

Significance
Regulated cell-surface proteolysis underpins processes of cellular migration in both physiological and pathological contexts. However, comprehending how multiple proteolytic events cohesively integrate to yield context-dependent cellular behavior remains a challenge. Here we present an experimental/computational paradigm for analyzing networks of protease activities that interface with signaling pathways to influence cellular migration in the invasive disease of endometriosis. We find that induced cellular migration is a quantitative consequence of positive feedback through ligand release and negative feedback through receptor shedding, which furthermore drives rapid resistance to kinase inhibitor treatment. Targeted clinical proteomics confirms dysregulated proteolysis in endometriosis.
Keywords: cell signaling networks, metalloproteinase activity, cue-signal-response analysis, amphiregulin
Abstract
A Disintegrin and Metalloproteinases (ADAMs) are the principal enzymes for shedding receptor tyrosine kinase (RTK) ectodomains and ligands from the cell surface. Multiple layers of activity regulation, feedback, and catalytic promiscuity impede our understanding of context-dependent ADAM “sheddase” function and our ability to predictably target that function in disease. This study uses combined measurement and computational modeling to examine how various growth factor environments influence sheddase activity and cell migration in the invasive disease of endometriosis. We find that ADAM-10 and -17 dynamically integrate numerous signaling pathways to direct cell motility. Data-driven modeling reveals that induced cell migration is a quantitative function of positive feedback through EGF ligand release and negative feedback through RTK shedding. Although sheddase inhibition prevents autocrine ligand shedding and resultant EGF receptor transactivation, it also leads to an accumulation of phosphorylated receptors (HER2, HER4, and MET) on the cell surface, which subsequently enhances Jnk/p38 signaling. Jnk/p38 inhibition reduces cell migration by blocking sheddase activity while additionally preventing the compensatory signaling from accumulated RTKs. In contrast, Mek inhibition reduces ADAM-10 and -17 activities but fails to inhibit compensatory signaling from accumulated RTKs, which actually enhances cell motility in some contexts. Thus, here we present a sheddase-based mechanism of rapidly acquired resistance to Mek inhibition through reduced RTK shedding that can be overcome with rationally directed combination inhibitor treatment. We investigate the clinical relevance of these findings using targeted proteomics of peritoneal fluid from endometriosis patients and find growth-factor–driven ADAM-10 activity and MET shedding are jointly dysregulated with disease.
A Disintegrin and Metalloproteinases (ADAMs), especially ADAM-10 and -17, are the principal mediators of proteolytic ectodomain shedding on the cell surface (1). ADAMs and the closely related matrix metalloproteinases (MMPs) work together as “sheddases” to cleave hundreds of diverse transmembrane substrates including growth factor ligands, receptor tyrosine kinases (RTKs), adhesion molecules, and even proteases themselves from the cell surface. Unfortunately, little is known regarding how such a broad palette of proteolytic activity integrates to modulate behaviors such as cellular motility. Furthermore, extensive cross-talk and complexity among signaling networks, proteases, and their substrates make understanding sheddase regulation on a component-by-component basis challenging (2). Therapeutics have targeted sheddases and their substrates for the treatment of invasive diseases such as cancer, yet many of these inhibitors have failed in clinical trials (3). Therefore, a need exists for understanding how the balance of sheddase-mediated degradation integrates multiple layers of signaling networks to coordinately influence cell behavior in various disease contexts.
Here we study how sheddase activity contributes to cell migration in the invasive disease of endometriosis, defined by the presence of endometrial-like tissue residing outside the uterus. Up to 10% of adult females and 40% of infertile women have the disease, which also exhibits comorbidity with several cancers (4, 5). Endometriosis currently has no cure: hormonal therapies merely manage the disease with significant side effects, and surgery provides only temporary relief for many, with recurrence rates as great as 40% within 5 y postoperation (6). Like cancer, endometriosis is associated with aberrant cell invasion into ectopic organ sites, and endometriotic tissues often exhibit dysregulated molecular pathways commonly perturbed in other invasive diseases. Mitogenic and inflammatory phospho-signaling [for example, phosphorylated extracellular-signal-related kinase 1/2 (p-Erk1/2), phosphorylated protein kinase B (p-Akt), and phosphorylated p38 mitogen-activated protein kinase (p-p38)], RTKs (including epidermal growth factor receptor, EGFR), and metalloproteinases have all been clinically associated with endometriosis (7, 8), and consequently represent attractive therapeutic strategies (9–11).
Many challenges in developing targeted therapeutics stem from network-level complexities such as compensatory feedback, and recent work has demonstrated how critical such mechanisms are to achieving therapeutic success, especially in cancer (12, 13). Computational models of systems-level biochemical networks have shown promise as tools to understand how multiple enzymatic reactions integrate to impact overall biological behavior, often with the goal of aiding the design of personalized or combination therapies (14, 15). Considering its complex role in disease, sheddase regulation represents an ideal application of such network-level approaches. In this work, we apply the “cue–signal–response” (CSR) paradigm (14, 15) (Fig. 1A) to examine how disease-implicated growth-factor cues interact with experimentally monitored phospho-protein and protease networks (collectively referred to as signals), ultimately to influence cellular migration response. Computational modeling elucidates quantitative and predictive relationships among multiple layers of experimental data and offers testable hypotheses of context-dependent behavior and signaling feedback. We find ADAM-10 and -17 to be critical regulators of motility that are dynamically controlled through several signaling pathways, thereby affecting cell behavior through both positive feedback from EGF ligand release and negative feedback from Hepatocyte Growth Factor Receptor (HGFR; MET), Human Epidermal Growth Factor Receptor 2 (HER2), and HER4 RTK shedding. We find kinase inhibition generally reduces ADAM-10 and -17 activities, reduces subsequent RTK shedding, and consequently allows the accumulated RTKs to enhance downstream c-Jun N-terminal kinase (Jnk) and p38 signaling. Thus, here we demonstrate an ADAM-10 and -17–based mechanism of rapidly acquired resistance to kinase inhibition through reduced RTK shedding that can be overcome with combination therapy. Targeted proteomic analysis of clinical samples from endometriosis patients indeed confirms growth-factor–driven ADAM-10 activity and consequent MET shedding are dysregulated with disease. Overall, our results have wide implications for designing combination therapies and identifying context-dependent personalized therapeutic strategies for both kinase and protease inhibitors.
Fig. 1.
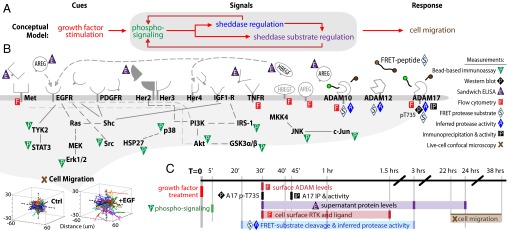
CSR study design. (A) CSR overview: we stimulate endometriotic cells with a panel of growth factor cues; record multiple downstream signals comprising measurements of phospho-signaling, sheddase regulation, and sheddase substrate regulation; and use computational modeling to map these observations onto cell migration responses. (B) Overview of signals and responses included in the CSR dataset. All receptors shown were directly measured and/or stimulated. (C) Experimental timeline of CSR study. Dark colored lines denote measurement time points. At lower left, cell migration is depicted as single-cell tracks, where initial cell positions were centered for visualization.
Results
Overview of CSR Study Design.
We use a CSR approach to understand the signaling-regulated impacts of sheddase activity on cell migration from a multivariate, network-level perspective (Fig. 1A). First, we stimulate the immortalized 12Z cell line, a commonly studied in vitro disease model established from an endometriotic biopsy (16), with a panel of growth factor cues (see SI Appendix, Table S1 for references of clinical associations): EGF, transforming growth factor alpha (TGFa), neuregulin beta–1 (NRG1b), hepatocyte growth factor (HGF), insulin-like growth factor 1 (IGF1), and platelet-derived growth factor–bb (PDGFbb). Poststimulation, we quantitatively monitor an array of downstream molecular features, or “signals” (Fig. 1 B and C). Nearly all “signals” in the CSR dataset are clinically associated with endometriosis (see SI Appendix, Table S1 for unabbreviated names and clinical evidence). To assess intracellular signaling, 5 min poststimulation we measured levels of 11 key phospho-proteins using bead-based sandwich immunoassays (SI Appendix, Fig. S2).
We performed multiple measurements of key ADAM sheddases, themselves, following growth-factor stimulation. To directly assess ADAM-17 activity, we immunoprecipitated the enzyme from whole-cell lysate, incubated the bound protein with a FRET substrate, and recorded cleavage rates by fluorimetry (SI Appendix, Fig. S3A). However, this approach disrupts protein complexes observed in the live-cell context. Therefore, we coadministered seven soluble FRET-based polypeptide substrates with growth factor treatments to assess regulation of general catalytic activity of proteases in live cells. Cleavage rates of these substrates were assessed by live-cell fluorimetry for several hours (SI Appendix, Fig. S3 B and C). However, the FRET substrates are by nature nonspecific. We therefore used Proteolytic Activity Matrix Analysis (PrAMA) as an inference algorithm to estimate the effective concentrations of catalytically active ADAM-10, -12, and -17, based on FRET substrate cleavage rates and prior knowledge of enzyme substrate specificities (17) (SI Appendix, Fig. S3 D and E). Additionally, we quantified ADAM-10 and -17 surface levels to assess protease trafficking (SI Appendix, Fig. S4 A–H) and monitored levels of ADAM-17–pT735, which is thought to impact activity (18) (SI Appendix, Fig. S4I).
We investigated regulation of membrane-bound substrates by quantifying their cell-surface trafficking and proteolysis. To measure short-term shedding of heparin-binding EGF (HBEGF, an EGF ligand), we transgenically overexpressed it with a Myc-tagged ectodomain and a GFP-tagged C terminus (19). Immunostaining enabled quantification of intact HBEGF on the cell surface relative to total levels. These measurements were averaged over three time points (30, 60, and 90 min) (SI Appendix, Fig. S5). For more highly expressed membrane-bound substrates (compared with HBEGF), we measured endogenous levels of surface-bound MET (also known as HGF receptor), tumor necrosis factor receptor 1 (TNFR1), and amphiregulin (AREG, an EGF ligand), averaged over three time points poststimulation (30, 60, and 90 min; SI Appendix, Fig. S6A). We also monitored supernatant levels of endogenous substrate at multiple times using enzyme-linked immunoassays (ELISAs), although not all analytes were detectable at early time points (SI Appendix, Fig. S6 B–G).
Finally, we used time-lapse confocal microscopy to assess features of cell migration as responses to the growth-factor cues and previously described molecular signals. Dye-labeled 12Z cultures suspended in collagen-I gels were individually tracked for 16 h, and various descriptions of cell movement including total path length, net displacement, and the “random motility coefficient” derived from a thermodynamic-based model of the persistent random walk were calculated as metrics of single-cell motility for each condition (20) (SI Appendix, Fig. S7).
CSR Modeling Suggests Parallel Ligand and Receptor Shedding Influence Cell Migration.
To glean information from the full CSR dataset (shown in Fig. 2A), we began by calculating correlation between pairs of measurements as they varied across the seven growth-factor treatment conditions. Significant pair-wise correlations were then graphically mapped in an unsupervised manner. This “correlation- network” encouragingly reflects several features of known biology (SI Appendix, Fig. S8A). For example, the greatest correlation among all phospho-signaling measurements lies between Jnk and its known substrate c-Jun (SI Appendix, Fig. S8B). ADAM-10 catalytic activity, as inferred by PrAMA, correlated very closely with supernatant accumulation of a known substrate, MET. Among the most negatively correlated measurements, cell-surface AREG was strongly anticorrelated with supernatant accumulation of AREG. At a higher level, the correlation network suggests modularity among the data, where highly interconnected phospho-signaling events link to early (30 min to 3 h) protease activity measurements primarily through ADAM-17 phosphorylation. These early markers of protease activity then correlate with supernatant accumulation of ligands and receptors by 24 h, which in turn are highly correlative with features of cell migration (SI Appendix, Fig. S8). Of all measurements in the CSR dataset, 3D cell migration features correlated most closely with ligand and receptor shedding. We tested if ligand/receptor shedding was affected by whether cells were cultured on 2D tissue culture plastic or in 3D collagen-I matrices. For those species included in the CSR dataset, we found significant agreement between results from these two cell culture models (SI Appendix, Fig. S9A), further suggesting that ligand/receptor shedding measurements made in 2D cultures sufficiently reflect shedding and migration behaviors observed in 3D cultures.
Fig. 2.
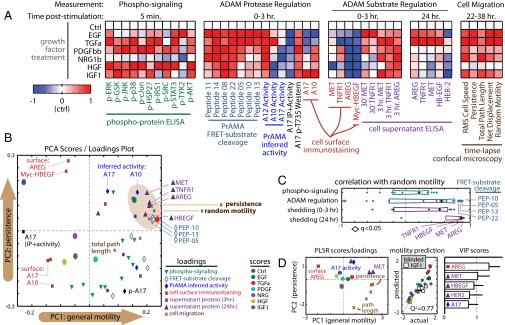
CSR modeling suggests dual ligand/receptor shedding influences cell migration. (A) Heat maps depict CSR dataset, at time points described in Fig. 1C. Data were normalized by control-centering and scaling max absolute value to 1. (B) PCA presents an unsupervised description of covariation within the CSR dataset values in A. The brown ellipse denotes measurements most associated with persistent cell migration. (C) AREG and MET shedding by 24 h poststimulation most closely correlate with random motility, among all measurements in the CSR dataset (q-value denotes multiple-hypothesis–corrected P value). Box-and-whisker bars show the first to third quartiles. (D) The reduced PLSR model describes features of cell migration as a function of select variables from the CSR dataset. The scores/loadings plot (Left) describes covariance among the descriptor variables and migration features. Cross-validation prediction accuracy (Q2 = 77%) was determined with IGF1 measurements blinded from the optimization routine (Center). Model descriptors were ranked by importance according to VIP score (Right). Error bars denote SE.
We performed principal components analysis (PCA) to describe measurements from the CSR dataset in terms of key axes of covariance, or principal components (PCs), as they varied across the growth factor treatments. The scores/loadings plot describes where each of the growth factor treatments and measurement variables fall along the first two PCs, which capture 40% and 25% of the total data variance, respectively (Fig. 2B; shown fully labeled in SI Appendix, Fig. S9B). Similar to results from the correlation network, modularity can also be observed in the PCA scores/loadings plot. The lower right quadrant is largely populated with phospho-protein levels and short-term metrics of substrate shedding, and these are anticorrelated with surface levels of endogenous substrate (AREG and Myc–HBEGF) in the upper left quadrant. The upper right quadrant associates with persistent migratory behavior, and is populated with ADAM-10 and -17 activities (inferred by PrAMA), along with levels of supernatant ligands/receptors at 24 h. Direct comparison of correlations between CSR dataset measurements and the random motility coefficient echo the PCA results: supernatant ligand/receptor at 24 h represent by far the most correlative indicators of cell migration, led by AREG and MET as the top two features (Fig. 2C).
Taken together, these results suggest that growth-factor stimulation directly regulates the effective concentration of catalytically active sheddases, which then leads to similar (although not identical) patterns of shedding across multiple endogenous and FRET-based substrates. Furthermore, these patterns of substrate proteolysis correlate extremely well with cell migration, suggesting that sheddases significantly influence motility.
Joint AREG and MET Shedding Predict Cell Migration.
Although individual shed analytes significantly correlate with features of cellular motility, single-variable relationships between shedding and motility fail to accurately predict motile responses under untested conditions in a sufficiently quantitative manner, with a prediction accuracy of Q2 < 50%. Consequently, we implemented partial least squares regression (PLSR) as a statistical method to distill the effects of multiple shedding events into key axes of control (PCs, as with PCA) that quantitatively combine to describe overall migration behavior. More specifically, we used an optimization algorithm to build a reduced PLSR model that optimally selects the minimal set of descriptor variables from the CSR dataset that predict migration with high accuracy. To improve model accuracy, we included additional measurements, made in the presence of a broad-spectrum metalloproteinase inhibitor (BB94) and an EGFR blocking antibody monoclonal antibody 225 (mab225), to determine the dependency of shed analyte accumulation on sheddase activity and EGFR endocytosis of autocrine ligand (data shown in SI Appendix, Fig. S6). Among all measurements in the “expanded” CSR dataset, metrics of AREG and MET shedding were the two most important variables chosen by the algorithm (Fig. 2D; fully labeled scores/loadings in SI Appendix, Fig. S9C). Although patterns of MET and AREG shedding closely correlate with each other, PLSR model accuracy significantly improves when both are included together, suggesting subtle underlying mechanisms of substrate specificity. Indeed, PLSR accuracy relies upon multiple PCs for accurate prediction accuracy (SI Appendix, Fig. S9D), implying multiple axes of substrate shedding regulation.
In addition to supernatant ligand/receptor accumulation, we also measured accumulation of MMPs and tissue inhibitor of metalloproteinases (TIMPs) across the panel of growth factor treatments (SI Appendix, Fig. S10A). The aims here were to investigate enzymes more associated with extracellular matrix degradation and to examine their ability to predict cell migration compared with ligand/receptor levels. In comparison with ligand/receptor shedding, however, MMP/TIMP levels generally did not significantly correlate with or help in prediction of cell migration (SI Appendix, Fig. S10 B and C). This indicates that, at least with respect to growth factor stimulation, cell motility is principally regulated outside modulation of MMP/TIMP expression.
Overall, the correlation network modeling, PCA results, and PLSR models all suggest that concomitant ligand and receptor shedding, and especially AREG and MET shedding, are key determinants of endometriotic cell migration in response to various growth factor cues. Based on this model, we elected to further experimentally investigate regulation of AREG/MET proteolysis along with its resultant functional and therapeutic consequences.
Positive Signaling Feedback via AREG Shedding Drives Cell Migration.
CSR modeling results predicted a role for AREG shedding in governing cell migration, and we next sought to investigate its potential role in mediating positive signaling feedback through EGFR. Experiments with BB94 demonstrated that AREG supernatant accumulation is metalloproteinase-dependent, and treatment with mab225 provided evidence that soluble AREG is actively being endocytosed via EGFR in an autocrine manner (Fig. 3A). Interestingly, we found that saturating levels of TGFa, which is known to exhibit higher binding affinity to EGFR compared with AREG (21) and likely inhibits AREG–EGFR binding, stimulates even greater AREG supernatant accumulation. This result suggested a positive feedback loop similar to those described previously (22), here with EGFR signaling promoting AREG shedding, which in turn enhances further EGFR signaling. Stimuli beside EGF ligands also stimulated AREG shedding, including the inflammatory cytokine TNFa (Fig. 3A), which transactivates EGFR in an EGF-ligand–dependent manner (Fig. 3B). Furthermore, TNFa stimulation enhanced the effect of mab225 treatment in reducing cellular migration (Fig. 3 C and D).
Fig. 3.
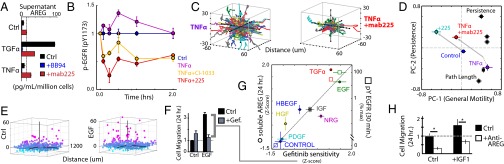
EGFR transactivation through AREG shedding sensitizes 12Z to EGFR inhibition. (A) AREG levels reveal induced shedding and autocrine uptake via EGFR (24 h posttreatment; ELISA; see SI Appendix, Fig. S6G for details). (B–D) TNFa stimulates EGFR pY1173 (B; bead immunoassay) and motility (C and D) in an EGFR-ligand–dependent manner. In D, cellular motility was described using PCA of time-lapse microscopy measurements 22–38 h poststimulation (shown as single-cell tracks in C, where initial cell positions were centered for visualization). Scores (circles) and loadings (squares) are plotted. (E) 12Z nuclei positions demonstrate EGF-stimulated migration into collagen-I gels as a function of distance from the plate bottom, shown quantified in F. Treatment with gefitinib blocks the effect of EGF added 1 h later. (G) Supernatant AREG (left axis; ELISA) and p-EGFR levels (right axis; bead immunoassay) correlate with the gefinitib efficacy in reducing cell migration. (H) Anti-AREG decoy Ab treatment inhibits basal and IGF1-stimulated cell migration in the endpoint assay. (*P < 0.05; single-tailed Student t test.) All error bars denote SE.
With evidence of AREG-mediated EGFR transactivation, we examined whether growth factor stimulation sensitized cell motility to EGFR kinase inhibition. For this and all subsequent cell migration experiments, we used a migration endpoint assay (SI Appendix, Fig. S11). Briefly, cells were seeded under collagen-I gels and exposed to bath application of growth factors after 1 h treatment with inhibitors. We quantified cellular migration into gels 24 h later (Fig. 3E). Although EGFR kinase inhibition using gefitinib (an EGFR inhibitor) and lapatinib (a dual EGFR/HER2 inhibitor) was ineffective at reducing cellular motility under basal conditions, nearly every tested growth factor sensitized cells to kinase inhibition (Fig. 3 F and G; SI Appendix, Fig. S11). We compared AREG shedding to gefitinib sensitivity across the growth-factor treatments and found significant correlation (Fig. 3G). AREG shedding is particularly enhanced with IGF1 treatment, and IGF1 sensitized cells most to gefitinib compared with other non-ErbB family (that is, non-EGFR/HER2/HER3/HER4 targeting) growth factors. Examination of phosphorylated EGFR (p-EGFR) in IGF1-treated cells confirmed EGFR transactivation (Fig. 3G). Treatment with an anti-AREG decoy antibody effectively reduced both basal and IGF1-induced cellular motility, confirming a specific role for AREG among other potential EGF ligands (Fig. 3H). In sum, these data provide further evidence for the role of AREG-mediated positive signaling feedback in endometriosis cell migration.
EGFR Autocrine Signaling Regulates ADAM-10 and -17 Catalytic Activities.
We next examined how AREG shedding itself is regulated by ADAM proteases, particularly in the context of EGFR signaling feedback. Direct examination of ADAM-10 and -17 catalytic activity in live cells using PrAMA revealed that the positive feedback via EGFR activity occurs at least in part through direct regulation of ADAM-10 and -17 catalytic activity (Fig. 4A). EGF and TGFa treatment led to an increase in FRET substrate proteolysis, while mab225 treatment led to a decrease (SI Appendix, Fig. S12A). These effects were also seen with endogenous sheddase substrates (besides AREG). For example, mab225 treatment led to an increase in surface TNFR1 and a decrease in its supernatant accumulation (SI Appendix, Fig. S12 B and C). However, the exact mechanisms of protease regulation remain unknown. Although EGF stimulation led to decreased ADAM-17 dimerization (SI Appendix, Fig. S4 J and K) and increased ADAM-17 pT735 (Fig. 2A), mab225 treatment did not elicit changes in ADAM-17 dimerization (SI Appendix, Fig. S12 D and E), ADAM-17 activity as measured after immunoprecipitation (SI Appendix, Fig. S12F), ADAM-17–pT735 (SI Appendix, Fig. S12G), or ADAM-17 surface levels (SI Appendix, Fig. S12H). Nonetheless, PrAMA results combined with decreased endogenous substrate shedding suggest decreasing ADAM-10 and -17 catalytic activities in response to mab225 treatment. Given these complex results, we decided to perform additional computational modeling to formulate testable hypotheses as to how proteases may regulate substrate shedding in response to various signaling cues.
Fig. 4.

Both ADAM-10 and -17 contribute to AREG shedding. (A) PrAMA indicates mab225 treatment reduces ADAM activities. Following 30 min of mab225 treatment, FRET substrates and growth factors were simultaneously added to serum-starved 12Z cultures, and protease activities were recorded by fluorimetry for 3 h poststimulation. (B) ADAM-10 inhibition only increases surface AREG under EGF stimulation, quantified by immunostaining following 24 h treatment with proADAM10 and EGF. (C) proADAM10 treatment significantly reduces levels of supernatant AREG in the presence of EGF (ELISA). (D) Both ADAM-10 and -17 siRNA treatment reduce supernatant AREG levels (ELISA). (E) ADAM-17 knockdown reduces basal ADAM-10 activity, but not vice versa (PrAMA). (F) ADAM-10 knockdown blocks cell migration in the endpoint assay. (G) proADAM10 treatment inhibits basal and IGF1-stimulated cell migration in the endpoint assay. All error bars denote SE. (*P < 0.05, Student t test.)
AREG Shedding Is Controlled by ADAM-10 and -17 in a Context-Dependent Manner.
We constructed reduced PLSR models to describe endogenous substrate shedding as a function of phospho-proteins, protease surface levels, and protease activity (including PrAMA and the immunoprecipitation and FRET-based activity, or IP+activity, assay). PLSR results decomposed substrate proteolysis along two PCs, with PC-1 describing overall shedding and PC-2 distinguishing ligands vs. receptors (SI Appendix, Fig. S13 A and B). Interestingly, the PLSR results suggested a concerted role for both ADAM-10 and -17, where each protease exhibits more or less influence depending on the growth-factor context (SI Appendix, Fig. S13 C and D). Indeed, knockdown of either ADAM-10 or -17 reduces shedding of all of the substrates tested (SI Appendix, Fig. S13E). One particular hypothesis from the PLSR modeling is that EGF and TGFa stimulation drive ADAM-10 activity more than ADAM-17 activity. These results were primary determined by observations that (i) EGF and TGFa lead to decreased activity measured in the ADAM-17 IP+activity assay, (ii) EGF and TGFa stimulate down-regulation of ADAM-17 surface levels, and (iii) PrAMA infers that EGF and TGFa stimulate significantly more ADAM-10 activity than ADAM-17 activity (Fig. 2A). Consequently, although AREG is predominantly thought of as an ADAM-17 substrate (23), PLSR results suggest that EGF-stimulated AREG shedding may actually be occurring via ADAM-10. Using recombinant ADAM-10 prodomain as a specific inhibitor, we found ADAM-10 inhibition to cause increased AREG surface levels under EGF-stimulated, but not basal, treatment conditions (Fig. 4B). Furthermore, ADAM-10 inhibition only decreased supernatant AREG accumulation after EGF stimulation (Fig. 4C). siRNA knockdown of ADAM-10 showed a greater inhibitory effect on AREG supernatant accumulation in EGF-stimulated cells (Fig. 4D). In contrast, ADAM-17 knockdown equally reduced AREG shedding under basal and EGF-stimulated conditions (Fig. 4D). Direct examination of specific ADAM activities in the siRNA-treated cells using PrAMA suggests that ADAM-10 does not impact ADAM-17 activity, further supporting a specific role for ADAM-10 in shedding AREG (Fig. 4E). Finally, Western blots show metalloproteinase-dependent, EGF-stimulated cleavage of pro-AREG in cell lysates (SI Appendix, Fig. S14 A–C), and digestion of immunoprecipitated pro-AREG with recombinant ADAM-10 and -17 demonstrated that both enzymes are capable of acting upon AREG and generating cleavage products similar to those seen in the EGF-stimulated lysate (SI Appendix, Fig. S14 D–F). Overall, these results provide evidence for EGF-stimulated ADAM-10 activity and a context-dependent dual role for ADAM-10 and -17 in regulating substrate shedding.
ADAM-10 Inhibition Reduces Cellular Migration.
Consistent with the CSR modeling results showing the importance of ADAM-10 activity (as inferred using PrAMA) for cell motility, we found that ADAM-10 knockdown substantially decreased basal motility (Fig. 4F). Additionally, a specific ADAM-10 inhibitor (proADAM10) significantly reduced basal and IGF1-stimulated cell motility (Fig. 4G). In contrast, ADAM-17 knockdown did not show an effect on basal cell motility (Fig. 4F), possibly due in part to previously reported adhesion-related protein functions (23–25). Notably, however, ADAM-17 activity (as inferred using PrAMA) did not significantly correlate with cell motility in a positive manner in the CSR dataset, and ADAM-17 IP+activity results significantly anticorrelated with features of cell migration. Taken together, these data demonstrate that ADAM-10 influences cellular migration, owing at least in part to its role in mediating AREG shedding and autocrine EGFR signaling.
Negative Signaling Feedback via RTK Shedding Reduces Jnk/p38 Signaling.
In addition to positive signaling feedback from AREG, CSR modeling suggested the shedding of RTKs (principally MET) also plays a role in governing cell migration. We hypothesized that RTK shedding functions as a mechanism of negative signaling feedback by attenuating receptor phospho-signaling. Indeed, direct protease inhibition using BB94 led to increased full-length p-HER2 and p-HER4 (Fig. 5A), total and p-MET (Fig. 5B), and p-p38 and p-cJun (Fig. 5C). Supernatant MET, HER2, and HER4 correspondingly decreased (SI Appendix, Figs. S2 and S15A). We also found that BB94-induced p-Jnk elevation could be blocked with MET knockdown, further suggesting that enhanced signaling was due to accumulation of MET on the cell surface (Fig. 5D). Experiments confirmed that protease-inhibition effects were related to ADAM-10, the principal MET sheddase. Specific inhibition of ADAM-10 led to an accumulation of cell-surface MET (Fig. 5E), and we found ADAM-10 knockdown led to an increase in p-p38 that could be blocked using the MET inhibitor foretinib (Fig. 5F). Overall, these results demonstrate that ADAM-10 mediated RTK shedding functions as a negative signaling feedback mechanism, and that direct inhibition of sheddase activity leads to the accumulation of HER2, HER4, and MET, along with enhanced signaling through p38/Jnk/cJun signaling pathways.
Fig. 5.

ADAMs mediate negative signaling feedback via RTK shedding. (A) Full-length p-HER2 and p-HER4 levels increase with 1.5 h BB94 treatment (bead immunoassay). Subsequent 30 min TGFa-treatment did not alter BB94 effects. (B) The 1.5 h BB94 treatment followed by 30 min treatments with NRG1b and HGF lead to higher total, full-length MET (x-axis) and METpY-1349 (y-axis) (Western blot; see SI Appendix, Fig. S15B for images). (C) BB94 increases p-cJun and p-p38 (bead immunoassay). Cells were stimulated with NRG1b and TNFa for 30 min following 1.5 h BB94 treatment. (D) BB94 increases p-Jnk levels in a MET-dependent manner. Following siRNA knockdown of MET, cells were treated with BB94 for 1.5 h and stimulated with NRG1b for 30 min (bead immunoassay). (E) The 24 h proA10 treatment increases surface MET levels, +/− cotreatment with EGF, detected by immunostaining. (F) ADAM-10 knockdown increases basal p-p38 levels in a MET-dependent manner, using 1.5 h treatment with the MET inhibitor foretinib (bead immunoassay). All error bars denote SE. (*P < 0.05, Student t test.)
Indirect Sheddase Down-Regulation via Kinase Inhibition Mediates Drug Resistance.
Given evidence that sheddase activity can be activated by multiple signaling pathways, we next interrogated the effects of various kinase inhibitors on protease activity. In general, treatment with various Mek, Jnk, p38, and PI3K inhibitors broadly reduced the accumulation of both receptor and ligand sheddase substrates in cellular supernatant (Fig. 6A). The Mek inhibitor U0126 was also found to reduce supernatant TNFR1 levels within 30 min of treatment (SI Appendix, Fig. S16A), and multiple kinase inhibitors led to increased surface TNFR1 levels after 1 h of treatment (SI Appendix, Fig. S16B). We found U0126 treatment to elicit no change in ADAM-17 dimerization (SI Appendix, Fig. S16C), phosphorylation (SI Appendix, Fig. S16D), surface levels (SI Appendix, Fig. S16 E and F), or ADAM-10 surface levels (SI Appendix, Fig. S16G), and the ADAM-17 IP+activity assay only showed a slight decrease with U0126 treatment (SI Appendix, Fig. S16H). In contrast, live-cell measurements using PrAMA indicated a substantial reduction in ADAM-10 and -17 catalytic activities with kinase inhibition (Fig. 6B). Based on these results, we hypothesized that indirect sheddase inhibition secondary to kinase inhibition could lead to compensatory signaling from reduced RTK shedding. Indeed, we found that pretreatment with Mek or Jnk inhibitors increased full-length p-HER2 levels following NRG1b stimulation (Fig. 6C).
Fig. 6.

Decreased ADAM activity via kinase inhibition mediates drug resistance. (A) The 24 h treatments with kinase inhibitors that target Mek (blue), Jnk (magenta), p38 (red), and PI3K (cyan) lead to reduced supernatant levels of ligands and RTKs (ELISA). Cells were also cotreated with EGF or mab225. (B) PrAMA-inferred sheddase activities decrease in response to 4 h Mek and Jnk inhibitor treatment. (C) NRG1b-induced p-HER2 increases following a 1.5 h pretreatment with Mek and Jnk inhibitors (bead immunoassay). (D) Mek and PI3K inhibitor efficacies depend on growth factor context, while Jnk and p38 inhibitors do not (24 h endpoint migration assay). (E) Data from D were combined with experiments using additional Jnk and Mek inhibitors, and results are plotted to highlight differences in inhibitor efficacy under EGF- vs. HGF/NRG1b-stimulated conditions. All error bars denote SE. (*P < 0.05, Student t test.)
We next tested whether kinase inhibitors, and their indirect effects on RTK shedding, would have an impact on cellular migration in response to various growth factor stimuli (Fig. 6D). Results show two distinct patterns of inhibitor efficacy: p38 and Jnk inhibitors strongly reduced 12Z motility under all growth factor treatment conditions, while in contrast, Mek and PI3K inhibitors demonstrated context-dependent efficacy (Fig. 6D). While Mek and PI3K inhibitors effectively reduced EGF and TGFa stimulated motility, they actually enhanced motile responses to NRG1b and HGF. We further tested context-dependency using alternative Mek and Jnk inhibitors, and found results to be consistent (Fig. 6E; SI Appendix, Fig. S16I). To explain these differences, we measured NRG1b-stimulated p-p38 levels after 1 h pretreatment with Jnk or Mek inhibitors. Results indicated that compared with Jnk inhibition, Mek inhibition was unsuccessful in reducing p-p38 (SI Appendix, Fig. S16J). Overall, these data suggest that compensatory signaling through unshed RTKs, primarily through p38 and Jnk signaling pathways, can lead to Mek inhibitor resistance. Moreover, this compensatory signaling can become amplified in the presence of ligands that stimulate ADAM substrate RTKs (such as NRG1b and HGF).
Combined MET–Mek Inhibition Blocks Motility Across Multiple Growth Factor Contexts.
Given our evidence that protease inhibition can enhance MET signaling and that Mek inhibitor resistance in part arises from reduced sheddase activity, we hypothesized that Mek insensitivity in the presence of HGF and NRG1b is mediated by enhanced MET signaling. Using foretinib as an inhibitor of MET (and several other ADAM substrate RTKs, including VEGFR-2), we found that combination Mek–MET inhibition was more effective than either inhibitor alone, under multiple growth-factor contexts (Fig. 7 A and B). Combination Mek–MET inhibition reduced basal p-Jnk levels more than either inhibitor alone (Fig. 7C). U0126 treatment only blocked NRG1b-stimulated migration when combined with MET siRNA treatment (Fig. 7D). Individual effects from MET siRNA and U0126 were not significant in this experiment. Overall, these results confirm the importance of alternative MET signaling in the context of Mek inhibition and reduced MET shedding.
Fig. 7.

Combination MET and Mek inhibition overcomes sheddase-mediated drug resistance. (A and B) The 1 h pretreatments with Mek (U0126) and MET (foretinib) inhibitors reduce 12Z migratory response to growth factor stimulation in the endpoint migration assay. Combination treatment exhibits enhanced efficacy. (C) The 1 h combination MET/Mek inhibition using foretinib and U0126 leads to reduced p-Jnk (bead immunoassay). (D) Combination MET knockdown and Mek inhibitor (U0126) synergistically block NRG1b-stimulated cell migration in the endpoint migration assay. Neither MET knockdown nor U0126 treatment had a significant effect individually. All error bars denote SE. (*P < 0.05, Student t test.)
Clinical Samples Suggest Dysregulated ErbB Signaling and ADAM-10 Activity with Disease.
Finally, to test for relevance of our in vitro findings to in vivo pathophysiology in human patients, we analyzed surgically obtained peritoneal fluid (PF) from patients with and without endometriosis. PF comprises a heterogeneous mixture of leukocytes, cell debris, and soluble proteins that interact with endometriotic lesions. We analyzed clarified PF samples using a targeted proteomics approach that used roughly the same reagents used in 12Z supernatant profiling experiments, assessing total protein levels using sandwich immunoassays and comparing these to previously reported proteolytic ADAM and MMP activities from the same patient samples (26) (Fig. 8A). Due to the large number of highly correlated measurements in each patient sample, we decomposed the data into an interpretable set of PCs using PCA. The first and third PCs best capture differences between control and disease PF samples (Fig. 8B; fully labeled in SI Appendix, Fig. S17A). Interestingly, disease samples fall into two distinct clusters in PC space, with one cluster defined by relatively high levels of ADAM-10 activity and high concentrations of ADAM-10 substrates including EGF, AREG, HER2, and HER4. In agreement with our in vitro finding that AREG is a substrate of ADAM-10 (Fig. 4), we observed significant correlation between ADAM-10 activity and concentrations of HER2 and AREG in the PF samples (SI Appendix, Fig. S17B). In contrast to the high ADAM-10 cluster of disease samples, the second cluster of disease samples exhibits relatively low ADAM-10 activity, higher levels of ADAM-10 inhibitors (TIMPs), and higher levels of ADAM-9 activity. Of note, ADAM-9 is not inhibited by TIMPs (27). The control samples form a nonoverlapping cluster between the two disease clusters. Although the sample size is small (n = 7 disease samples), PCA results suggest multiple disease states in endometriosis that are defined principally by dysregulation of ADAM-10 activity and corresponding changes in ADAM-10 substrate accumulation.
Fig. 8.

Clinical PF samples suggest dysregulated ErbB-driven ADAM-10 activity. (A) PF samples from patients with and without endometriosis were profiled for protein levels (bead immunoassay) and protease activities (PrAMA). (B) PCA scores and loading plot decomposes patient samples along PCs of covariation and separates samples into three nonoverlapping clusters in an unsupervised manner (fully labeled in SI-17A). (C) The ratio of MET to TIMP1 increases with disease (*P = 0.014, Wilcoxon rank-sum).
We then used supervised partial least squares discriminant analysis (PLS-DA) to classify patient samples as falling into one of the three patient clusters based on a minimal number of protein measurements. PLS-DA shows that combined measurement of ADAM-9 activity and three ADAM-10 substrates (HER2, AREG, and HBEGF) can sufficiently classify patients with high (>95%) accuracy (SI Appendix, Fig. S17 C and D). We also used PLS-DA to classify patient samples into just two groups, disease and control, and observed that combined measurements of MMP-2 activity along with MET and TIMP1 levels sufficiently classify samples as either disease or control with high (>95%) accuracy (SI Appendix, Fig. S17 E and F). We analyzed the simple ratio of MET to TIMP1 levels for a more interpretable result, and observed a significant increase with disease (Fig. 8C). To identify the likely cellular source of increased MET shedding in the PF samples, we analyzed various cell populations from healthy and endometriotic patients, including eutopic endometrial fibroblasts and PF mononuclear cells (PFMCs). Compared with 12Z, PFMCs shed only 10% the relative levels of MET (SI Appendix, Fig. S18A). In contrast, endometrial fibroblasts, which generally express significant MET (28), shed similar levels of MET as 12Z (SI Appendix, Fig. S18B). Experiments with primary endometrial fibroblasts demonstrate that EGF stimulates dual EGF ligand and MET shedding in other relevant endometrial cell populations (SI Appendix, Fig. S18 B and C). Therefore, both endometriotic lesions and endometrial fibroblasts represent significant sources of total MET observed in the PF, particularly in those patients with elevated ErbB ligand present. Consequently, the ratio of MET to TIMP1 may be a good surrogate marker of ADAM-10 activity on endometrial and endometriotic tissue. Overall, these results suggest that joint dysregulation of ADAM-10 activity, ErbB signaling, and corresponding RTK shedding play an important role in disease progression.
Discussion
Understanding systematic regulation of ectodomain shedding has been challenging to accomplish on a component-by-component basis for multiple reasons. The web of protease–substrate interactions involves significant overlap and cross-talk: proteases (i) degrade potentially hundreds of often shared substrates (29), (ii) interact with and regulate each other through direct proteolysis (30), and (iii) respond to and modulate signaling pathways (18). These multiple layers of complexity compel quantitative and multivariate approaches, and here we use integrative experimental/computational methodologies to understand how ADAM sheddases interact with signaling networks to direct overall cellular behavior.
Network-Level Insights into Sheddase Regulation of Cell Migration.
In this work, we combine quantitative experimental measurements with network-inference methods to build computational models of signaling-mediated protease regulation and motility. The CSR approach successfully identifies canonical biochemical interactions, for example between Jnk and c-Jun (SI Appendix, Fig. S8), while simultaneously providing unique insight into mechanisms of sheddase regulation. Shedding is generally considered a function of both protease catalytic activity and substrate availability, yet the balance of protease and substrate regulation remains unclear (19). Here we directly assess proteolytic activity in a relatively “substrate-independent” manner using PrAMA, and find close correlation between proteolysis of both soluble FRET peptides and multiple endogenous membrane-bound ligands and receptors. Moreover, these measurements best described cell migration among all other measurements in the CSR dataset, including phospho-signaling responses proximal to the growth-factor receptors that were being stimulated. Overall, these results (i) provide evidence for significant regulation of the sheddases themselves, (ii) clearly underscore how joint ligand and RTK shedding are concomitantly controlled, and (iii) suggest a prominent role for ectodomain shedding in governing cell migration.
Although we found strong correlations among multiple substrate shedding reactions, we also identified ample evidence that broad patterns of sheddase activity are governed by more than just a single regulatory pathway. This could be seen, for example, by the marked differences in surface-level changes among the various ligands and receptors, as they varied across the growth factor treatments (Fig. 2B). Reflecting this observation, predictive modeling of substrate shedding and motility required multiple descriptors and PCs to achieve sufficient accuracy. Furthermore, CSR modeling results emphasize that sheddase regulation is a dynamic process. For example, PCA and correlation-network results pointed to modularity within the CSR dataset, characterized by early phospho-signaling events linked to ectodomain shedding primarily through ADAM-17 phosphorylation (SI Appendix, Fig. S8). Although complex, the network-inference results nevertheless converge upon AREG and MET as key regulators of cell migration, where they are defined as central components in predictive models of motility (Fig. 2D).
Based on computational modeling results, we investigated a mechanism of shedding defined by the coordinated, context-specific action of both ADAM-10 and -17. Multiple computational and experimental results suggested that EGF and TGFa primarily stimulate ADAM-10 activity (Fig. 4A) and lead to the down-regulation of ADAM-17 surface levels within 30 min of treatment (SI Appendix, Fig. S4). Surprisingly, these results also suggested that EGF-induced AREG shedding may be occurring through the activity of ADAM-10, even though AREG has traditionally been considered an ADAM-17 substrate. We confirmed that ADAM-10 had the potential to cleave AREG using recombinant protease (SI Appendix, Fig. S14 D and F), and also found that ADAM-10 inhibition affected AREG shedding to a much greater degree under EGF treatment conditions (Fig. 4 B–E). Furthermore, siRNA knockdown of ADAM-10 and -17 confirm the dual dependency of multiple other substrates on both ADAM-10 and -17 activities, in agreement with previous work (31). Overall, these results demonstrate how sheddases dynamically interact with multiple signaling pathways to govern overlapping ectodomain shedding events, and emphasize the difficulty in selectively manipulating the proteolysis of specific substrates through kinase and protease inhibitors.
Implications of RTK Ectodomain Shedding in Modulating Drug Response.
Although sheddase involvement in ErbB ligand shedding makes them compelling drug targets in ErbB-driven disease, the biological consequences of ADAM-10 and -17–mediated RTK shedding continue to be poorly understood. In HER2+ breast cancer, ADAM-10 inhibition reduces HER2 shedding, which generally has been described as beneficially limiting the accumulation of the membrane-bound HER2 fragment (p95HER2) that remains after ectodomain proteolysis (32). However, it remains unclear how p95HER2 activity compares to full-length HER2, especially after ligand stimulation. Furthermore, soluble HER2 ectodomain has been shown to inhibit signaling (33). For other RTKs including HER4 and MET, shedding likely reduces RTK signaling at the cell surface (34, 35). TIMP1 inhibition of MET shedding in breast cancer enhances MET signaling and increases liver metastasis (36). In this work we demonstrate that cellular motility is an integrative process that depends not just on AREG shedding, but also on the combined and quantitative effect of multiple proteolytic reactions, including RTK shedding. We find that ADAM-10 and -17–mediated receptor shedding down-regulates HER2, HER4, and MET signaling (Fig. 5). Reduced sheddase activity and RTK cleavage, either through metalloproteinase inhibition (Fig. 5) or indirectly through signaling pathway inhibition (Fig. 6), leads to accumulation of intact RTKs on the cell surface. RTK accumulation potentiates the signaling response to HGF and NRG1b, and causes enhanced RTK phosphorylation (Figs. 5B and 6C) and downstream activation of Jnk and p38 (Fig. 5 C–F). Consequently, Mek and PI3K inhibitors actually enhance the motile response of endometriotic cells to NRG1b and HGF treatment by inhibiting RTK shedding while failing to block the compensatory p38 and Jnk activity that results from signaling of accumulated RTKs (Fig. 6 D and E). Previous studies implicate Jnk and p38 in endometriosis (37, 38), and our results show that Jnk and p38 inhibitors effectively reduce ADAM activity while also blocking the compensatory signaling and motility regardless of the growth factor environment (Fig. 6 D and E; SI Appendix, Fig. S16J). Overall, these results have significant implications for the design of combination therapies involving the numerous signaling pathways that affect ADAM activity, and complement previous studies that stress the importance of Jnk/p38 pathways in cell migration (39).
The emergence of secondary resistance to targeted kinase inhibition represents a major obstacle in developing successful therapeutics, and in this work we identify a unique sheddase-mediated mechanism of rapidly acquired inhibitor resistance that has potential applications for a variety of kinase and protease inhibitor therapies. In the context of breast cancer, secondary resistance to Mek inhibitors has been well documented and arises from up-regulation of RTKs that are known sheddase substrates, including PDGFRb, MET, and AXL (40). Furthermore, the presence of growth factors that activate known ADAM substrate RTKs, for example MET, facilitates the emergence of resistant populations (12). Consistent with these results, here we present that Mek inhibitor resistance arises through multiple up-regulated RTKs, many of which have been implicated in other reports including MET and HER2. In this work we demonstrate that sheddases play a role in the acute up-regulation of receptor levels, and this is particularly relevant in the presence of growth factors that have been previously implicated as prosurvival and promigration microenvironmental cues (12, 41, 42). In endometriosis, kinase inhibitors are in the earlier stages of testing and acquired inhibitor resistance is not yet a clear problem. Nonetheless, we demonstrate that the logic of combination therapies can be successful in our in vitro model for overcoming compensatory signaling pathways that arise secondarily from inhibitor treatment.
Clinical Evidence of Dysregulated Sheddase Activity and Therapeutic Implications.
Analysis of clinical samples from endometriosis patients helped demonstrate the relevance and inherent overlap of sheddase-mediated proteolysis and RTK signaling dysregulation in disease progression. Although many previous studies have examined ErbB signaling and metalloproteinase levels individually (SI Appendix, Table 1), here we present a multivariate analysis of systemic interaction between ErbB ligands, RTK shedding, and metalloproteinase dysregulation. Furthermore, we use measurements from a recently developed microfluidic device to analyze protease activity directly and relate these observations to corresponding protease substrate levels observed in the same patient sample (26). Clinical results confirm many of the observations made in vitro, for example demonstrating significant correlation between ADAM-10 activity and accumulation of known ADAM-10 substrates such as HER2, EGF, and AREG (SI Appendix, Fig. S17). This clinical correlation supports in vitro evidence that AREG shedding is sustained through a positive feedback loop involving ADAM-10 activity, EGFR signaling, and multiple cell types including endometriotic epithelium (12Z), endometrial fibroblasts, and PFMCs (SI Appendix, Fig. S18). Furthermore, this positive feedback loop drives persistent cellular migration and enhances cellular sensitivity to various kinase inhibitors in vitro. Interestingly, we find that disease PF samples comprise two distinct clusters defined in large part by the balance between ADAM-9 and ADAM-10 activities (Fig. 8B). ADAM-10 is a known ADAM-9 substrate, and ADAM-9 has been observed to down-regulate ADAM-10 activity on the cell surface (30). Common among both clusters of disease samples, however, was the observation that the ratio of MET shedding to TIMP1 concentration increased with endometriosis, thereby confirming the relevance of MET signaling in designing therapeutic strategies that may impact ADAM-10 activity (Fig. 8C). Previous work has shown TIMP1 to inhibit the establishment of endometriosis in a mouse model, and these effects were primarily assumed to be MMP-related (43). However, in this work we demonstrate the critical role of ADAM-10 in mediating in vitro cellular migration, and our clinical evidence associates TIMP1 with ADAM-10 activity via its relation with MET shedding.
Conclusions.
We have presented an integrative paradigm for analyzing how complex networks of protease activities work in concert with signaling pathways to influence overall cell response to various disease-relevant environments and therapeutic interventions. In the future, we anticipate that this approach may be useful to explore other facets of sheddase regulation (such as osmotic stress), cell phenotypes (including proliferation and apoptosis), and sheddase-related diseases (such as breast cancer). In this work, we found that ADAM-10 and -17 tune cellular signaling by concomitantly shedding ligand and receptor ectodomains from the cell surface, and we demonstrate here how this competing signaling feedback determines context-dependent cell migration and drug response (Fig. 9).
Fig. 9.
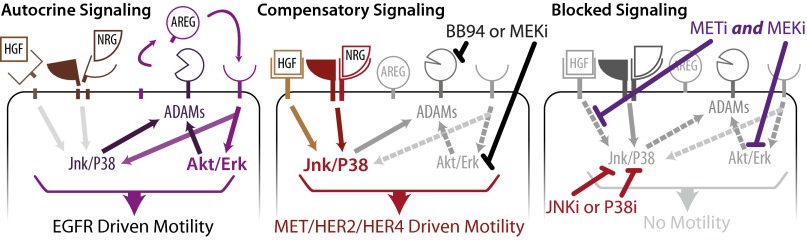
Ectodomain shedding exerts pro- and antimigratory effects depending on context. (Left) EGFR drives motility in an Erk/Akt-dependent manner. High sheddase activity leads to HER2, HER4, and MET shedding along with concomitant positive feedback via AREG release. (Center) Suppression of sheddase activity directly or indirectly via Mek inhibition confers reduced RTK shedding and enhanced response to NRG and HGF through Jnk and p38. (Right) p38, Jnk, or combination Mek/MET inhibition blocks motility.
Full appreciation of the many competing roles of sheddase activity will be essential for understanding their function in development and disease, and has wide implications for designing therapeutic strategies in a broad range of pathologies.
Materials and Methods
Full description of the materials used in this work can be found in SI Appendix, Section S19. Briefly, the 12Z cell line was generously provided by Anna Starzinski-Powitz (University of Frankfurt, Frankfurt, Germany) by way of Steve Palmer (EMD Serono). The 12Z were serum-starved for 4 h before all experiments, with the exception of cell migration experiments, which were performed in serum. Throughout the article, phospho-protein and supernatant protein measurements were made using commercially available sandwich immunoassays (often Luminex bead-based) from Bio-Rad, EMD4Biosciences, Millipore, and R&D Systems, with the exception of measurements of MET phosphorylation and ADAM-17 phosphorylation by Western blot. Throughout the article, all experiments were performed at least twice, from separate biological samples, and all reported error bars indicate SEM unless otherwise stated.
For live-cell 3D migration assessment, cells were labeled with a cell-tracker dye (CMPTX; Invitrogen), mixed with DMEM + 2.2 mg/mL pH-neutralized collagen-I (BD Biosciences) at 500,000 cells per mL, placed in a glass-bottom multiwell plate (MatTek), polymerized for 30 min at 37 °C, and then overlaid with full serum media overnight. Cells were stimulated 4 h before imaging on an environment-controlled Nikon TE2000 microscope. Image stacks of 70-3 μm slices were obtained every 60 min for 16 h. Cells were tracked using Bitplane Imaris. Each of the cell tracks was fit to a persistent random walk model to calculate the random motility coefficient (44). For endpoint migration assays and 3D shedding assays, unlabeled cells were mixed with DMEM plus collagen on ice, immediately placed in a standard 96-well tissue culture plate, spun for 5min at 300 × g, and polymerized for 30 min at 37 °C. Gels were then bathed in 50 μL full serum media containing inhibitors for 60 min, followed by the addition of 50 μL full serum media containing growth factors. After 24 h incubation, supernatant was collected, clarified, and frozen for later analysis. Gels were fixed with 1% paraformaldehyde, stained with YO-PRO-1 (Invitrogen), and imaged at 5× with a CARVII confocal imager (BD) every 3 μm. Images were analyzed using a modified spot finding algorithm (45) in Matlab (Mathworks).
Supplementary Material
Acknowledgments
We thank Andreas Herrlich (Harvard Medical School, Boston, MA), Steve Palmer, and Anna Starzinski-Powitz for reagents. We thank Marcia Moss and Fred Rasmussen (Biozyme, Inc., Apex, NC) for reagents and advice. We acknowledge the Swanson Biotechnology Center for use of microscopy and flow cytometry facilities. We acknowledge National Institutes of Health Grants R01-EB10246 and U54-CA112967 for funding support, along with the Koch Institute Frontier Research Program Initiator Award and the Repligen Fellowship in Cancer Research (to A.S.M.).
Footnotes
The authors declare no conflict of interest.
*This Direct Submission article had a prearranged editor.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1222387110/-/DCSupplemental.
References
- 1.Blobel CP. ADAMs: Key components in EGFR signalling and development. Nat Rev Mol Cell Biol. 2005;6(1):32–43. doi: 10.1038/nrm1548. [DOI] [PubMed] [Google Scholar]
- 2.Morrison CJ, Butler GS, Rodríguez D, Overall CM. Matrix metalloproteinase proteomics: Substrates, targets, and therapy. Curr Opin Cell Biol. 2009;21(5):645–653. doi: 10.1016/j.ceb.2009.06.006. [DOI] [PubMed] [Google Scholar]
- 3.Fingleton B. MMPs as therapeutic targets—Still a viable option? Semin Cell Dev Biol. 2008;19(1):61–68. doi: 10.1016/j.semcdb.2007.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Melin A, Sparén P, Bergqvist A. The risk of cancer and the role of parity among women with endometriosis. Hum Reprod. 2007;22(11):3021–3026. doi: 10.1093/humrep/dem209. [DOI] [PubMed] [Google Scholar]
- 5.Ozkan S, Murk W, Arici A. Endometriosis and infertility: Epidemiology and evidence-based treatments. Ann N Y Acad Sci. 2008;1127:92–100. doi: 10.1196/annals.1434.007. [DOI] [PubMed] [Google Scholar]
- 6.Vercellini P, et al. Post-operative endometriosis recurrence: A plea for prevention based on pathogenetic, epidemiological and clinical evidence. Reprod Biomed Online. 2010;21(2):259–265. doi: 10.1016/j.rbmo.2010.04.018. [DOI] [PubMed] [Google Scholar]
- 7.Uzan C, et al. Status of HER1 and HER2 in peritoneal, ovarian and colorectal endometriosis and ovarian endometrioid adenocarcinoma. Virchows Arch. 2009;454(5):525–529. doi: 10.1007/s00428-009-0755-5. [DOI] [PubMed] [Google Scholar]
- 8.Inagaki M, et al. Association study between epidermal growth factor receptor and epidermal growth factor polymorphisms and endometriosis in a Japanese population. Gynecol Endocrinol. 2007;23(8):474–478. doi: 10.1080/09513590701521057. [DOI] [PubMed] [Google Scholar]
- 9.Ngô C, et al. Protein kinase inhibitors can control the progression of endometriosis in vitro and in vivo. J Pathol. 2010;222(2):148–157. doi: 10.1002/path.2756. [DOI] [PubMed] [Google Scholar]
- 10.Hoeflich KP, et al. In vivo antitumor activity of MEK and phosphatidylinositol 3-kinase inhibitors in basal-like breast cancer models. Clin Cancer Res. 2009;15(14):4649–4664. doi: 10.1158/1078-0432.CCR-09-0317. [DOI] [PubMed] [Google Scholar]
- 11.Yoshino O, et al. FR 167653, a p38 mitogen-activated protein kinase inhibitor, suppresses the development of endometriosis in a murine model. J Reprod Immunol. 2006;72(1-2):85–93. doi: 10.1016/j.jri.2005.02.004. [DOI] [PubMed] [Google Scholar]
- 12.Turke AB, et al. Preexistence and clonal selection of MET amplification in EGFR mutant NSCLC. Cancer Cell. 2010;17(1):77–88. doi: 10.1016/j.ccr.2009.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Muranen T, et al. Inhibition of PI3K/mTOR leads to adaptive resistance in matrix-attached cancer cells. Cancer Cell. 2012;21(2):227–239. doi: 10.1016/j.ccr.2011.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lee MJ, et al. Sequential application of anticancer drugs enhances cell death by rewiring apoptotic signaling networks. Cell. 2012;149(4):780–794. doi: 10.1016/j.cell.2012.03.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Miller-Jensen K, Janes KA, Brugge JS, Lauffenburger DA. Common effector processing mediates cell-specific responses to stimuli. Nature. 2007;448(7153):604–608. doi: 10.1038/nature06001. [DOI] [PubMed] [Google Scholar]
- 16.Zeitvogel A, Baumann R, Starzinski-Powitz A. Identification of an invasive, N-cadherin-expressing epithelial cell type in endometriosis using a new cell culture model. Am J Pathol. 2001;159(5):1839–1852. doi: 10.1016/S0002-9440(10)63030-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Miller MA, et al. Proteolytic Activity Matrix Analysis (PrAMA) for simultaneous determination of multiple protease activities. Integr Biol (Camb) 2011;3(4):422–438. doi: 10.1039/c0ib00083c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Xu P, Derynck R. Direct activation of TACE-mediated ectodomain shedding by p38 MAP kinase regulates EGF receptor-dependent cell proliferation. Mol Cell. 2010;37(4):551–566. doi: 10.1016/j.molcel.2010.01.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dang M, et al. Epidermal growth factor (EGF) ligand release by substrate-specific A Disintegrin and Metalloproteases (ADAMs) involves different protein kinase C (PKC) isoenzymes depending on the stimulus. J Biol Chem. 2011;286(20):17704–17713. doi: 10.1074/jbc.M110.187823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Meyer AS, et al. 2D protrusion but not motility predicts growth factor-induced cancer cell migration in 3D collagen. J Cell Biol. 2012;197(6):721–729. doi: 10.1083/jcb.201201003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shoyab M, Plowman GD, McDonald VL, Bradley JG, Todaro GJ. Structure and function of human amphiregulin: A member of the epidermal growth factor family. Science. 1989;243(4894 Pt 1):1074–1076. doi: 10.1126/science.2466334. [DOI] [PubMed] [Google Scholar]
- 22.Joslin EJ, et al. Structure of the EGF receptor transactivation circuit integrates multiple signals with cell context. Mol Biosyst. 2010;6(7):1293–1306. doi: 10.1039/c003921g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dang M, et al. 2013. Proteolytic release of life-essential growth factors is regulated by substrate-selecting signaling pathways, not by inducing protease activity Proc Natl Acad Sci USA, 10.1073/pnas.1307478110.
- 24.Simpson KJ, et al. Identification of genes that regulate epithelial cell migration using an siRNA screening approach. Nat Cell Biol. 2008;10(9):1027–1038. doi: 10.1038/ncb1762. [DOI] [PubMed] [Google Scholar]
- 25.Maretzky T, et al. Migration of growth factor-stimulated epithelial and endothelial cells depends on EGFR transactivation by ADAM17. Nat Commun. 2011;2:229. doi: 10.1038/ncomms1232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chen CH, et al. 2012. A multiplexed bioassay using droplet-based microfluidics for protease activity in clinical samples: application to endometriosis J Am Chem Soc 135(5):1645–8.
- 27.Amour A, et al. The enzymatic activity of ADAM8 and ADAM9 is not regulated by TIMPs. FEBS Lett. 2002;524(1-3):154–158. doi: 10.1016/s0014-5793(02)03047-8. [DOI] [PubMed] [Google Scholar]
- 28.Yoshida S, et al. Hepatocyte growth factor/Met system promotes endometrial and endometriotic stromal cell invasion via autocrine and paracrine pathways. J Clin Endocrinol Metab. 2004;89(2):823–832. doi: 10.1210/jc.2003-030874. [DOI] [PubMed] [Google Scholar]
- 29.Butler GS, Dean RA, Morrison CJ, Overall CM. Identification of cellular MMP substrates using quantitative proteomics: Isotope-coded affinity tags (ICAT) and isobaric tags for relative and absolute quantification (iTRAQ) Methods Mol Biol. 2010;622:451–470. doi: 10.1007/978-1-60327-299-5_26. [DOI] [PubMed] [Google Scholar]
- 30.Moss ML, et al. ADAM9 inhibition increases membrane activity of ADAM10 and controls α-secretase processing of amyloid precursor protein. J Biol Chem. 2011;286(47):40443–40451. doi: 10.1074/jbc.M111.280495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Le Gall SM, et al. ADAMs 10 and 17 represent differentially regulated components of a general shedding machinery for membrane proteins such as transforming growth factor alpha, L-selectin, and tumor necrosis factor alpha. Mol Biol Cell. 2009;20(6):1785–1794. doi: 10.1091/mbc.E08-11-1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Xia W, Liu L-H, Ho P, Spector NL. Truncated ErbB2 receptor (p95ErbB2) is regulated by heregulin through heterodimer formation with ErbB3 yet remains sensitive to the dual EGFR/ErbB2 kinase inhibitor GW572016. Oncogene. 2004;23(3):646–653. doi: 10.1038/sj.onc.1207166. [DOI] [PubMed] [Google Scholar]
- 33.Ghedini GC, et al. Shed HER2 extracellular domain in HER2-mediated tumor growth and in trastuzumab susceptibility. J Cell Physiol. 2010;225(1):256–265. doi: 10.1002/jcp.22257. [DOI] [PubMed] [Google Scholar]
- 34.Naresh A, et al. The ERBB4/HER4 intracellular domain 4ICD is a BH3-only protein promoting apoptosis of breast cancer cells. Cancer Res. 2006;66(12):6412–6420. doi: 10.1158/0008-5472.CAN-05-2368. [DOI] [PubMed] [Google Scholar]
- 35.Foveau B, et al. Down-regulation of the met receptor tyrosine kinase by presenilin-dependent regulated intramembrane proteolysis. Mol Biol Cell. 2009;20(9):2495–2507. doi: 10.1091/mbc.E08-09-0969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schelter F, et al. Tumor cell-derived Timp-1 is necessary for maintaining metastasis-promoting Met-signaling via inhibition of Adam-10. Clin Exp Metastasis. 2011;28(8):793–802. doi: 10.1007/s10585-011-9410-z. [DOI] [PubMed] [Google Scholar]
- 37.Yoshino O, et al. Possible pathophysiological roles of mitogen-activated protein kinases (MAPKs) in endometriosis. Am J Reprod Immunol. 2004;52(5):306–311. doi: 10.1111/j.1600-0897.2004.00231.x. [DOI] [PubMed] [Google Scholar]
- 38.Wagner EF, Nebreda ÁR. Signal integration by JNK and p38 MAPK pathways in cancer development. Nat Rev Cancer. 2009;9(8):537–549. doi: 10.1038/nrc2694. [DOI] [PubMed] [Google Scholar]
- 39.Huang C, Rajfur Z, Borchers C, Schaller MD, Jacobson K. JNK phosphorylates paxillin and regulates cell migration. Nature. 2003;424(6945):219–223. doi: 10.1038/nature01745. [DOI] [PubMed] [Google Scholar]
- 40.Duncan JS, et al. Dynamic reprogramming of the kinome in response to targeted MEK inhibition in triple-negative breast cancer. Cell. 2012;149(2):307–321. doi: 10.1016/j.cell.2012.02.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wilson TR, et al. Widespread potential for growth-factor-driven resistance to anticancer kinase inhibitors. Nature. 2012;487(7408):505–509. doi: 10.1038/nature11249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Goswami S, et al. Macrophages promote the invasion of breast carcinoma cells via a colony-stimulating factor-1/epidermal growth factor paracrine loop. Cancer Res. 2005;65(12):5278–5283. doi: 10.1158/0008-5472.CAN-04-1853. [DOI] [PubMed] [Google Scholar]
- 43.Bruner KL, Matrisian LM, Rodgers WH, Gorstein F, Osteen KG. Suppression of matrix metalloproteinases inhibits establishment of ectopic lesions by human endometrium in nude mice. J Clin Invest. 1997;99(12):2851–2857. doi: 10.1172/JCI119478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kim HD, et al. Epidermal growth factor-induced enhancement of glioblastoma cell migration in 3D arises from an intrinsic increase in speed but an extrinsic matrix- and proteolysis-dependent increase in persistence. Mol Biol Cell. 2008;19(10):4249–4259. doi: 10.1091/mbc.E08-05-0501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Santella A, Du Z, Nowotschin S, Hadjantonakis AK, Bao Z. A hybrid blob-slice model for accurate and efficient detection of fluorescence labeled nuclei in 3D. BMC Bioinformatics. 2010;11:580. doi: 10.1186/1471-2105-11-580. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.