

Significance
Lon protease degrades unfolded or damaged proteins as well as numerous cellular regulatory proteins. How these different classes of substrates are recognized is poorly understood. We find that Lon hexamers assemble via a matrix of N-domain interactions to form a dodecamer with altered substrate-degradation properties. Access of protein substrates to the degradation machinery in the dodecamer appears to require passage through equatorial portals. As a consequence, large substrates that are efficiently degraded by hexamers seem to be preferentially excluded from dodecamers. This gating mechanism allows the substrate repertoire of Lon to be adjusted by its assembly state.
Keywords: ATP-dependent protease, EM structure, IbpB, substrate gating, regulated proteolysis
Abstract
Lon is an ATPase associated with cellular activities (AAA+) protease that controls cell division in response to stress and also degrades misfolded and damaged proteins. Subunits of Lon are known to assemble into ring-shaped homohexamers that enclose an internal degradation chamber. Here, we demonstrate that hexamers of Escherichia coli Lon also interact to form a dodecamer at physiological protein concentrations. Electron microscopy of this dodecamer reveals a prolate structure with the protease chambers at the distal ends and a matrix of N domains forming an equatorial hexamer–hexamer interface, with portals of ∼45 Å providing access to the enzyme lumen. Compared with hexamers, Lon dodecamers are much less active in degrading large substrates but equally active in degrading small substrates. Our results support a unique gating mechanism that allows the repertoire of Lon substrates to be tuned by its assembly state.
Protein quality control is vital under stress conditions that promote protein unfolding and aggregation. Escherichia coli Lon degrades many unfolded proteins (1–3) and also degrades folded proteins, including SulA (supressor of Lon protein) and the inclusion-body binding proteins A and B (IbpA and B) (4–6). In E. coli and many other bacteria, Lon is up-regulated under numerous stress conditions (7–10). In mitochondria, Lon helps combat oxidative stress (11–14), and human mitochondrial Lon was recently identified as a potential antilymphoma target (15). It is widely believed that a major role of Lon in all organisms is to degrade misfolded proteins (2, 10, 16).
Lon subunits consist of an N domain, a central ATPase associated with cellular activities (AAA+) ATPase module, and a C-terminal peptidase domain. Although early reports suggested that Lon might be a tetramer (17), it is now clear that six subunits of the E. coli enzyme assemble into a hexamer with an internal degradation chamber accessible via an axial pore in the AAA+ ring (18, 19). Lon substrates are recognized, unfolded if necessary by ATP-dependent reactions mediated by the AAA+ ring, and then translocated through the pore and into the peptidase chamber for degradation (20).
In many families of ATP-dependent proteases, the AAA+ unfolding/translocation ring and the self-compartmentalized peptidase are encoded by distinct polypeptides, which assemble into independent oligomers before interacting to form the functional protease (21, 22). For example, the ClpXP protease consists of AAA+ ClpX hexamers, which dock with the self-compartmentalized ClpP peptidase. This interaction suppresses the ATPase rate of ClpX and enhances the peptidase activity of ClpP (22). Lon activity cannot be controlled in this way because the ATPase and protease domains are always physically attached. Little is currently known about how Lon activity is regulated, although mutational studies show that the AAA+ and peptidase domains influence each other’s activities (23–25). In some cases, the function of the two domains also appears to be linked via allosteric communication mediated by substrate binding (26, 27).
Here, we demonstrate that Lon forms dodecamers that equilibrate with hexamers at physiological concentrations. A structure determined by EM at low resolution reveals a unique protease architecture with the degradation chambers of each hexamer at opposite ends of a prolate ellipsoid. Near the equator of this structure, the arrangement of N domains creates portals, which could serve as entry sites for protein substrates. Formation of the dodecamer suppresses proteolysis of large but not small protein substrates, suggesting that the dodecamer uses a gating mechanism that allows the repertoire of Lon substrates to be tuned by its state of assembly.
Results
Lon Exists in Multiple Oligomeric Forms.
In the process of characterizing E. coli Lon by size-exclusion chromatography (SEC) and multiangle laser light scattering (MALS), we observed enzyme complexes with properties similar to those expected for hexamers (calculated MR 525 kDa) and dodecamers (calculated MR 1,050 kDa). For example, SEC-MALS of the LonS679A variant, which had an active-site mutation in the peptidase domain to prevent autodegradation (28), revealed two major species corresponding to molecular weights of 565 ± 13 and 930 ± 5 kDa (Fig. 1A). Based on previous characterization of Lon as a hexamer (18, 19), the simplest interpretation of these results is that the larger species is a Lon dodecamer, which may dissociate to some extent during the SEC run.
Fig. 1.

Lon assembles into dodecamers as well as hexamers. (A) Catalytically inactive LonS679A (24-µM loading concentration) formed hexamers (expected MR ∼525 kDa) and dodecamers (expected MR ∼1,050 kDa) in SEC-MALS experiments. The variation in apparent MR over both peaks suggests that dodecamers and hexamers are in equilibrium. Dashed lines represent an error of 5% in measurement of molecular weight. Chromatography was performed at room temperature in 50 mM Hepes (pH 7.6), 150 mM NaCl, 20 mM MgCl2, 10% (vol/vol) glycerol, and 0.1 mM TCEP. (B) Concentration-dependent changes in the population of LonS679A dodecamers, hexamers, and monomers in SV-AUC c(s20,w) distributions. Traces at each concentration were offset on the y axis for clarity. Experiments were performed at 20 °C in 50 mM Hepes-KOH (pH 7.5), 150 mM NaCl, 0.01 mM EDTA, 0.1 mM TCEP, 1 mM MgCl2, and 0.1 mM ATPγS.
To characterize assembly further, we used sedimentation velocity-analytical ultracentrifugation (SV-AUC) at multiple concentrations of LonS679A in the presence of 100 μM ATPγS (Fig. 1B). Both the large (dodecamer) and smaller (hexamer) assemblies were clearly detectable at multiple LonS679A concentrations, as was a smaller species, which appeared to be a monomer. As expected, the dodecamer was more highly populated at higher concentrations, and the hexamer and presumed monomer populations increased at lower concentrations. The hexamer and dodecamer were both populated at concentrations that are physiologically relevant (see below).
To confirm that wild-type Lon also formed dodecamers, we used analytical gel filtration (Fig. S1), which is rapid and minimizes autoproteolysis. In addition, because nucleotide can affect Lon assembly, experiments were performed with ATPγS or without nucleotide. Under the conditions tested, Lon appeared to chromatograph as a mixture of hexamers and dodecamers, and nucleotide had little effect on the distribution of these species (Fig. S1).
Dodecamers Should Exist at Intracellular Concentrations.
To investigate the potential for Lon dodecamers to form in vivo, we determined intracellular concentrations using quantitative Western blots. A dilution series of purified Lon was analyzed on the same membrane as Lon from cells grown at 30 °C (Fig. 2A). The concentration of Lon in monomer equivalents ranged from 1.7 to 3.7 µM over four measurements and averaged 2.5 ± 0.5 µM (SEM) for cells grown at 30 °C. Lon forms hexamers and dodecamers at these concentrations in vitro. Following a temperature increase to 42 °C, a modest increase in Lon levels was observed by Western blots (∼1.2-fold) (Fig. 2B).
Fig. 2.

Quantitative Western blots of Lon levels in E. coli are consistent with dodecamer formation. (A) Western blot following SDS-PAGE of different concentrations of purified Lon and an E. coli lysate before heat shock. The intracellular concentration of Lon was 2.5 ± 0.5 µM or ∼1,500 ± 300 Lon monomers per cell at 30 °C (n = 4). (B) Lon concentrations increased slightly in cells grown after a temperature increase to at 42 °C compared with cells grown at 30 °C (n = 3). Values are averages ±1 SEM.
EM Dodecamer Structure.
EM images of negatively stained Lon complexes showed two major populations (Fig. 3A). Classification of over 4,000 particles revealed that one major species was a dodecamer with roughly sixfold symmetry (Fig. 3B). These dodecamers preferentially assumed a side-view orientation on the grid (Fig. 3B). Multiple views of hexamers were also observed. The side view of the hexamer corresponded to roughly half of the density observed in the side view of the dodecamer (Fig. 3B).
Fig. 3.
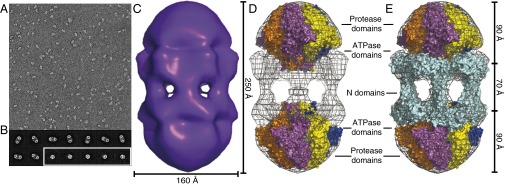
Lon dodecamers and hexamers as visualized by negative-stain EM. (A) Representative field-of-view of electron micrographs of single wild-type Lon particles shows two major populations, differing by roughly twofold in size. (29,000× magnification) (B) Representative class images of dodecamers (Upper and Lower Left) and hexamers (Lower Right, enclosed in white box). Samples were preincubated at 37 °C and following dilution onto grids for imaging contained 0.24 µM Lon (subunit equivalents), 2 µM sul20 peptide, and 50 µM ATPγS. (C) Three-dimensional reconstruction of the Lon dodecamer. In the orientation shown, the dodecamer (displayed in surface representation) is ∼250 Å high and ∼160 Å wide. (D) The Upper and Lower portions of the electron-density map were fit well by hexameric models of the peptidase and ATPase domains from a crystal structure of B. subtilis Lon (3M6A.pdb). (E) Same as D except the equatorial density was fit using a dimeric model of the N domain from E. coli Lon (3LJC.pbd). The equatorial portals between N domains are ∼45 Å in diameter. These figures were generated with PyMOL (v1.2r3pre; Schrödinger) using mesh level 4 and surface level 3.
Following sixfold (C6) averaging, a 3D reconstruction of the dodecamer classes revealed face-to-face hexameric rings connected by six strands of density (Fig. 3C). The dodecamer was ∼250 Å long and ∼160 Å wide and roughly the shape of a prolate ellipsoid. We created planar hexameric models of the ATPase and the protease domains from a crystal structure including these portions of a Bacillus subtilis Lon subunit (29) (Experimental Procedures) and manually placed them into the density map (Fig. 3 D and E). The protease domains fit well into the density at the distal ends, with the adjacent ATPase domains closer to the equator. There were some clashes between the protease and ATPase domains, but the low resolution of the structure and uncertainty about the quality of our hexameric models precluded better fitting. The extra density near the equator was fit as a matrix of interacting E. coli Lon N domains, arranged as overlapping dimers through a coiled-coil region, which create bridges between the two halves of the structure (Fig. 3E). Strikingly, portals with diameters of ∼45 Å were clearly visible between the N domains. As discussed below, these portals may exclude entry of large substrates into the lumen of the enzyme, where the degradation machinery resides.
Hexamers Have Higher Basal ATPase Activity than Dodecamers.
Because equilibration precluded isolation of pure hexamers or dodecamers for functional assays, we measured rates of ATP hydrolysis over the same range of Lon concentrations that altered the dodecamer/hexamer ratio in the SV-AUC studies. In control experiments, we confirmed that hexamers and dodecamers were still present as the major species in buffers and under conditions that mimicked our enzyme-assay conditions (Fig. S2). When rates of ATP hydrolysis were normalized for the total number of Lon subunits in each reaction, basal hydrolysis slowed substantially as the dodecamer/hexamer ratio increased (Fig. 4 and Fig. S3A). The concentration dependence of the ATPase activity was fitted best by a KD of 3.3 ± 1.5 µM for the hexamer–dodecamer interaction, a hydrolysis rate of 23 ± 3.6 subunit−1 min−1 for the hexamer, and a hydrolysis rate of 1.8 ± 1.3 subunit−1 min−1 for the dodecamer (Fig. S3A). Thus, the hexamer hydrolyzes ATP ∼10-fold faster than the dodecamer in the absence of protein substrates.
Fig. 4.

Lon hydrolyzes ATP more slowly at higher concentrations (calculated in subunit equivalents), where the dodecamer/hexamer ratio increases. Data are plotted as averages ±1 SEM (n = 3). Reactions were performed at 37 °C and contained 4 mM ATP, 5 mM MgCl2, 5 mM KCl, 2% (vol/vol) DMSO, 360 mM potassium glutamate, 12% (wt/vol) sucrose, and 50 mM Hepes-KOH (pH 8).
Dodecamers Degrade “Large” Substrates Poorly and “Small” Substrates Well.
To evaluate degradation of different substrates by the dodecamer, we assayed proteolysis using a range of Lon concentrations. The first substrate was an inclusion-body binding protein, specifically E. coli IbpB, which contains a native α-crystallin domain that is recognized by Lon (6). Although IbpB monomers are relatively small (∼16 kDa), they assemble into large cage-like oligomers (30, 31). For example, IbpB runs at an apparent MR > 670 kDa in gel-filtration chromatography (Fig. S4A) (6). We determined initial rates of Lon degradation of 35S-labeled IbpB by assaying the production of acid-soluble peptides and normalized these rates by the total concentration of Lon subunits (Fig. 5A). Importantly, IbpB was degraded more slowly at higher Lon concentrations, where more dodecamer was present. Fig. 5B shows Michaelis–Menten plots for IbpB degradation using 1.5 and 6 µM Lon. KM was similar at both Lon concentrations, but Vmax was substantially lower at the higher Lon concentration. Fitting the Lon-concentration dependence of IbpB degradation gave a KD of 1.8 ± 5 µM for the hexamer–dodecamer interaction, a maximum degradation rate of 0.057 ± 0.058 subunit−1 min−1 for the hexamer, and a rate of 0.00095 ± 0.021 subunit−1 min−1 for the dodecamer (Fig. S3B). Thus, the Lon dodecamer degrades IbpB ∼60-times more slowly than the hexamer.
Fig. 5.

High concentrations of Lon degrade IbpB and FITC-casein less efficiently. (A) Normalized initial rates of IbpB degradation (45 µM) decreased as the Lon concentration increased. (B) Michealis–Menten plots of normalized rates of steady-state degradation of IbpB by 1.5 or 6 µM Lon. The lines are fits to the Michealis–Menten equation. (C) Normalized initial rates of degradation of FITC–β-casein (50 µM) also decreased with increasing Lon concentration. In all panels, data are plotted as averages ±1 SEM (n = 3), and reactions were performed at 37 °C and contained 4 mM ATP, 5 mM MgCl2, 5 mM KCl, 2% (vol/vol) DMSO, 360 mM potassium glutamate, 12% (wt/vol) sucrose, and 50 mM Hepes-KOH (pH 8).
Next, we examined degradation of fluorescein isothiocyanate (FITC)-conjugated β-casein (monomer MR ∼25 kDa) by different concentrations of Lon. β-Casein is unstructured but can form micelles and even as a monomer has a radius of gyration much larger than expected for a compact structure (Fig. S4A) (32, 33). Normalized initial rates of FITC-casein degradation were determined by changes in fluorescence, and decreased substantially as the Lon concentration and dodecamer/hexamer ratio increased (Fig. 5C). Fitting the Lon dependence of degradation gave a KD of 1.3 ± 0.6 µM for the hexamer–dodecamer interaction, a maximum degradation rate of 1.8 ± 0.4 subunit−1 min−1 for the hexamer, and a rate of 0.00001 ± 0.038 subunit−1 min−1 for the dodecamer (Fig. S3C). Thus, as with IbpB, the Lon dodecamer degrades this substrate far more slowly than does the Lon hexamer.
Finally, we used the appearance of acid-soluble peptides to assay Lon degradation of 35S-labeled titin-I27 proteins (MR ∼12 kDa) with appended N- or C-terminal sul20 or β20 degrons (27, 34). We also assayed degradation of some titin-I27 substrates following cysteine carboxymethylation, which unfolds the protein (35). Notably, Lon concentration had little effect on degron-tagged titin-I27 degradation, whether substrates were native or denatured or contained N- or C-terminal degradation tags (Fig. 6 A and B). Moreover, in the presence of carboxymethylated titin-I27–sul20, the rate of ATP hydrolysis by Lon showed a much smaller dependence on enzyme concentration (Fig. 6C), compared with the rate of ATP hydrolysis in the absence of a protein substrate (Fig. 4). We considered the possibility that binding of the sul20 degron to Lon might result in dodecamer dissociation. However, dodecamers were present in the EM experiments performed in the presence of sul20 peptide (Fig. 3), and addition of this peptide did not detectably change the dodecamer/hexamer ratio in gel-filtration experiments (distributions were similar to Fig. S1). In contrast to the case with the Ibps, the model degron substrates stimulate the rate of ATP-hydrolysis by Lon ∼3- to 10-fold (Fig. 6C). Nonetheless, the substrate-stimulated ATPase rate (normalized by Lon concentration) was lowest at the highest Lon concentrations (6 and 12 μM) (Fig. 6C), indicating that both degron-enzyme interactions and the hexamer-dodecamer equilibrium influence ATP hydrolysis.
Fig. 6.

Lon hexamers and dodecamers degrade degron-tagged titin-I27 substrates with similar efficiencies. (A) Degradation of native substrates with C-terminal sul20 degrons (gray), C-terminal β20 degrons (blue), and N-terminal β20 degrons (orange). (B) Degradation of denatured titin-I27 substrates. Degron colors are the same as in A. (C) In the presence of carboxymethylated titin-I27-sul20 (40 µM), ATPase rates decreased only marginally at high Lon concentrations. In all panels, data are plotted as averages ±1 SEM (n = 3). Reactions were performed at 37 °C and contained 25 mM Tris⋅HCl (pH 8.0), 100 mM KCl, 10 mM MgCl2, 1 mM DTT, and 2 mM ATP.
Taken together, our results support a model in which Lon hexamers and dodecamers are both active proteases. Importantly, however, the dodecamer only efficiently degraded the degron-tagged titin-I27 substrates, which behaved as much smaller species than the IbpB and β-casein substrates (Fig. S4). As we discuss below, the portals created by dodecamer assembly may provide a “gating” mechanism that prevents larger substrates from entering the luminal chamber and being degraded.
Discussion
Our results show that hexamers of E. coli Lon assemble into a dodecamer that displays different enzymatic properties. Hexamers and dodecamers are both populated at low micromolar concentrations in vitro, and the Lon concentration in vivo is ∼2.5 µM. Thus, the hexamer-dodecamer equilibrium is likely to be a physiologically relevant factor in controlling Lon activity in cells.
Our EM structure of the Lon dodecamer reveals a face-to-face association of hexamers in which the N domains appear to be largely responsible for stabilizing the complex. This architecture positions the degradation chambers of each hexamer at the distal ends of the complex and has not been observed in other AAA+ proteases. As observed in the crystal structure of an archaeal LonB hexamer (36), the degradation chambers in the dodecamer appear to be sequestered from bulk solution. Thus, degradation still requires substrate unfolding/translocation by the Lon AAA+ ring, an apparently universal feature of AAA+ proteases (20).
The Lon N domains have been implicated in substrate recognition (37–39). Here we show an additional role for the N domain in dodecamer assembly. Thus, formation of the dodecamer may alter the substrate-recognition properties of the enzyme by creating or occluding substrate-binding sites. Furthermore, we find that when the N domains interact with each other, and/or with the ATPase domain of the opposite hexamer in the dodecamer, the rate of ATP hydrolysis by Lon can be suppressed. A similar suppression of ATPase activity is observed when ClpX interacts ClpP (22). Understanding the molecular basis of the suppression of ATPase activity upon dodecamer assembly, and the activation of ATP hydrolysis by some Lon substrates is likely to provide important insight into the allosteric mechanisms that are used by substrates to control Lon’s enzymatic activities (26, 27).
A notable feature of the dodecamer is the presence of six portals, each ∼45 Å in diameter, spaced around the equator of the structure. The size of these portals should prevent entry of large substrates into the lumen of the dodecamer (Fig. 7). Indeed, we found that IbpB and β-casein substrates, which have large radii of gyration, were degraded very slowly by Lon dodecamers compared with hexamers. In fact, given the confidence of data fitting, we cannot exclude the possibility that the dodecamer cannot degrade IbpB or β-casein. In contrast, we found that smaller degron-tagged titin-I27 substrates in both folded and unfolded states were degraded at similar rates by Lon hexamers and dodecamers. These results support a model in which a major determinant of substrate degradation by the Lon dodecamer is the ability of a substrate to diffuse or be pulled through the portals to allow engagement by the degradation machinery. In principle, substrate size, charge, hydrophobicity, stability, or the length of the disordered degron tag could all contribute to determining whether specific substrates could enter the lumen and be degraded by Lon dodecamers.
Fig. 7.

Potential roles of Lon hexamers and dodecamers in intracellular protein degradation.
At present, we can only speculate about possible biological functions for the Lon dodecamer. One possibility is that cellular stress results in very high concentrations of misfolded proteins that form large aggregates or bind the IbpA/IbpB chaperones, with degradation of such substrates by Lon hexamers swamping the enzyme’s ability to degrade regulatory proteins and therefore inhibiting the recovery from stress. These regulatory proteins include SulA (19 kDa, C-terminal degron), which is rapidly degraded during recovery after DNA damage to allow cell division to resume (40). Similarly, SoxS (13 kDa, N-terminal degron), a transcriptional activator of the superoxide-response regulon (41), and UmuD (30-kDa dimer, N-terminal degron), a subunit of the lesion-bypass DNA polymerase (42) need to be degraded by Lon specifically as cells recover from stress. In this scenario, Lon dodecamers could carry out regulatory degradation, whereas hexamers triage and perform quality-control degradation of damaged/unfolded proteins. It is also possible, that dodecamers are more highly populated under stress conditions, reducing the rate of IbpA/IbpB degradation and increasing their concentration to suppress aggregation and promote refolding in cooperation with ClpB and DnaK (43, 44). Alternatively, equatorial interfaces in Lon dodecamers, which are absent in hexamers, might allow recognition and degradation of additional important substrates. ATP is abundant during exponential growth but scarce when nutrients are lacking. In principle, Lon dodecamer formation might serve to conserve ATP, as we find that basal hydrolysis by the dodecamer is much lower than by the hexamer. Thus, if Lon were predominantly dodecameric during stationary-phase growth, when substrates became available they could still be degraded to provide amino acids or for regulatory purposes but at a lower cost in terms of overall ATP consumption. Testing these models will require the design of Lon variants that are exclusively hexameric or dodecameric. We are pursuing higher resolution structures of the dodecamer to guide these efforts. B. subtilis and Mycobacterium smegmatis Lon have also been reported to form oligomers larger than hexamers (23, 29, 45). Thus, hexamer-dodecamer equilibria may be a relatively conserved feature of this important intracellular protease.
Experimental Procedures
Protein Purification.
Wild-type E. coli Lon (9) and the LonS679A variant (28) were purified as described previously (34) with minor changes. After lysis by sonication or French press, lysates were incubated with benzonase (Merck) for 1 h at 4 °C. Following elution from P11 phosphocellulose (Whatman, GE Healthcare), the sample was concentrated and buffer exchanged to 2 mL either in 25 mM Hepes [4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid]-KOH (pH 7.5), 2 M NaCl, 1 mM EDTA (ethylenediaminetetraacetic acid), 0.1 mM TCEP [tris(2-carboxyethyl)phosphine], and 10% (vol/vol) glycerol, or 50 mM Hepes-KOH (pH 7.5), 2 M NaCl, and 0.1 mM TCEP, passed through a 0.45-µm filter, and purified on a HR 10/300 Superose 6 gel-filtration column (GE Healthcare). Fractions containing Lon at >95% purity, as judged by SDS-PAGE (sodium dodecyl sulfate polyacrylamide gel electrophoresis) and 280/260 nm absorbance, were combined, dialyzed against Lon storage buffer [50 mM Hepes-KOH (pH 7.5), 150 mM NaCl, 10% (vol/vol) glycerol, 1 mM EDTA, and 0.1 mM TCEP], concentrated, flash frozen in aliquots, and stored at −80 °C.
E. coli IbpB was purified as previously described (6), dialyzed against Ibp storage buffer [50 mM Hepes-KOH (pH 8), 600 mM potassium glutamate, 20% (wt/vol) sucrose, and 0.1 mM TCEP], flash frozen in aliquots, and stored at −80 °C. His6-tagged titin-I27 proteins with sul20 or β20 degrons were purified as previously described (34).
SEC-MALS.
SEC was performed on a Wyatt WTC-030S5 size-exclusion column using an Agilent HPLC. MALS was measured in line using a Wyatt DAWN-HELEOS instrument; concentrations were determined using a Wyatt Optilab rEX instrument. Standard Zimm-plot analysis was preformed with the ASTRA software 5.3.4 (Wyatt Technology). LonS679A samples (loading concentrations 24, 12, and 6 μM) were run in 50 mM Hepes-KOH (pH 7.6), 150 mM NaCl, 20 mM MgCl2, 10% (vol/vol) glycerol, and 0.1 mM TCEP at room temperature.
Ultracentrifugation.
SV-AUC experiments for LonS679A were performed using a Beckman OptimaXL-I analytical ultracentrifuge (Biophysical Instrumentation Facility, Massachusetts Institute of Technology, Cambridge, MA). Samples were dialyzed overnight against 50 mM Hepes-KOH (pH 7.5), 150 mM NaCl, 0.01 mM EDTA, and 0.1 mM TCEP. Before loading the cells, 1 mM MgCl2 and 0.1 mM ATPγS were added to the samples. Samples were loaded in dual-sector charcoal-filled epon centerpieces and centrifuged at 16,000 rpm in a Beckman An50-Ti rotor at 20 °C. SEDFIT (46) was used to calculate the continuous distribution of sedimentation coefficients from 1S to 60S at resolutions of 200 or 100 scans per concentration with a confidence level (F-ratio) of 0.95. Calculations were performed using a density of 1.00831, a viscosity of 0.010475, and a Lon partial specific volume of 0.7431 (SEDNTERP; J. Philo; www.jphilo.mailway.com).
Western Blotting.
E. coli W3110 cells were grown at 30 °C in M9 medium supplemented with 0.4% (wt/vol) glucose, 100 μM CaCl2, 2 mM MgSO4, 0.2% (wt/vol) thiamin, and 0.2% (wt/vol) casamino acids. At an OD600 of 0.3, cultures were split, additional medium at 54 °C was added to the heat-shock sample, and additional medium at 30 °C was added to the control. The final temperature of the heat-shock sample was 42 °C. Aliquots of 1 mL were taken at each time point from both samples, OD600 was recorded, cells were pelleted by centrifugation, and the supernatant was removed. Pellets were stored at –20 °C until resuspended to 2.5 OD600 equivalents with 5× SDS loading dye. Resuspended samples were heated at 99 °C for 10 min while shaking, and then cooled. The samples (10 µL) were loaded on Mini-PROTEAN TGX 4–20% (wt/vol) polyacrylamide precast gels (Bio-Rad). The gels were transferred onto filter paper using a wet-transfer apparatus (Bio-Rad), probed with anti-Lon polyclonal antibody (produced by Covance Research Products) at a 1:2,000 dilution for 4 h at room temperature, incubated with goat anti-rabbit IgG-AP conjugate (Bio-Rad) at a 1:10,000 dilution for 1 h at room temperature, and developed with alkaline phosphatase dephosphorylates ECF Substrate (GE Healthcare). The blots were exposed with a blue laser and quantified with ImageQuant software (GE Healthcare).
The intracellular concentration of Lon was calculated from the Western blot using purified LonS679A as a standard; 1.9 ± 0.36 ng of Lon was present in a sample containing 3.55 ± 0.15 × 108 cells per OD (as determined by counting colony-forming units under the conditions of the experiment), giving a value of 2.2 ± 0.6 × 10−16 g per cell. Errors represent ±1 SEM (n = 4) and were propagated through all calculations. The intracellular Lon concentration was calculated using a subunit MR of 87.5 kDa and a cell volume of 10−15 L (47). The fold increase in Lon concentration following a temperature increase from 30 to 42 °C was determined by normalizing against a nonspecific band at the bottom of the Western to correct for any changes in cell density over the time course and dividing the intensity of the bands from cells grown at 42 °C by the intensity of the bands from cells grown at 30 °C.
Single-Particle EM Data Collection and Analysis.
Wild-type Lon (24 µM subunit equivalents in storage buffer) was incubated at 37 °C with 0.1 mM ATP, and 10 mM MgCl2 for 4 min; sul20 peptide (synthesized and purified in house) was added to a final concentration of 200 µM and incubated for 1 min; ATPγS was added to a final concentration of 5 mM, the sample was diluted ∼100-fold with storage buffer without glycerol, and immediately negatively stained with uranyl acetate (1% vol/vol) on continuous carbon-film grids. Electron micrographs of single particles were recorded with a 2Kx2K CCD camera on a TF20 electron microscope at 29,000× nominal magnification. A total of 4,235 oligomeric Lon particles were then boxed into a single stack, from which the particle images were 2×-binned to 7.92 Å per pixel. The dataset was subjected to “direct classification” using PARTICLE (www.image-analysis.net/EM), which is free of alignment error or reference bias.
For the dodecamer 3D reconstruction, a set of single-particle tomographic volumes were first collected and averaged to establish an unbiased initial model, which served as the reference to align the class averages in single-particle reconstruction and refinement. The model was further validated by the tilt-pair technique (48), using 86 pairs at 30°-tilt separation, in which the average angular deviation from the controlled tilting angle was less than 7°. Data analysis (particle screening, classification, single-particle tomography, 3D reconstruction, and model validation) was performed in the PARTICLE software package.
The 3M6A.pdb crystal structure includes the protease and ATPase domains of B. subtilis Lon in an indefinite spiral conformation (29). To make a planar hexameric model for fitting EM density, we aligned six copies of chain A from 3M6A.pdb to the protease domains in the hexameric structure of the E. coli Lon protease domain (1RR9.pdb). To fit the six equatorial bridges, we placed two E. coli Lon N domains (3LCJ.pdb) in each bridge with the coiled-coil regions crossing and rotated the globular domains to fit the density.
ATPase Assays.
ATP-hydrolysis rates were measured at 37 °C in a plate reader using an NADH enzyme-linked assay (49, 50) in buffer containing 4 mM ATP, 5 mM MgCl2, 5 mM KCl, 2% (vol/vol) DMSO (dimethyl sulfoxide), 360 mM potassium glutamate, 12% (wt/vol) sucrose, and 50 mM Hepes-KOH (pH 8).
Degradation Assays.
IbpB degradation reactions contained 60% (vol/vol) Ibp storage buffer, 5% (vol/vol) Lon buffer, 5 mM MgCl2, 5 mM KCl, and 2% (vol/vol) DMSO. An ATP-regeneration system, containing a final concentration of 4 mM ATP, 100 mg⋅mL−1 creatine kinase, and 10 mM creatine phosphate was added to initiate the reaction. Degradation was monitored at 37 °C by the formation of radioactive peptides soluble in trichloroacetic acid as described (6, 51). FITC-casein type III (Sigma-Aldrich) was resuspended in Ibp storage buffer. Degradation was monitored by the increase of fluorescence at 525 nm with excitation at 365 nm in a plate reader at 37 °C using final conditions as described for IbpB degradation.
Degradation of titin-I27 constructs was carried out at 37 °C in buffer containing 25 mM Tris-HCl (pH 8.0), 100 mM KCl, 10 mM MgCl2, 1 mM DTT (dithiothreitol), 2 mM ATP, 20 mM phosphoenolpyruvate, and 10 U/mL pyruvate kinase. Kinetics were determined using a mixture of 5% (mol/mol) 35S-labeled substrate and 95% (mol/mol) unlabeled substrate, as previously described (51).
Supplementary Material
Acknowledgments
We thank S. Bissonnette and E. Gur for reagents; members of our laboratories for helpful discussions; D. Pheasant at the Massachusetts Institute of Technology Biophysical Instrumentation Center for help with the analytical ultracentrifugation experiments; and the electron-microscopy facility at Harvard Medical School, which is supported by National Institutes of Health Grant P01 GM62580. This work was supported by National Institutes of Health Grants GM-49224 and AI-16892; a National Institutes of Health National Research Service Award postdoctoral Fellowship F32GM094994 (to E.F.V.); and a National Science Foundation Graduate Research Fellowship (to M.L.W.). T.A.B. is an employee of the Howard Hughes Medical Institute.
Footnotes
The authors declare no conflict of interest.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1307066110/-/DCSupplemental.
References
- 1.Fredriksson A, Ballesteros M, Dukan S, Nyström T. Defense against protein carbonylation by DnaK/DnaJ and proteases of the heat shock regulon. J Bacteriol. 2005;187(12):4207–4213. doi: 10.1128/JB.187.12.4207-4213.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kowit JD, Goldberg AL. Intermediate steps in the degradation of a specific abnormal protein in Escherichia coli. J Biol Chem. 1977;252(23):8350–8357. [PubMed] [Google Scholar]
- 3.Shineberg B, Zipser D. The ion gene and degradation of beta-galactosidase nonsense fragments. J Bacteriol. 1973;116(3):1469–1471. doi: 10.1128/jb.116.3.1469-1471.1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gottesman S, Halpern E, Trisler P. Role of sulA and sulB in filamentation by lon mutants of Escherichia coli K-12. J Bacteriol. 1981;148(1):265–273. doi: 10.1128/jb.148.1.265-273.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mizusawa S, Gottesman S. Protein degradation in Escherichia coli: The lon gene controls the stability of sulA protein. Proc Natl Acad Sci USA. 1983;80(2):358–362. doi: 10.1073/pnas.80.2.358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bissonnette SA, Rivera-Rivera I, Sauer RT, Baker TA. The IbpA and IbpB small heat-shock proteins are substrates of the AAA+ Lon protease. Mol Microbiol. 2010;75(6):1539–1549. doi: 10.1111/j.1365-2958.2010.07070.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Goff SA, Casson LP, Goldberg AL. Heat shock regulatory gene htpR influences rates of protein degradation and expression of the lon gene in Escherichia coli. Proc Natl Acad Sci USA. 1984;81(21):6647–6651. doi: 10.1073/pnas.81.21.6647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Phillips TA, VanBogelen RA, Neidhardt FC. lon gene product of Escherichia coli is a heat-shock protein. J Bacteriol. 1984;159(1):283–287. doi: 10.1128/jb.159.1.283-287.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Goff SA, Goldberg AL. Production of abnormal proteins in E. coli stimulates transcription of lon and other heat shock genes. Cell. 1985;41(2):587–595. doi: 10.1016/s0092-8674(85)80031-3. [DOI] [PubMed] [Google Scholar]
- 10.Van Melderen L, Aertsen A. Regulation and quality control by Lon-dependent proteolysis. Res Microbiol. 2009;160(9):645–651. doi: 10.1016/j.resmic.2009.08.021. [DOI] [PubMed] [Google Scholar]
- 11.Bender T, Lewrenz I, Franken S, Baitzel C, Voos W. Mitochondrial enzymes are protected from stress-induced aggregation by mitochondrial chaperones and the Pim1/LON protease. Mol Biol Cell. 2011;22(5):541–554. doi: 10.1091/mbc.E10-08-0718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Venkatesh S, Lee J, Singh K, Lee I, Suzuki CK. Multitasking in the mitochondrion by the ATP-dependent Lon protease. Biochim Biophys Acta. 2012;1823(1):56–66. doi: 10.1016/j.bbamcr.2011.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ngo JK, Davies KJ. Mitochondrial Lon protease is a human stress protein. Free Radic Biol Med. 2009;46(8):1042–1048. doi: 10.1016/j.freeradbiomed.2008.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ngo JK, Pomatto LC, Bota DA, Koop AL, Davies KJ. Impairment of lon-induced protection against the accumulation of oxidized proteins in senescent wi-38 fibroblasts. J Gerontol A Biol Sci Med Sci. 2011;66(11):1178–1185. doi: 10.1093/gerona/glr145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bernstein SH, et al. The mitochondrial ATP-dependent Lon protease: A novel target in lymphoma death mediated by the synthetic triterpenoid CDDO and its derivatives. Blood. 2012;119(14):3321–3329. doi: 10.1182/blood-2011-02-340075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rosen R, et al. Protein aggregation in Escherichia coli: Role of proteases. FEMS Microbiol Lett. 2002;207(1):9–12. doi: 10.1111/j.1574-6968.2002.tb11020.x. [DOI] [PubMed] [Google Scholar]
- 17.Goldberg AL, Moerschell RP, Chung CH, Maurizi MR. ATP-dependent protease La (lon) from Escherichia coli. Methods Enzymol. 1994;244:350–375. doi: 10.1016/0076-6879(94)44027-1. [DOI] [PubMed] [Google Scholar]
- 18.Botos I, et al. The catalytic domain of Escherichia coli Lon protease has a unique fold and a Ser-Lys dyad in the active site. J Biol Chem. 2004;279(9):8140–8148. doi: 10.1074/jbc.M312243200. [DOI] [PubMed] [Google Scholar]
- 19.Park SC, et al. Oligomeric structure of the ATP-dependent protease La (Lon) of Escherichia coli. Mol Cells. 2006;21(1):129–134. [PubMed] [Google Scholar]
- 20.Sauer RT, Baker TA. AAA+ proteases: ATP-fueled machines of protein destruction. Annu Rev Biochem. 2011;80:587–612. doi: 10.1146/annurev-biochem-060408-172623. [DOI] [PubMed] [Google Scholar]
- 21.Sauer RT, et al. Sculpting the proteome with AAA(+) proteases and disassembly machines. Cell. 2004;119(1):9–18. doi: 10.1016/j.cell.2004.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Joshi SA, Hersch GL, Baker TA, Sauer RT. Communication between ClpX and ClpP during substrate processing and degradation. Nat Struct Mol Biol. 2004;11(5):404–411. doi: 10.1038/nsmb752. [DOI] [PubMed] [Google Scholar]
- 23.Roudiak SG, Seth A, Knipfer N, Shrader TE. The lon protease from Mycobacterium smegmatis: Molecular cloning, sequence analysis, functional expression, and enzymatic characterization. Biochemistry. 1998;37(1):377–386. doi: 10.1021/bi971732f. [DOI] [PubMed] [Google Scholar]
- 24.Starkova NN, Koroleva EP, Rumsh LD, Ginodman LM, Rotanova TV. Mutations in the proteolytic domain of Escherichia coli protease Lon impair the ATPase activity of the enzyme. FEBS Lett. 1998;422(2):218–220. doi: 10.1016/s0014-5793(98)00012-x. [DOI] [PubMed] [Google Scholar]
- 25.van Dijl JM, et al. The ATPase and protease domains of yeast mitochondrial Lon: Roles in proteolysis and respiration-dependent growth. Proc Natl Acad Sci USA. 1998;95(18):10584–10589. doi: 10.1073/pnas.95.18.10584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Waxman L, Goldberg AL. Selectivity of intracellular proteolysis: Protein substrates activate the ATP-dependent protease (La) Science. 1986;232(4749):500–503. doi: 10.1126/science.2938257. [DOI] [PubMed] [Google Scholar]
- 27.Gur E, Sauer RT. Degrons in protein substrates program the speed and operating efficiency of the AAA+ Lon proteolytic machine. Proc Natl Acad Sci USA. 2009;106(44):18503–18508. doi: 10.1073/pnas.0910392106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Van Melderen L, Gottesman S. Substrate sequestration by a proteolytically inactive Lon mutant. Proc Natl Acad Sci USA. 1999;96(11):6064–6071. doi: 10.1073/pnas.96.11.6064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Duman RE, Löwe J. Crystal structures of Bacillus subtilis Lon protease. J Mol Biol. 2010;401(4):653–670. doi: 10.1016/j.jmb.2010.06.030. [DOI] [PubMed] [Google Scholar]
- 30.Jiao W, Qian M, Li P, Zhao L, Chang Z. The essential role of the flexible termini in the temperature-responsiveness of the oligomeric state and chaperone-like activity for the polydisperse small heat shock protein IbpB from Escherichia coli. J Mol Biol. 2005;347(4):871–884. doi: 10.1016/j.jmb.2005.01.029. [DOI] [PubMed] [Google Scholar]
- 31.Shearstone JR, Baneyx F. Biochemical characterization of the small heat shock protein IbpB from Escherichia coli. J Biol Chem. 1999;274(15):9937–9945. doi: 10.1074/jbc.274.15.9937. [DOI] [PubMed] [Google Scholar]
- 32.Farrell HM, Jr, et al. Molten globule structures in milk proteins: Implications for potential new structure-function relationships. J Dairy Sci. 2002;85(3):459–471. doi: 10.3168/jds.S0022-0302(02)74096-4. [DOI] [PubMed] [Google Scholar]
- 33.Lucey JA, Srinivasan M, Singh H, Munro PA. Characterization of commercial and experimental sodium caseinates by multiangle laser light scattering and size-exclusion chromatography. J Agric Food Chem. 2000;48(5):1610–1616. doi: 10.1021/jf990769z. [DOI] [PubMed] [Google Scholar]
- 34.Gur E, Sauer RT. Recognition of misfolded proteins by Lon, a AAA(+) protease. Genes Dev. 2008;22(16):2267–2277. doi: 10.1101/gad.1670908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kenniston JA, Baker TA, Fernandez JM, Sauer RT. Linkage between ATP consumption and mechanical unfolding during the protein processing reactions of an AAA+ degradation machine. Cell. 2003;114(4):511–520. doi: 10.1016/s0092-8674(03)00612-3. [DOI] [PubMed] [Google Scholar]
- 36.Cha SS, et al. Crystal structure of Lon protease: Molecular architecture of gated entry to a sequestered degradation chamber. EMBO J. 2010;29(20):3520–3530. doi: 10.1038/emboj.2010.226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Roudiak SG, Shrader TE. Functional role of the N-terminal region of the Lon protease from Mycobacterium smegmatis. Biochemistry. 1998;37(32):11255–11263. doi: 10.1021/bi980945h. [DOI] [PubMed] [Google Scholar]
- 38.Melnikov EE, et al. Limited proteolysis of E. coli ATP-dependent protease Lon—A unified view of the subunit architecture and characterization of isolated enzyme fragments. Acta Biochim Pol. 2008;55(2):281–296. [PMC free article] [PubMed] [Google Scholar]
- 39.Chir JL, Liao JH, Lin YC, Wu SH. The N-terminal sequence after residue 247 plays an important role in structure and function of Lon protease from Brevibacillus thermoruber WR-249. Biochem Biophys Res Commun. 2009;382(4):762–765. doi: 10.1016/j.bbrc.2009.03.109. [DOI] [PubMed] [Google Scholar]
- 40.Ishii Y, Amano F. Regulation of SulA cleavage by Lon protease by the C-terminal amino acid of SulA, histidine. Biochem J. 2001;358(Pt 2):473–480. doi: 10.1042/0264-6021:3580473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shah IM, Wolf RE., Jr Sequence requirements for Lon-dependent degradation of the Escherichia coli transcription activator SoxS: Identification of the SoxS residues critical to proteolysis and specific inhibition of in vitro degradation by a peptide comprised of the N-terminal 21 amino acid residues. J Mol Biol. 2006;357(3):718–731. doi: 10.1016/j.jmb.2005.12.088. [DOI] [PubMed] [Google Scholar]
- 42.Gonzalez M, Frank EG, Levine AS, Woodgate R. Lon-mediated proteolysis of the Escherichia coli UmuD mutagenesis protein: In vitro degradation and identification of residues required for proteolysis. Genes Dev. 1998;12(24):3889–3899. doi: 10.1101/gad.12.24.3889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mogk A, Deuerling E, Vorderwülbecke S, Vierling E, Bukau B. Small heat shock proteins, ClpB and the DnaK system form a functional triade in reversing protein aggregation. Mol Microbiol. 2003;50(2):585–595. doi: 10.1046/j.1365-2958.2003.03710.x. [DOI] [PubMed] [Google Scholar]
- 44.Mogk A, et al. Refolding of substrates bound to small Hsps relies on a disaggregation reaction mediated most efficiently by ClpB/DnaK. J Biol Chem. 2003;278(33):31033–31042. doi: 10.1074/jbc.M303587200. [DOI] [PubMed] [Google Scholar]
- 45.Rudyak SG, Brenowitz M, Shrader TE. Mg2+-linked oligomerization modulates the catalytic activity of the Lon (La) protease from Mycobacterium smegmatis. Biochemistry. 2001;40(31):9317–9323. doi: 10.1021/bi0102508. [DOI] [PubMed] [Google Scholar]
- 46.Brown PH, Schuck P. Macromolecular size-and-shape distributions by sedimentation velocity analytical ultracentrifugation. Biophys J. 2006;90(12):4651–4661. doi: 10.1529/biophysj.106.081372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ali Azam T, Iwata A, Nishimura A, Ueda S, Ishihama A. Growth phase-dependent variation in protein composition of the Escherichia coli nucleoid. J Bacteriol. 1999;181(20):6361–6370. doi: 10.1128/jb.181.20.6361-6370.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Henderson R, et al. Tilt-pair analysis of images from a range of different specimens in single-particle electron cryomicroscopy. J Mol Biol. 2011;413(5):1028–1046. doi: 10.1016/j.jmb.2011.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nørby JG. Coupled assay of Na+,K+-ATPase activity. Methods Enzymol. 1988;156:116–119. doi: 10.1016/0076-6879(88)56014-7. [DOI] [PubMed] [Google Scholar]
- 50.Lindsley JE. Use of a real-time, coupled assay to measure the ATPase activity of DNA topoisomerase II. Methods Mol Biol. 2001;95:57–64. doi: 10.1385/1-59259-057-8:57. [DOI] [PubMed] [Google Scholar]
- 51.Gottesman S, Roche E, Zhou Y, Sauer RT. The ClpXP and ClpAP proteases degrade proteins with carboxy-terminal peptide tails added by the SsrA-tagging system. Genes Dev. 1998;12(9):1338–1347. doi: 10.1101/gad.12.9.1338. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.