

Abstract
Bacterial RNA degradation often begins with conversion of the 5′-terminal triphosphate to a monophosphate, creating a better substrate for subsequent ribonuclease digestion. For example, in Bacillus subtilis and related organisms, removal of the gamma and beta phosphates of primary transcripts by the RNA pyrophosphohydrolase RppH triggers rapid 5′-exonucleolytic degradation by RNase J. However, the basis for the selective targeting of a subset of cellular RNAs by this pathway has remained largely unknown. Here we report that purified B. subtilis RppH requires at least two unpaired nucleotides at the 5′ end of its RNA substrates and prefers three or more. The second of these 5′-terminal nucleotides must be G, whereas a less strict preference for a purine is evident at the third position, and A is slightly favored over G at the first position. The same sequence requirements are observed for RppH-dependent mRNA degradation in B. subtilis cells. By contrast, a parallel pathway for 5′-end–dependent RNA degradation in that species appears to involve an alternative phosphate-removing enzyme that is relatively insensitive to sequence variation at the first three positions.
Keywords: BsRppH, YtkD, yhxA, glpP
Of the mechanisms that control gene expression in all living organisms, mRNA degradation is among the least well understood. Within the same cell, the half-lives of distinct mRNAs can differ by up to two orders of magnitude (seconds to an hour in bacteria, minutes to more than a day in higher eukaryotes), with proportionate effects on mRNA levels (1). In addition, the lifetimes of many messages can be modulated in response to environmental signals.
Although it was originally assumed that Escherichia coli could serve as a paradigm for mRNA degradation in all bacteria, it is now clear that many bacterial species use a different set of ribonucleases and distinct mechanisms to degrade mRNA. A gamma proteobacterium, E. coli generally relies on the essential endonuclease RNase E to cleave transcripts internally, generating RNA fragments that are then degraded to mononucleotides by a combination of further RNase E cleavage and 3′-exonuclease digestion (1). Remarkably, despite its crucial role in E. coli, RNase E is entirely absent from a large number of other bacteria, including many Gram-positive species and even some Gram-negative proteobacteria (2). Instead, those species generally contain one or more ribonucleases that are absent from E. coli. For example, Bacillus subtilis, Staphylococcus aureus, and Helicobacter pylori contain two such ribonucleases—RNase Y, a membrane-associated endonuclease, and RNase J, a 5′-monophosphate–dependent 5′ exonuclease that is also capable of acting as an endonuclease—in addition to a number of 3′ exonucleases (2–6).
Because primary transcripts in B. subtilis are protected from exonucleolytic degradation by their 5′-terminal triphosphate and 3′-terminal stem loop, it was initially believed that message degradation in that species bypasses those termini and begins with endonucleolytic cleavage, generating RNA fragments with unprotected ends that would be susceptible to rapid exonuclease attack. However, we have recently described an alternative degradation pathway in B. subtilis in which the decay of primary transcripts is triggered by the RNA pyrophosphohydrolase RppH (previously known as YtkD), a Nudix hydrolase that removes two of the three 5′-terminal phosphates to generate a full-length intermediate that is monophosphorylated and therefore vulnerable to rapid 5′-exonucleolytic digestion by RNase J (7). A related 5′-end–dependent pathway is also present in bacterial species that lack RNase J, such as E. coli, where pyrophosphate removal triggers rapid cleavage by RNase E, the endonucleolytic activity of which is stimulated by a 5′ monophosphate (8–10).
Among the B. subtilis mRNAs targeted by the RppH-dependent pathway is the 2.2-kb transcript of the yhxA-glpP operon, which encodes a transcription antiterminator (GlpP) important for regulating the synthesis of glycerol-3-phosphate dehydrogenase (11). The lifetime of this transcript can be prolonged in B. subtilis by deleting the rppH gene, by depleting the 5′-exonuclease subunit of RNase J, or by adding a 5′-terminal stem loop, an RNA modification that prevents 5′ attack by both RppH (7) and RNase J (4). However, apart from an unpaired 5′ end, nothing was known about the features of transcripts like yhxA-glpP that cause them to be targeted by RppH.
We have now investigated the substrate specificity of B. subtilis RppH, both in vitro and in vivo. These studies not only have determined the number of unpaired 5′-terminal nucleotides required by this enzyme but also have revealed a pivotal yet heretofore unsuspected influence of their sequence on reactivity. Together, these findings have identified some of the key distinguishing characteristics of mRNAs that are degraded by this important 5′-end–dependent pathway.
Results
Requirement for Unpaired Nucleotides at the 5′ Terminus.
Previous studies have shown that B. subtilis RppH is unable to convert triphosphorylated RNA to monophosphorylated RNA when the 5′ end is base paired (7). To determine the minimum number of unpaired 5′-terminal nucleotides required for phosphate removal by this enzyme, we designed an RNA substrate whose secondary structure was unambiguous. This substrate (A8) comprised an 8-nucleotide single-stranded segment beginning with A and followed by two stem-loop structures (Fig. 1). Each stem was topped by a GNRA loop, the first of which contained the only U in the entire molecule. When synthesized by in vitro transcription in the presence of [γ-32P]ATP and fluorescein-12-UTP, A8 acquired a γ radiolabel within its 5′-terminal triphosphate and a single fluorescein label at the apex of the first stem loop. As an internal standard, we also synthesized a related RNA substrate (A8XL) that differed from A8 only in having an additional stem loop at the 3′ end (Fig. 1).
Fig. 1.

In vitro RNA substrates. The sequence and expected secondary structure of A8, A4, A3, A2, A1, A1+3, G8, and A8XL RNA are shown. Each bore a 5′-terminal triphosphate (ppp), a γ-32P radiolabel (*) at the 5′ end, and a fluorescein label at the top of the first stem loop. Truncated derivatives of A8 (A4, A3, A2, and A1) and G8 (G4, G3, G2, G1, and G0) lacked 4–8 nucleotides from the 3′ boundary of the 5′-terminal single-stranded segment (Fig. S2). In each RNA name, the letter indicates the identity of the 5′-terminal nucleotide, and the numeral indicates the number of unpaired nucleotides at the 5′ end. A1+3 was identical to A1 except for three additional nucleotides at the 3′ end.
We first compared the activity of RppH and RppH-E68Q, a catalytically inactive mutant, by adding equal amounts of each purified protein to doubly labeled A8 and A8XL and monitoring the reactions as a function of time (Fig. S1). As expected, only the wild-type enzyme released radioactivity from these RNA substrates, confirming that it was the sole source of catalytic activity in the purified protein preparation. The reactivity of A8 and A8XL toward RppH was then compared by normalizing the amount of radiolabeled RNA remaining at each time point to the total RNA remaining, as determined from fluorescence intensity. The γ radiolabel was lost from both substrates at a similar rate, demonstrating that the third stem loop of A8XL had a negligible effect on its reactivity.
To ascertain how many unpaired 5′-terminal nucleotides are required by RppH, we truncated the single-stranded segment of A8 from its 3′ boundary to create RNA substrates with only one to four unpaired nucleotides at the 5′ end (A4, A3, A2, and A1; Fig. 1 and Fig. S2). The reactivity of each of these doubly labeled RNAs toward RppH was then compared in the presence of the internal standard A8XL. A4 and A3 underwent phosphate removal nearly as fast as A8, whereas A2 reacted at a noticeably slower rate and A1 was entirely unreactive (Fig. 2 A and B). To prove that the greater reactivity of A4 versus A1 was a consequence of the number of unpaired nucleotides at the 5′ end and not the overall length, we added three unpaired nucleotides to the 3′ end of A1 to create A1+3 (Fig. 1). As expected, A1+3 was no more reactive than A1. These findings demonstrate that RppH requires substrates with at least two unpaired nucleotides at the 5′ terminus and that it prefers three or more.
Fig. 2.

Effect of the length of the 5′-terminal single-stranded segment on reactivity with RppH in vitro. (A) Representative gel images. In vitro transcribed A4 and A1 bearing a γ-32P radiolabel and an internal fluorescein label were mixed with labeled A8XL and treated with purified RppH (8 nM), and the radioactivity (P-32) and fluorescence (Fluor) of each RNA were monitored as a function of time by gel electrophoresis. (B and C) Graphs. RppH-catalyzed phosphate removal from A8, A4, A3, A2, A1, and A1+3 or from G8, G4, G3, G2, G1, and G0 was monitored as in A and quantified by normalizing the radioactivity remaining in each RNA to the corresponding fluorescence intensity. Each time point is the average of two or more independent measurements. Error bars correspond to SDs.
Effect of 5′-Terminal RNA Sequence in Vitro.
The requirement for at least two to three unpaired 5′-terminal nucleotides suggests that RppH likely makes intimate contact with the first two to three nucleotides of its substrates, raising the possibility of sequence preferences at those positions. To begin to examine this possibility, we synthesized a set of RNA substrates identical to A8, A4, A3, A2, and A1 but with G instead of A at the 5′ end (G8, G4, G3, G2, and G1, as well as G0, which had no unpaired nucleotides at the 5′ end; Fig. 1 and Fig. S2) and then monitored the rate of RppH-catalyzed 5′-phosphate removal from each in the presence of A8XL. The relative reactivity of these substrates was the same as for the A series: G8, G4, and G3 were all highly reactive, G2 exhibited intermediate reactivity, and G1 and G0 were completely unreactive (Fig. 2C). Furthermore, similar reaction rates were measured for A4 and G4 and for A8 and G8, the RNAs starting with G being only marginally less reactive than their A-initiated counterparts (Fig. 3A and Fig. S3). These results indicate that, of the two nucleotides usually present at the 5′ end of primary transcripts in bacterial cells (12), RppH has a slight preference for A over G and that this small bias does not affect the number of unpaired 5′-terminal nucleotides required by the enzyme.
Fig. 3.
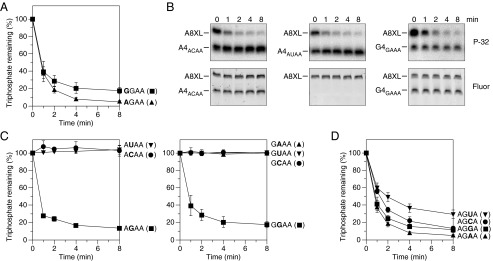
Effect of the sequence of the first three RNA nucleotides on reactivity with RppH in vitro. (A) Position 1. The reactivity of A4AGAA and G4GGAA was compared as in Fig. 2. The subscript in each RNA name indicates the sequence of the four unpaired nucleotides at the 5′ end. Consequently, A4AGAA and G4GGAA were equivalent to A4 and G4, respectively. (B and C) Position 2. The reactivity of A4AGAA, A4ACAA, and A4AUAA and of G4GGAA, G4GAAA, G4GCAA, and G4GUAA was compared. To avoid modifying the second nucleotide, A4AUAA and G4GUAA were not labeled with fluorescein; instead, the fluorescence of fluorescein-labeled A8XL was used to normalize the data from each time point. The synthesis of A4AAAA was not successful. (D) Position 3. The reactivity of A4AGAA, A4AGGA, A4AGCA, and A4AGUA was compared. To avoid modifying the third nucleotide, A4AGUA was not labeled with fluorescein.
We next varied the identity of the second nucleotide of A4 and G4, hereafter renamed A4AGAA and G4GGAA, respectively, to reflect the sequence of unpaired nucleotides at the 5′ terminus. In every case, the effect of these substitutions on phosphate removal was striking. Regardless of the sequence context, replacing the G at position 2 with any other nucleotide (A, C, or U) abolished the reactivity of the substrate (Fig. 3 B and C). We conclude that, under these reaction conditions, B. subtilis RppH can act on RNA substrates only if the second nucleotide is G.
Finally, we changed the identity of the third nucleotide of A4AGAA, replacing the A at that position with G, C, or U. Among these changes, the substitution of U had the greatest effect, significantly slowing but not abolishing phosphate removal by RppH, the substitution of C had a more modest effect (the impediment being most noticeable at early reaction times), and the substitution of G had only a small effect (Fig. 3D).
Effect of 5′-Terminal RNA Sequence in B. subtilis.
To ascertain whether, in its natural milieu, RppH exhibits sequence preferences resembling those observed for the purified protein, we examined its influence on sequence variants of a known mRNA target in B. subtilis. Previously, we have shown that the transcript of the dicistronic yhxA-glpP operon is degraded in B. subtilis by a 5′-end–dependent mechanism in which the 5′-terminal triphosphate is converted by RppH to a monophosphate, thereby exposing the transcript to rapid degradation by the 5′ exonuclease RNase J (7). We have further shown that a truncated version of this transcript (mini yhxA-glpP), comprising the 5′ untranslated region (UTR) and first 34 codons of yhxA fused in-frame to the last eight codons of glpP and the 3′ UTR, decays at the same rate and by an identical mechanism as the full-length yhxA-glpP transcript, irrespective of gene dosage.
The sequence of the yhxA-glpP transcript and its truncated derivative begins 5′-AGG. We changed the identity of each of these three nucleotides in the context of plasmid-encoded mini yhxA-glpP mRNA. None of the sequence changes altered the site of transcription initiation, as judged by RNA ligase-mediated RACE analysis of cellular RNA that had been treated with tobacco acid pyrophosphatase to convert all triphosphates to monophosphates. The half-life of each sequence variant was then compared in B. subtilis cells that contained or lacked RppH by monitoring the rate of disappearance of the mini transcript following the inhibition of transcription with rifampicin. In vivo, RppH had a significant (∼3-fold) destabilizing effect not only on mini yhxA-glpP mRNA but also on the variant in which A was substituted for G at position 3 (3A) (Fig. 4). By contrast, it failed to accelerate the decay of sequence variants bearing any nucleotide other than G at position 2 (2A, 2C, or 2U; 0.9- to 1.1-fold rate enhancement) and had a diminished destabilizing effect when the G at position 3 was replaced with a pyrimidine (3C or 3U; <2-fold rate enhancement) or the A at position 1 was replaced with G (1G; ∼2.2-fold rate enhancement). These results are entirely consistent with the specificity observed for RppH in vitro.
Fig. 4.

Effect of the sequence of the first three RNA nucleotides on RppH-dependent mRNA degradation in B. subtilis. The half-lives of mini yhxA-glpP mRNA and derivatives thereof with a substitution at the first, second, or third position were compared in isogenic B. subtilis cells containing or lacking an rppH gene. The numeral in the name of each variant indicates the nucleotide position (1, 2, or 3), and the letter indicates the nucleotide identity (A, G, C, or U). In every case, the identity of the unspecified nucleotides was the same as in the 5′ UTR of the wild-type transcript: A at position 1, G at position 2, and G at position 3; therefore, 1A, 2G, and 3G are all equivalent to mini yhxA-glpP with a wild-type 5′ UTR. (A) Representative time courses. The decay of mini yhxA-glpP mRNA with G or U at position 2 (2G or 2U) was monitored by Northern blot analysis of RNA extracted from wild-type or ∆rppH cells at time intervals after arresting transcription with rifampicin. Band intensities were plotted semilogarithmically as a function of time, and best-fit lines were calculated by linear regression. (B) Half-lives. Each mRNA half-life is the average of two or more independent measurements. Error bars correspond to SDs.
Promiscuity of an Alternative 5′-End–Dependent Pathway in B. subtilis.
Multiple lines of evidence suggest that, in addition to RppH, B. subtilis contains a second, as yet unidentified RNA pyrophosphohydrolase capable of triggering 5′-end–dependent mRNA degradation (7). Like the RppH-dependent pathway, degradation involving the alternative pyrophosphohydrolase appears to be inhibited by 5′-terminal base pairing, and because the two enzymes contribute additively to the decay rate of mini yhxA-glpP mRNA, appending a stem loop to the 5′ end has a greater stabilizing influence (approximately fivefold) than does simply deleting the rppH gene (approximately threefold) (7). To ascertain whether the mini yhxA-glpP sequence variants that were insensitive to the presence of RppH (2A, 2C, and 2U) remained susceptible to degradation by this parallel pathway, we measured their rate of decay in ∆rppH cells after adding a 5′-terminal stem loop to impede degradation triggered by the other pyrophosphohydrolase. In every case, the half-life of the transcript was prolonged by this modification (Fig. 5). The ability of a 5′ stem loop to stabilize these mutant transcripts in the absence of RppH indicates that the nucleotide substitutions specifically inhibited phosphate removal by RppH without blocking 5′-end–dependent degradation by the alternative pathway.
Fig. 5.

mRNA stabilization by a 5′-terminal stem loop in B. subtilis cells lacking RppH. Total RNA was extracted at time intervals after rifampicin addition to B. subtilis ∆rppH cells that contained a plasmid encoding mini yhxA-glpP mRNA (2G) or a sequence variant (2A, 2C, or 2U), with (+hp) or without (−hp) a 5′-terminal stem loop, and mRNA half-lives were compared. Error bars correspond to SDs.
Discussion
Previous studies have shown that mRNA degradation by a 5′-end–dependent pathway requires a single-stranded 5′ terminus in both B. subtilis and E. coli (7, 10). However, the number of unpaired 5′-terminal nucleotides required for degradation by this mechanism had not been determined in these or any other species. Nor was it realized that the identity of those nucleotides, particularly at internal positions, could influence the rate of 5′-end–dependent decay. Here we have shown that 5′-terminal phosphate removal by purified B. subtilis RppH requires at least two unpaired nucleotides at the 5′ end and prefers three or more. Furthermore, B. subtilis RppH has a marked sequence dependence, requiring G at position 2 of its substrates, preferring a purine at position 3, and slightly favoring an A at position 1, both in vitro and in vivo. The requirements for at least two unpaired 5′-terminal nucleotides and a G at the second position can be explained by the array of amino acid side chains and an ordered water molecule with which that guanine interacts when bound to RppH and by the distance of this binding pocket from the catalytic center, as reported in the companion article by Piton et al. (13).
The RppH substrates compared in vitro were designed so as to minimize the potential for conformational ambiguity that could otherwise cause any differences in reactivity to be misinterpreted. For 17 of the 21 substrates, the expected base-paired conformation was calculated to be at least 15 kcal/mol more stable than any other conformation with fewer unpaired nucleotides at the 5′ end, leaving little doubt as to their secondary structure. Even among the four exceptions, the expected conformation was significantly favored over the next most stable conformation (calculated ∆∆G = 7 kcal/mol for G4GCAA and A4AGCA, 5 kcal/mol for G4GUAA, and 3 kcal/mol for A4AGUA). Despite somewhat less certainty as to the conformation of G4GCAA and G4GUAA, the critical importance of the second nucleotide for their lack of reactivity was corroborated by the equally poor reactivity of their counterparts A4ACAA and A4AUAA. Moreover, the excellent correlation between the effect of 5′-proximal nucleotide substitutions on the reactivity of A4AGAA variants in vitro and the sensitivity of mini yhxA-glpP variants to RppH in B. subtilis supports the conclusion that the observed disparities in reactivity were due to sequence differences and not to unintended conformational changes.
In principle, the specificity of the RppH-dependent pathway for mRNA decay in B. subtilis should be governed by the specificity of both RppH and RNase J, their relative influence being determined by which enzyme has more stringent substrate requirements and possibly also by which step (phosphate removal or subsequent exonucleolytic degradation) is rate limiting. Purified RNase J appears to require at least 9–10 unpaired nucleotides at the 5′ end for maximum 5′-exonuclease activity and 4–5 unpaired nucleotides for exonucleolytic processivity (4, 14), more than the number needed for phosphate removal by RppH. Therefore, the length requirements of RNase J are likely to predominate even though phosphate removal by RppH often appears to be rate determining [as judged from the low steady-state ratio of the monophosphorylated to triphosphorylated forms of transcripts targeted by RppH (7)]. Conversely, whereas RppH has strict sequence requirements, the 5′-exonuclease activity of RNase J exhibits little if any sequence dependence (14), as would be expected for an enzyme that must rapidly degrade a great variety of decay intermediates to mononucleotides. Consequently, the specificity of RppH-dependent mRNA degradation in B. subtilis should be defined by the length requirements of RNase J and the sequence requirements of RppH. Our data have now confirmed that the sequence requirements of this pathway in B. subtilis match those of RppH in vitro. Consistent with this observation, both of the transcripts previously shown to be targeted by B. subtilis RppH (7) have a G at position 2 and a purine at position 3 (yhxA-glpP: 5′-AGG…; ∆ermC: 5′-GGA…). Although the B. subtilis transcriptome has not yet been examined systematically to identify other targets of this enzyme, they too are expected to conform to the strict requirement for a G at the second position.
The results of prior experiments suggest that, in addition to RppH, B. subtilis contains a second enzyme capable of triggering 5′-end–dependent mRNA degradation by converting 5′-terminal triphosphates to monophosphates (7). For example, adding a stem loop to the 5′ end or depleting the exonuclease subunit of RNase J from B. subtilis cells has a greater stabilizing effect on mini yhxA-glpP mRNA than does deleting the rppH gene. Moreover, in ∆rppH cells, the steady-state percentage of yhxA-glpP and ∆ermC transcripts that are monophosphorylated remains one-quarter to one-half as high as in wild-type cells. Finally, extracts prepared from B. subtilis mutants that lack RppH retain ∼30% of the RNA pyrophosphohydrolase activity of wild-type cell extracts. The observation that the already long lifetimes of the 2A, 2U, and 2C mutants of mini yhxA-glpP mRNA could be further prolonged in ∆rppH cells by adding a 5′-terminal stem loop indicates that those messages continued to be susceptible to 5′-end–dependent degradation triggered by the other RNA pyrophosphohydrolase. Furthermore, the similar half-lives of all of the mini yhxA-glpP sequence variants in ∆rppH cells (6–9 min) imply that phosphate removal by that other RNA pyrophosphohydrolase is relatively insensitive to sequence variation at positions 1, 2, and 3. These findings suggest that B. subtilis contains at least two RNA pyrophosphohydrolases, one of which (RppH) requires a G at position 2 and the other of which is sequence independent. This apparent difference in specificity raises the possibility that the overall sequence specificity of 5′-end–dependent mRNA degradation in B. subtilis might vary under different growth conditions, depending upon the relative availability of those two enzymes.
Methods
RNA Synthesis and Purification.
To prepare double-stranded DNA templates for in vitro transcription, pairs of partially complementary oligodeoxynucleotides (20 pmol each; Table S1) were annealed and then extended with the Klenow fragment of DNA polymerase (5 units; NEB) and four deoxyNTPs (1 mM each) in NEB buffer 3 (final volume of 20 µL). The fully double-stranded product was phenol extracted, ethanol precipitated, and dissolved in water. Templates for the synthesis of RNAs beginning with 5′-AG contained a T7 ϕ2.5 promoter (15); all other templates contained a T7 ϕ6.5 promoter.
Doubly labeled A8XL, A8, A4 (A4AGAA), A3, A2, A1, A4AGGA, and A4AGCA were synthesized by combining the corresponding double-stranded DNA template (1 pmol) with ATP, GTP, and CTP (0.1 mM each), [γ-32P]ATP (40 µCi), fluorescein-12-UTP (0.1 mM; Roche), DTT (10 mM), rRNasin (40 units, Promega), and T7 RNA polymerase (100 units; NEB) in NEB T7 RNA polymerase buffer (final volume of 40 µL). After 6 h at 37 °C, the RNA transcripts were phenol extracted, ethanol precipitated, and purified by electrophoresis on a 16% (wt/vol) polyacrylamide/8 M urea gel. Fluorescent bands were excised, eluted overnight at 37 °C in RNA elution buffer (0.3 M sodium acetate pH 5.2, 1 mM EDTA, 1% SDS), and ethanol precipitated. Each RNA pellet was dissolved in water (40 µL) and stored at –20 °C.
A mutant form of T7 RNA polymerase (T7RNAP-A6-36.4), bearing a hexahistidine tag and several amino acid substitutions (K93T A136T S228G S397R S684Y) (16), was used instead of T7 RNA polymerase for the synthesis of A4ACAA and A4AUAA. [γ-32P]GTP (60 µCi) was used instead of [γ-32P]ATP for the synthesis of G8, G4 (G4GGAA), G3, G2, G1, G0, G4GAAA, G4GCAA, and G4GUAA. UTP (0.1 mM) was used instead of fluorescein-12-UTP for the synthesis of A4AUAA, A4AGUA, and G4GUAA.
RNA secondary structure was predicted by using the mfold Web server (17).
Purification of RppH.
Wild-type and catalytically inactive forms of B. subtilis RppH bearing an amino-terminal hexahistidine tag were produced in E. coli strain BL21(DE3) ∆rppH ∆rna containing plasmid pPlac-BsRppH or pPlac-BsRppH-E68Q. Protein synthesis was induced by adding isopropyl β-D-1-thiogalactopyranoside (1 mM) to a log-phase culture (A600 = 0.5) growing in LB medium (1,000 mL) at 37 °C. After 3 h, the cells were pelleted, resuspended in 10 mL of buffer E [10 mM Hepes pH 7.5, 0.3 M sodium chloride, 0.25% Genapol, 0.1 mM PMSF, and Complete EDTA-free protease inhibitor (Roche)], and disrupted in an EmulsiFlex-C3 high pressure homogenizer (Avestin). The cell lysates were cleared twice by centrifugation at 14,500 × g for 15 min at 4 °C, and the supernatants were incubated with 2 mL of BD TALON metal affinity resin (Clontech) for 2 h at 4 °C with gentle agitation. The resin was washed five times with buffer E containing 0–20 mM imidazole, and RppH or RppH-E68Q was then eluted with buffer E containing 500 mM imidazole. The proteins were further purified by size exclusion chromatography on a HiPrep 16/60 Sephacryl S-200 HR column (GE Healthcare) equilibrated and eluted with buffer E. Peak fractions were pooled, concentrated to 1–10 µM by ultrafiltration in an Amicon Ultra-4 centrifugal filter device (molecular weight cut-off: 10,000; Millipore), and stored at –80 °C in buffer E containing 25% (vol/vol) glycerol.
Monitoring Pyrophosphate Removal by RppH in Vitro.
Equal amounts of the 5′ γ-32P–labeled RNA to be tested and 5′ γ-32P–labeled A8XL (0.4 pmol each) were prewarmed to 37 °C for 3 min in B. subtilis RppH (BsRppH) reaction buffer (20 mM Hepes pH 7.5, 5 mM magnesium chloride, 1 mM DTT, 1% glycerol; total volume of 99 µL). A 0-min sample (9 µL) was removed, mixed with 8 µL of loading buffer [90 mM EDTA, 82% (vol/vol) formamide], and cooled on ice. RppH (10 µL of an 80 nM solution in BsRppH reaction buffer) was added to the remainder (final RppH concentration of 8 nM), and 10 µL samples were withdrawn at time intervals (1, 2, 4, and 8 min) and quenched on ice with 8 µL of loading buffer. (Under these conditions, the reaction rate is limited by the concentration of RppH.) All of the samples were then heated to 95 °C for 3 min and subjected to electrophoresis on a 13.5% (wt/vol) polyacrylamide/8 M urea gel. After visualizing the fluorescence of the RNA bands with a Typhoon Trio scanner (GE Healthcare), the gel was dried, and the radioactivity of the bands was visualized with a Storm 820 PhosphorImager (Molecular Dynamics). Band intensities were quantified by using ImageQuant software, and radioactivity was normalized to fluorescence.
Measurement of mRNA Lifetimes in B. subtilis.
mRNA half-life measurements were performed in B. subtilis strain 168 (18) or a derivative in which the rppH gene had been deleted and replaced with a spectinomycin resistance gene (7). Mini yhxA-glpP mRNA and mutants bearing 5′-proximal nucleotide substitutions and/or a 5′-terminal stem loop (AGGGCCGAAGCUUCGGCCCU) were encoded by plasmid pBYGmini (7) and derivatives thereof.
B. subtilis cells were grown to midlog phase at 37 °C in defined basal limitation medium (19) containing fructose [0.75% (wt/vol)], potassium phosphate (0.6 mM), and tryptophan (50 µg/mL). At time intervals after inhibiting transcription with rifampicin (0.2 mg/mL), total RNA was extracted from lysozyme-treated cells by the hot-phenol procedure (20), and equal amounts were analyzed by Northern blotting to determine the level of mini yhxA-glpP mRNA and tRNACys remaining (7). mRNA half-lives were then calculated by linear regression analysis of the data.
Identifying 5′ Termini by RNA Ligase-Mediated RACE.
Samples of total RNA from B. subtilis containing plasmid pBYGmini or a derivative thereof (5 μg) were treated with tobacco acid pyrophosphatase (2 units; Epicentre) and then ligated to a synthetic DNA-RNA chimeric oligonucleotide (1 μg; Table S1) by treatment with T4 RNA ligase 1 (5 units; NEB) and ATP (1 mM) for 1 h at 37 °C. The ligation products were reverse transcribed by treatment with SuperScript III reverse transcriptase (200 units; Invitrogen) and a mini yhxA-glpP–specific primer (5 pmol; Table S1) for 1 h at 55 °C, and the resulting cDNA was amplified by successive rounds of PCR with nested pairs of primers surrounding the transcription initiation site (Table S1). Finally, the PCR products were purified by agarose gel electrophoresis and sequenced.
Supplementary Material
Acknowledgments
We thank Kevin Esvelt and David Liu for providing DNA encoding T7RNAP-A6-36.4, and Tricia Foley for helpful experimental advice. This investigation was supported by a postdoctoral fellowship (to J.R.) from the Vilcek Endowment and by Research Grant R01GM035769 (to J.G.B.) from the National Institutes of Health.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission. B.F.L. is a guest editor invited by the Editorial Board.
See Commentary on page 8765.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1222670110/-/DCSupplemental.
References
- 1.Belasco JG. All things must pass: Contrasts and commonalities in eukaryotic and bacterial mRNA decay. Nat Rev Mol Cell Biol. 2010;11(7):467–478. doi: 10.1038/nrm2917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Shahbabian K, Jamalli A, Zig L, Putzer H. RNase Y, a novel endoribonuclease, initiates riboswitch turnover in Bacillus subtilis. EMBO J. 2009;28(22):3523–3533. doi: 10.1038/emboj.2009.283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Even S, et al. Ribonucleases J1 and J2: Two novel endoribonucleases in B.subtilis with functional homology to E.coli RNase E. Nucleic Acids Res. 2005;33(7):2141–2152. doi: 10.1093/nar/gki505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mathy N, et al. 5′-to-3′ exoribonuclease activity in bacteria: role of RNase J1 in rRNA maturation and 5′ stability of mRNA. Cell. 2007;129(4):681–692. doi: 10.1016/j.cell.2007.02.051. [DOI] [PubMed] [Google Scholar]
- 5.Bechhofer DH. Messenger RNA decay and maturation in Bacillus subtilis. Prog Mol Biol Transl Sci. 2009;85:231–273. doi: 10.1016/S0079-6603(08)00806-4. [DOI] [PubMed] [Google Scholar]
- 6.Mathy N, et al. Bacillus subtilis ribonucleases J1 and J2 form a complex with altered enzyme behaviour. Mol Microbiol. 2010;75(2):489–498. doi: 10.1111/j.1365-2958.2009.07004.x. [DOI] [PubMed] [Google Scholar]
- 7.Richards J, et al. An RNA pyrophosphohydrolase triggers 5′-exonucleolytic degradation of mRNA in Bacillus subtilis. Mol Cell. 2011;43(6):940–949. doi: 10.1016/j.molcel.2011.07.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mackie GA. Ribonuclease E is a 5′-end-dependent endonuclease. Nature. 1998;395(6703):720–723. doi: 10.1038/27246. [DOI] [PubMed] [Google Scholar]
- 9.Celesnik H, Deana A, Belasco JG. Initiation of RNA decay in Escherichia coli by 5′ pyrophosphate removal. Mol Cell. 2007;27(1):79–90. doi: 10.1016/j.molcel.2007.05.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Deana A, Celesnik H, Belasco JG. The bacterial enzyme RppH triggers messenger RNA degradation by 5′ pyrophosphate removal. Nature. 2008;451(7176):355–358. doi: 10.1038/nature06475. [DOI] [PubMed] [Google Scholar]
- 11.Glatz E, Nilsson RP, Rutberg L, Rutberg B. A dual role for the Bacillus subtilis glpD leader and the GlpP protein in the regulated expression of glpD: Antitermination and control of mRNA stability. Mol Microbiol. 1996;19(2):319–328. doi: 10.1046/j.1365-2958.1996.376903.x. [DOI] [PubMed] [Google Scholar]
- 12.Saecker RM, Record MT, Jr, Dehaseth PL. Mechanism of bacterial transcription initiation: RNA polymerase - promoter binding, isomerization to initiation-competent open complexes, and initiation of RNA synthesis. J Mol Biol. 2011;412(5):754–771. doi: 10.1016/j.jmb.2011.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Piton J, et al. Bacillus subtilis RNA deprotection enzyme RppH recognizes guanosine in the second position of its substrates. Proc Natl Acad Sci USA. 2013;110:8858–8863. doi: 10.1073/pnas.1221510110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dorléans A, et al. Molecular basis for the recognition and cleavage of RNA by the bifunctional 5′-3′ exo/endoribonuclease RNase J. Structure. 2011;19(9):1252–1261. doi: 10.1016/j.str.2011.06.018. [DOI] [PubMed] [Google Scholar]
- 15.Coleman TM, Wang G, Huang F. Superior 5′ homogeneity of RNA from ATP-initiated transcription under the T7 φ 2.5 promoter. Nucleic Acids Res. 2004;32(1):e14. doi: 10.1093/nar/gnh007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Esvelt KM, Carlson JC, Liu DR. A system for the continuous directed evolution of biomolecules. Nature. 2011;472(7344):499–503. doi: 10.1038/nature09929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zuker M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003;31(13):3406–3415. doi: 10.1093/nar/gkg595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zeigler DR, et al. The origins of 168, W23, and other Bacillus subtilis legacy strains. J Bacteriol. 2008;190(21):6983–6995. doi: 10.1128/JB.00722-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stülke J, Hanschke R, Hecker M. Temporal activation of β-glucanase synthesis in Bacillus subtilis is mediated by the GTP pool. J Gen Microbiol. 1993;139(9):2041–2045. doi: 10.1099/00221287-139-9-2041. [DOI] [PubMed] [Google Scholar]
- 20.Celesnik H, Deana A, Belasco JG. PABLO analysis of RNA: 5′-phosphorylation state and 5′-end mapping. Methods Enzymol. 2008;447:83–98. doi: 10.1016/S0076-6879(08)02205-2. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.