Leduc D, et al. Functional evidence for active site location of tetrameric thymidylate synthase X at the interphase of three monomers. Proc Natl Acad Sci USA. 2004;101(19):7252–7257. doi: 10.1073/pnas.0401365101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chang C-W, et al. Mapping solvation dynamics at the function site of flavodoxin in three redox states. J Am Chem Soc. 2010;132(36):12741–12747. doi: 10.1021/ja1050154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Leenders R, Van Hoek A, Van Iersel M, Veeger C, Visser AJWG. Flavin dynamics in oxidized Clostridium beijerinckii flavodoxin as assessed by time-resolved polarized fluorescence. Eur J Biochem. 1993;218(3):977–984. doi: 10.1111/j.1432-1033.1993.tb18456.x. [DOI] [PubMed] [Google Scholar]
- 27.Marcus RA, Sutin N. Electron transfers in chemistry and biology. Biochim Biophys Acta. 1985;811(3):265–322. [Google Scholar]
- 28.Moser CC, Anderson JLR, Dutton PL. Guidelines for tunneling in enzymes. Biochim Biophys Acta. 2010;1797(9):1573–1586. doi: 10.1016/j.bbabio.2010.04.441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bastiaens PI, van Hoek A, Benen JA, Brochon JC, Visser AJ. Conformational dynamics and intersubunit energy transfer in wild-type and mutant lipoamide dehydrogenase from Azotobacter vinelandii. A multidimensional time-resolved polarized fluorescence study. Biophys J. 1992;63(3):839–853. doi: 10.1016/S0006-3495(92)81659-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Aubert C, Vos MH, Mathis P, Eker APM, Brettel K. Intraprotein radical transfer during photoactivation of DNA photolyase. Nature. 2000;405(6786):586–590. doi: 10.1038/35014644. [DOI] [PubMed] [Google Scholar]
- 31.Gauden M, et al. On the role of aromatic side chains in the photoactivation of BLUF domains. Biochemistry. 2007;46(25):7405–7415. doi: 10.1021/bi7006433. [DOI] [PubMed] [Google Scholar]
- 32.Gauden M, et al. Hydrogen-bond switching through a radical pair mechanism in a flavin-binding photoreceptor. Proc Natl Acad Sci USA. 2006;103(29):10895–10900. doi: 10.1073/pnas.0600720103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Costentin C, Louault C, Robert M, Savéant J-M. The electrochemical approach to concerted proton—electron transfers in the oxidation of phenols in water. Proc Natl Acad Sci USA. 2009;106(43):18143–18148. doi: 10.1073/pnas.0910065106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Song N, Stanbury DM. Proton-coupled electron-transfer oxidation of phenols by hexachloroiridate(IV) Inorg Chem. 2008;47(24):11458–11460. doi: 10.1021/ic8015595. [DOI] [PubMed] [Google Scholar]
- 35.Dhananjeyan MR, Annapoorani R, Renganathan R. A comparative study on the TiO2 mediated photo-oxidation of uracil, thymine and 6-methyluracil. J Photochem Photobiol A. 1997;109(2):147–153. [Google Scholar]
- 36.Chosrowjan H, Taniguchi S, Mataga N, Tanaka F, Visser AJWG. The stacked flavin adenine dinucleotide conformation in water is fluorescent on picosecond timescale. Chem Phys Lett. 2003;378:354–358. [Google Scholar]
- 37.Chosrowjan H, et al. Conformational heterogeneity in pyranose 2-oxidase from Trametes multicolor revealed by ultrafast fluorescence dynamics. J Photochem Photobiol A. 2012;234(3-4):44–48. [Google Scholar]
- 38.Graziani S, et al. Catalytic mechanism and structure of viral flavin-dependent thymidylate synthase ThyX. J Biol Chem. 2006;281(33):24048–24057. doi: 10.1074/jbc.M600745200. [DOI] [PubMed] [Google Scholar]
- 39.Sampathkumar P, et al. Structure of the Mycobacterium tuberculosis flavin dependent thymidylate synthase (MtbThyX) at 2.0A resolution. J Mol Biol. 2005;352(5):1091–1104. doi: 10.1016/j.jmb.2005.07.071. [DOI] [PubMed] [Google Scholar]
- 40.Agrawal N, Lesley SA, Kuhn P, Kohen A. Mechanistic studies of a flavin-dependent thymidylate synthase. Biochemistry. 2004;43(32):10295–10301. doi: 10.1021/bi0490439. [DOI] [PubMed] [Google Scholar]
- 41.Koehn EM, et al. An unusual mechanism of thymidylate biosynthesis in organisms containing the thyX gene. Nature. 2009;458(7240):919–923. doi: 10.1038/nature07973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chernyshev A, Fleischmann T, Kohen A. Thymidyl biosynthesis enzymes as antibiotic targets. Appl Microbiol Biotechnol. 2007;74(2):282–289. doi: 10.1007/s00253-006-0763-1. [DOI] [PubMed] [Google Scholar]
- 43. Laptenok SP, Nuernberger P, Lukacs A, Vos MH (2013) Fluorescence Spectroscopy and Microscopy: Methods and Protocols, eds Engelborghs Y, Visser A (Springer, Berlin), in press.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.