

Abstract
Cyclin-dependent kinases (Cdks) coordinate cell division, and their activities are tightly controlled. Phosphorylation of threonine 14 (T14) and tyrosine 15 (Y15) inhibits Cdks and regulates their activities in numerous physiologic contexts. Although the roles of Cdk1 inhibitory phosphorylation during mitosis are well described, studies of Cdk2 inhibitory phosphorylation during S phrase have largely been indirect. To specifically study the functions of Cdk2 inhibitory phosphorylation, we used gene targeting to make an endogenous Cdk2 knockin allele in human cells, termed Cdk2AF, which prevents Cdk2 T14 and Y15 phosphorylation. Cdk2AF caused premature S-phase entry, rapid cyclin E degradation, abnormal DNA replication, and genome instability. Cdk2AF cells also exhibited strikingly abnormal responses to replication stress, accumulated irreparable DNA damage, and permanently exited the cell cycle after transient exposure to S-phase inhibitors. Our results reveal the specific and essential roles of Cdk2 inhibitory phosphorylation in the successful execution of the replication stress checkpoint response and in maintaining genome integrity.
Keywords: Wee1, cyclin A
Cyclin-dependent kinases (Cdks) are regulated by positive and negative phosphorylations (1). T14/Y15 phosphorylation by the Wee1 and Myt1 kinases inhibits Cdks by preventing ATP binding, and these conserved phosphorylations are highly regulated during the cell cycle and by signaling pathways (1, 2). Wee1 and Myt1 are opposed by cell division cycle 25 (Cdc25) phosphatases, which, in mammals, comprise three related enzymes that dephosphorylate T14/Y15 to promote Cdk activity (2–4).
Wee1 and Cdc25 functions in mammals are best understood in G2/M, where Cdk1 inhibitory phosphorylation is required for mitotic progression and DNA damage responses. Cdk1 regulates Wee1 and Cdc25C by phosphorylation, which results in feedback loops that produce switch-like mitotic transitions (5). DNA damage pathways also use Cdk1 T14/Y15 phosphorylation to prevent mitotic entry by inhibiting Cdk1 (6, 7).
Cdk2 is activated by cyclin E and cyclin A; cyclin E-Cdk2 regulates G1 progression and S-phase entry, whereas cyclin A-Cdk2 acts later in S phase and mitosis (8, 9). Cdk2 is also regulated by Wee1 and Cdc25A, but the consequences of Cdk2 inhibitory phosphorylation are less well understood than for Cdk1 (10-13). Studies of Wee1 and Cdc25A have implicated Cdk2 inhibition in DNA replication and S-phase DNA damage checkpoints (14–17). However, the contributions of Cdk1 vs. Cdk2 to these phenotypes have been difficult to disentangle because manipulations such as Wee1 inhibition concurrently impact multiple Cdks. Indeed, a recent mouse model unexpectedly found that Cdk2 T14/Y15 phosphorylation is not required for DNA damage-induced cell cycle arrest, but instead has roles in S-phase entry and centrosome duplication (18).
Cyclin E-Cdk2 activity peaks at the G1-S transition, and its periodicity results from regulated transcription and cyclin E degradation by the SCF (Skp1/Cul1/F-box protein)-Fbw7 (F-box and WD repeat domain containing 7) ubiquitin ligase (8, 19–21). Deregulated cyclin E activity causes proliferative defects, chromosomal instability, and carcinogenesis (22–26). Because cyclin E degradation is triggered by autophosphorylation, Fbw7 only targets cyclin E in active Cdk2 complexes (27, 28). We previously found that cyclin E-Cdk2 activity is inversely correlated with Cdk2 T14/Y15 phosphorylation and hypothesized that Cdk2 inhibitory phosphorylation regulates cyclin E stability (29).
Here, we examined the functions of Cdk2 inhibitory phosphorylation in S phase by constructing an endogenous Cdk2 knockin allele in human cells that prevents its inhibitory phosphorylation while leaving the rest of the Wee1/Cdc25 pathway intact. We found that Cdk2 inhibitory phosphorylation not only directly regulates cyclin E degradation, but is also essential for maintaining genome stability. Moreover, the inability to inhibit Cdk2 by phosphorylation during replication stress caused irreparable DNA damage and permanent cell cycle exit. Our data demonstrate the specific and essential roles of Cdk2 inhibitory phosphorylation during S phase and replication stress.
Results
Cdk2 Inhibitory Phosphorylation Regulates Cyclin E Stability.
We used adeno-associated virus gene targeting to make a Cdk2AF allele in Hct116 cells in which Cdk2 T14 and Y15 are converted to alanine and phenylalanine, respectively (Fig. S1A) (30). We obtained homozygous Cdk2AF/AF cells by retargeting Cdk2+/AF cells and verified the resulting genotypes (Fig. S1B). As expected, Cdk2 inhibitory phosphorylation was undetectable in Cdk2AF/AF cells (Fig. 1A and Fig. S2). Consistent with a previous study, Cdk2AF/AF cells had normal asynchronous cell cycle profiles (Fig. S1C) but entered S phase prematurely and contained elevated cyclin E-Cdk2 kinase activity when released from quiescence (Fig. 1 B and C) (18). Cyclin E abundance decreased more rapidly in Cdk2AF/AF cells progressing through the cell cycle than in Cdk2+/+ cells (Fig. 1C), which could reflect either decreased transcription or increased degradation. Cdk2AF did not affect cyclin E mRNA abundance (Fig. S1D), but pulse–chase analyses revealed accelerated cyclin E degradation in Cdk2AF/AF cells (t1/2 = 1.4 h) compared with Cdk2+/+ cells (t1/2 = 2.1 h; Fig. 1D). These data support the idea that Cdk2 inhibitory phosphorylation regulates cyclin E stability. However, because Cdk2AF/AF cells enter S phase prematurely, their increased cyclin E turnover could also be a secondary consequence of cell cycle kinetics.
Fig. 1.
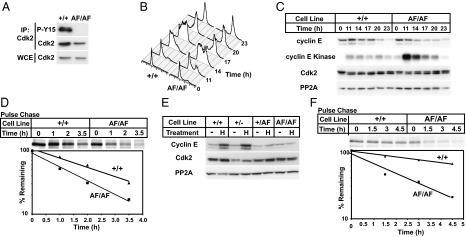
Cdk2AF/AF cells display early S-phase entry and accelerated cyclin E turnover. (A) Lysates were harvested from Cdk2+/+ and Cdk2AF/AF cells. Phospho-Y15 Cdk2 and total Cdk2 abundance were measured by Western blotting. (B) Cdk2+/+ and Cdk2AF/AF cells were arrested in G0/G1 by serum/leucine starvation and released into media containing nocodazole. Samples were harvested as indicated, and DNA content was analyzed by flow cytometry. (C) Cyclin E-Cdk2 abundance and kinase activity were assayed in the samples harvested in B. (D) Cyclin E half-life was determined by pulse–chase analysis beginning 13 h after serum/leucine release. (E) The indicated cell lines were treated with HU as indicated and assayed for cyclin E and Cdk2 levels. (C and E) PP2A-loading control. (F) Cyclin E turnover was measured in HU-arrested Cdk2+/+ and Cdk2AF/AF cells by pulse–chase analysis.
To determine whether Cdk2 inhibitory phosphorylation directly regulates cyclin E stability, we studied cyclin E during S-phase arrest, when its stability is isolated from cell cycle-induced changes. Moreover, because DNA damage response pathways destabilize Cdc25A during replication stress, Cdk2 inhibitory phosphorylation is high (31, 32). We thus examined cyclin E degradation in cells arrested in S phase by hydroxyurea (HU). HU treatment increased Cdk2 T14/Y15 phosphorylation in Cdk2+/+ and Cdk2+/− cells, but not in Cdk2+/AF or Cdk2AF/AF cells (Fig. S2). Interestingly, the Cdk2AF allele dominantly inhibited wt-Cdk2 phosphorylation in Cdk2+/AF cells (Fig. S2). This response is reminiscent of feedback by Cdk1 to prevent its own inhibition by phosphorylating Wee1 and Cdc25C, and suggests similar feedback between Cdk2 and Wee1/Cdc25A, because Cdk2 phosphorylates Wee1 and Cdc25A (33, 34). Cyclin E abundance normally increases in S phase-arrested cells, and we found that cyclin E accumulated to high levels in HU-treated Cdk2+/+ and Cdk2+/− cells (Fig. 1E). However, cyclin E did not accumulate in HU-arrested Cdk2+/AF or Cdk2AF/AF cells (Fig. 1E). Pulse–chase studies revealed that cyclin E was stabilized in Cdk2+/+ cells (t1/2 = 7.1 h), but remained labile in arrested Cdk2AF/AF cells (t1/2 = 2.1 h; Fig. 1F). We conclude that Cdk2 inhibitory phosphorylation directly regulates cyclin E stability.
Loss of Cdk2 Inhibitory Phosphorylation Causes Genomic Instability.
Cyclin E overexpression triggers a homeostatic p53-dependent induction of the p21 (Cip1) Cdk inhibitor, which feeds back to inactivate cyclin E-Cdk2 and suppress genomic instability (23, 24, 35). To determine whether Cdk2 hyperactivity caused by Cdk2AF has similar consequences, we examined p21 expression in Cdk2AF/AF cells. Indeed, Cdk2AF expression induced a similar response, as shown by increased p21 expression in Cdk2AF/AF cells, and increased cyclin E- and cyclin A-associated kinase activity after shRNA-mediated p21 depletion (Fig. S3A). To determine whether Cdk2AF expression actively drives p21 induction, we used allele-specific siRNA to silence Cdk2AF in Cdk2+/AF cells, which restored p21 abundance to near normal levels (Fig. S3B). Cyclin E deregulation causes genetic instability that is manifested by micronuclei formation (MN). This instability requires Cdk2 activity and is suppressed by p21 (24, 26, 35, 36). We thus determined whether Cdk2AF expression also caused genetic instability. Cdk2AF/AF cells exhibited increased MN compared with controls that was exacerbated by p21 silencing (% cells with MN—Cdk2+/+, 2.3 ± 1.4; Cdk2AF/AF: 6.9 ± 1.1; Cdk2AF/AF + p21 depletion: 11.3 ± 0.6; Fig. S3C), or p53 silencing (Cdk2+/+, 1.6 ± 0.2 in; Cdk2AF/AF, 4.6 ± 1.1; Cdk2AF/AF + p53 depletion, 9.4 ± 0.5; Fig. S3D). Endogenous Cdk2AF expression thus causes genetic instability that is suppressed by the p53-p21 pathway, indicating that Cdk2 inhibitory phosphorylation is required to maintain genome integrity in proliferating cells.
Cdk2 Inhibitory Phosphorylation Is Required for Recovery from Replication Stress.
We examined the role of Cdk2 inhibitory phosphorylation during recovery from replication stress by releasing Cdk2+/+ or Cdk2AF/AF cells from HU or aphidicolin (APH) arrest. Control cells treated with HU or APH readily progressed to G2/M after the inhibitors were removed, whereas most Cdk2AF/AF cells failed to exit S phase (Fig. 2 A and B). To further investigate the requirement for Cdk2 inhibitory phosphorylation in recovery from S-phase arrest, we treated Cdk2+/+ or Cdk2AF/AF cells with HU or APH for 24 h and examined their proliferation after inhibitor removal (Fig. 2C). Remarkably, transient HU or APH treatment prevented Cdk2AF/AF proliferation even 3 d after inhibitor removal, as assayed by both Alamar Blue (Fig. 2 D and E) and Crystal Violet (Fig. S4 A and B). Cdk2AF/AF cells also exhibited exquisite hypersensitivity to reduced concentrations of both inhibitors (Fig. 2 D and E and Fig. S4 A and B). To eliminate possible clonal selection artifacts, we similarly examined two independent Cdk2+/AF clones, both of which also failed to reenter the cell cycle and exhibited inhibitor hypersensitivity (Fig. S4 A and B).
Fig. 2.

Cdk2 inhibitory phosphorylation is required for recovery from replication stress. (A) Cdk2+/+ or Cdk2AF/AF cells were arrested in HU and released into media containing nocodazole. Samples were harvested as indicated, and DNA content was analyzed by flow cytometry. (B) Same as in A, but cells were arrested in Aphidicolin. (C) Experimental setup for D and E: On day 0 cells were seeded; on day 1, HU (D) or Aphidicolin (E) were added and were removed on day 2. Proliferation was assayed 3 d later by using Alamar Blue. Percent proliferation is calculated as percentage of proliferation relative to untreated cells. (F) Annexin V staining of Cdk2+/+ and Cdk2AF/AF cells treated with HU or staurosporine for 18 h. (STS, positive control). (G) Cdk2+/+ and Cdk2AF/AF cells were treated as in C and assayed for senescence-associated β-galactosidase activity 3 d after HU removal. Error bars indicate SD from three independent data points.
Because cells exit the cell cycle via several pathways, we examined Cdk2AF/AF cells for markers of apoptosis and senescence. Cdk2AF/AF cells exposed to HU or APH did not exhibit signs of apoptosis (annexin V staining), but instead stained strongly for senescence-associated β-galactosidase (Fig. 2 F and G). Cdk2 inhibitory phosphorylation is thus required for recovery from replication stress, and transient exposure to replication inhibitors initiated a Cdk2AF/AF cell senescence program.
Cdk2AF Causes DNA Damage During Replication Stress.
Normal replication intermediates are prone to damage, and stalled forks during S-phase arrest are particularly fragile. We thus determined whether Cdk2AF/AF cells accumulated DNA damage during S-phase arrest by examining phosphorylated histone H2AX (γH2AX), which marks double-strand breaks (DSBs) and other DNA lesions. Immunostaining revealed that proliferating Cdk2AF/AF cells exhibited normal levels of γH2AX staining, but that HU-arrested Cdk2AF/AF cells accumulated very high levels of γH2AX (Fig. 3A). We used flow cytometry to more quantitatively examine γH2AX abundance and found that Cdk2AF/AF cells rapidly accumulated excess γH2AX after HU treatment (Fig. 3 B and C and Fig. S5A). Finally, we used Western blotting to confirm the rapid and large increase in γH2AX (Fig. 3D). Additional experiments in arrested Cdk2AF/AF cells revealed that (i) increased γH2AX abundance also occurred during APH treatment (Fig. S5B), (ii) Cdk2AF/+ cells displayed increased γH2AX abundance during arrest (Fig. S5B), and (iii) the increased γH2AX abundance could not be resolved after inhibitor removal, suggesting that Cdk2AF resulted in irreparable DNA damage during replication stress (Fig. S5C).
Fig. 3.
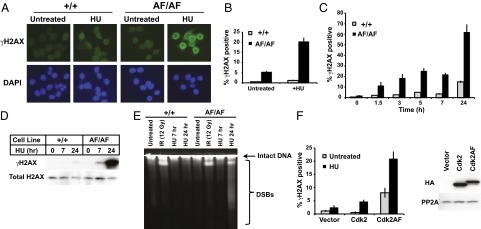
Loss of Cdk2 inhibitory phosphorylation causes DNA damage during replication stress. (A) Cdk2+/+ or Cdk2AF/AF cells were treated with HU (7 h) as indicated and stained with anti-γH2AX antibody and DAPI (to visualize nuclei). (B) Cdk2+/+ and Cdk2AF/AF cells were treated with HU (7 h) as indicated, and cells were harvested and analyzed by flow cytometry. The bar graph shows γH2AX staining that exceeded basal γH2AX induction in HU. (see Fig. S5A for γH2AX gating). (C) Cdk2+/+ and Cdk2AF/AF cells were treated with HU for increasing times and γH2AX-positive cells were identified by flow cytometry. (D) Cdk2+/+ and Cdk2AF/AF cells were treated with HU as indicated and γH2AX abundance and total H2AX abundance were analyzed by immunoblotting. (E) PFGE of Cdk2+/+ or Cdk2AF/AF cells that were untreated, treated with ionizing radiation (12 Gy), or HU. The migration of DNA containing DSBs is indicated. (F, Left) U2OS cells were transfected with vectors expressing Cdk2, Cdk2AF, or empty vector and treated with HU as shown. γH2AX abundance was assayed by flow cytometry. (F, Right) Ectopic Cdk2 or Cdk2AF protein expression. Error bars indicate SD from three independent data points.
We used pulsed-field gel electrophoresis (PFGE) to confirm that the γH2AX staining in Cdk2AF/AF cells reflected DSBs. Whereas gamma-irradiation caused DSBs in both cell types, HU caused DSBs in only the Cdk2AF/AF cells (Fig. 3E). Interestingly, we observed DSBs in Cdk2AF/AF cells after 24 h of HU treatment, but not after 7 h. Whether DSBs occur at 7 h and are below the level of PFGE detection or the γH2AX accumulation precedes DSB formation remains to be determined. Because our gene targeting studies used only Hct116 cells, we transiently transfected U2OS cells with either wt-Cdk2 or Cdk2AF vectors and found that Cdk2AF expression in U2OS cells led to elevated γH2AX levels after HU treatment (Fig. 3F), indicating that the Cdk2AF DNA damage phenotype is not unique to Hct116 cells. We conclude that Cdk2 inhibitory phosphorylation is essential for cellular recovery from replication stress, and that, in its absence, cells accumulate DNA damage and senesce.
Cyclin A-Cdk2 Activity Mediates the Cdk2AF Replication Stress Phenotype.
To test whether Cdk2 activity is required for DNA damage in Cdk2AF/AF cells, we pretreated cells with Roscovitine, a pharmacologic Cdk2 inhibitor, for 1 h before adding HU. Roscovitine greatly reduced γH2AX staining in HU-treated Cdk2AF/AF cells, indicating that Cdk2 activity is required to generate DNA damage (Fig. 4A). Because kinases mediate DNA damage signaling cascades, we postulated that additional kinases might be required for Cdk2AF-induced DNA damage. To this end, we performed an RNAi screen to identify kinases required for the Cdk2AF arrest phenotype that, when depleted, allowed HU-treated Cdk2AF/AF cells to reenter the cell cycle. Remarkably, Cdk2 was the top-scoring hit among ∼700 kinases tested (Fig. S6A), and two independent Cdk2 siRNAs almost completely restored Cdk2AF/AF cell cycle progression after HU release (Fig. 4B). Cdk2 expression and activity are thus required for the Cdk2AF DNA damage and cell cycle arrest phenotypes.
Fig. 4.

Constitutive Cyclin A-Cdk2 activity drives aberrant S-phase responses to replication stress. (A) Cdk2+/+ and Cdk2AF/AF cells were treated with HU for 6 h and Roscovitine as indicated, and γH2AX staining was analyzed by flow cytometry. (B, Upper) Cdk2AF/AF cells transfected with either control or two Cdk2 siRNAs were treated with 0.5 mM HU for 24 h, and percent proliferation relative to untreated controls was measured by Alamar Blue 3 d after HU release. (B, Lower) Cdk2 abundance after siRNA transfection. (C) Cdk2AF/AF cells transduced with control or lentiviruses expressing cyclin E or cyclin A shRNAs were grown asynchronously or treated with HU, and γH2AX abundance was assayed by flow cytometry. (D) Cyclin E and cyclin A abundance in samples from C.
Both cyclin E and cyclin A activate Cdk2, and we tested the role of each cyclin in the Cdk2AF DNA damage phenotype. Two independent shRNAs that resulted in near-complete cyclin E depletion did not alter γH2AX staining in HU-treated Cdk2AF/AF cells (Fig. 4 C and D). In contrast, shRNAs targeting cyclin A greatly reduced γH2AX staining, despite only partially depleting cyclin A (Fig. 4 C and D). Importantly, cyclin A depletion did not alter the S-phase fraction of HU-arrested Cdk2AF/AF cells (Fig. S6B). Thus, cyclin A-Cdk2, and not cyclin E-Cdk2, mediates the Cdk2AF-induced DNA damage.
Previous studies showed that DNA damage caused by Wee1 depletion requires the Mus81-Eme1 (essential meiotic endonuclease 1 homolog) endonuclease (15, 16) and we tested whether Mus81 contributed to the Cdk2AF/AF phenotype. Surprisingly, Mus81 depletion did not reduce Cdk2AF-induced γH2AX staining (Fig. S7 A and B). Although this discrepancy could reflect cell type-specific differences, we suspect that Mus81 is not required for Cdk2AF-induced DNA damage during replication stress. Collectively, these results indicate that failure to inhibit cyclin A-Cdk2 activity by inhibitory phosphorylation causes DNA damage during replication stress.
Cdk2AF/AF Cells Display Aberrant Replication Dynamics.
Cdk2 has roles in replication initiation and elongation. Therefore, we determined how Cdk2 inhibitory phosphorylation regulates replication dynamics in asynchronous cells, during HU arrest, and after HU release. We used microfluidics-assisted replication track analysis together with pulse labeling with thymidine analogs to analyze replication dynamics in Cdk2AF/AF cells (37, 38). We sequentially pulsed asynchronously growing cells with 5-ethynyldeoxyuridine (EdU) and 5-iododeoxyuridine (IdU) (Fig. 5A and Fig. S8A) and found that Cdk2AF/AF cells exhibited two distinct anomalies: (i) increased replication origin firing as shown by an increased percentage of active origins during the labeling period (Cdk2+/+ vs. CdkAF/AF: 2.1 ± 0.75% vs. 5.0 ± 2.1%; P = 0.042; Fig. 5B), and (ii) decreased fork speed as shown by shorter replication tracks (Cdk2+/+ vs. Cdk2AF/AF: 1.28 ± 0.12 kb/min vs. 0.93 ± 0.10 kb/min; P = 0.009; Fig. 5C). Cdk2 inhibitory phosphorylation is thus required for normal DNA replication dynamics in proliferating cells. Our results also suggest that the DNA replication defects due to Wee1 depletion, which show similar phenotypes, result from Cdk2 hyperactivity (15, 16).
Fig. 5.
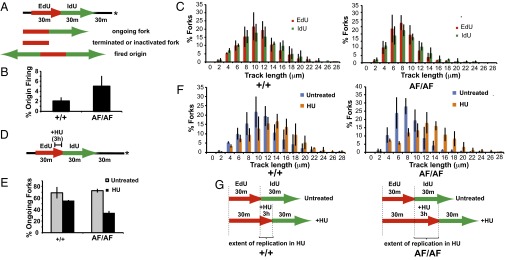
Cdk2AF/AF cells display aberrant replication dynamics. (A) Diagram of replication analysis by sequential labeling with EdU and IdU. Asterisk indicates time of harvest. Representative forks are shown. (B) Bar graph shows that origin firing during the labeling period is increased in Cdk2AF/AF cells (P = 0.042; Materials and Methods) (C) Ongoing forks progress slower in Cdk2AF/AF cells. Bar graphs show the percentages of replication forks with indicated EdU and IdU segment lengths. (D) Labeling scheme for HU arrest/release experiments. HU (2mM) was added at the end of a 30-min EdU labeling interval. After 3 h, HU and EdU were removed, and IdU was added. After 30 min, IdU was removed, and samples were harvested 30 min later. (E) Ongoing forks (EdU-IdU tracks) and terminated or inactivated forks (EdU only) were counted to determine percentage of ongoing forks. Cdk2AF/AF cells have significantly reduced fork restart rate after HU treatment (P = 0.0002). (F) Bar graphs show the percentages of replication forks with indicated EdU segment lengths in either untreated or HU-treated cells. (G) Graphic summary showing that Cdk2AF/AF cells display increased replication in HU. Brackets indicate the increased replication in HU in Cdk2AF/AF cells compared with Cdk2+/+ cells. Data in B, C, E, and F represent averages (with SD) from three experiments.
We next used DNA track analysis to examine replication dynamics in response to replication stress (Fig. 5D). After only 3 h of HU treatment, Cdk2AF/AF cells exhibited a significant defect in fork restart after HU release, as measured by the percentage of ongoing forks that resumed DNA synthesis after HU release (Fig. 5E and Fig. S8B, P = 0.0002 for HU-treated Cdk2+/+ and Cdk2AF/AF). Moreover, we found that Cdk2AF/AF cells replicated more DNA during HU arrest than Cdk2+/+ cells, as shown by the increased track lengths in HU-treated Cdk2AF/AF cells, compared with untreated cells (Fig. 5 F and G; see Materials and Methods for details on statistical analysis). The persistent fork progression in HU-treated Cdk2AF/AF cells indicates that Cdk2 inhibitory phosphorylation is required for normal execution of the S-phase checkpoint induced by replication stress. We conclude that Cdk2 inhibitory phosphorylation is required for normal replication dynamics in asynchronous and arrested cells.
Discussion
Our study reveals essential roles for Cdk2 T14/Y15 phosphorylation in maintaining genome integrity and in preventing DNA damage when S phase is stalled. Specifically, we have shown that Cdk2 inhibitory phosphorylation: (i) controls cyclin E degradation during S phase, (ii) acts in concert with p21 to control Cdk2 activity and protect against genomic instability, (iii) is required for the prevention of irreparable DNA damage during replication stress, and (iv) is responsible for ensuring proper replication dynamics in normal S phase and during replication stress. We cannot yet determine whether the high levels of γH2AX and DSBs in Cdk2AF/AF cells reflect increased DNA damage, impaired DNA repair, or both. Cdk2AF/AF cells share features with fission yeast checkpoint mutants (39) and budding yeast chromosomal instability mutants (40), where persistent DNA synthesis in HU is thought to cause replication fork collapse. A similar sequence of events may lead to irreparable DNA damage in Cdk2AF/AF cells.
Various perturbations of the pathways that impinge upon Cdk2 inhibitory phosphorylation have implicated Cdk2 inhibition as a critical step in checkpoint responses and DNA damage signaling during S phase. These perturbations include studies of Wee1 inhibition, Cdc25 overexpression, and DNA damage response kinases such as Chk1, which increase Cdk inhibitory phosphorylation by disabling Cdc25 (14–17, 41, 42). Our work now definitively demonstrates the essential role of Cdk2 inhibition in the replication stress checkpoint. However, although the Cdk2AF phenotype shares many features with those described with Wee1 and Chk1 inhibition, they are not identical. Most notably, we find that unlike Wee1 inhibition, Mus81 is not required for Cdk2AF-induced DNA damage during replication stress. Other differences include the accumulation of DNA damage in proliferating Wee1-depleted cells but not Cdk2AF cells, and the finding that Chk1 depletion causes apoptosis in arrested Hct116 cells, whereas Cdk2AF leads to senescence (14–16, 43). Although some of these discrepancies may simply reflect experimental conditions, it is likely that targets of Wee1 and Chk1 in addition to Cdk2 also contribute to these phenotypic differences.
The Wee1/Cdc25 axis is an attractive target for chemotherapy (44, 45). Indeed, Wee1 inhibitors thought to act primarily on Cdk1 and mitotic control are already in clinical trials. Cdc25 overexpression in cancers is associated with poor outcome and tumor aggressiveness (44), and causal roles for Cdc25 in carcinogenesis and genome instability have been demonstrated in cultured cells and mice (46, 47). Because Cdc25 overexpression is likely to phenocopy the Cdk2AF allele with respect to Cdk2 activity, we suggest that cancers with elevated Cdc25 expression may also exhibit hypersensitivity and persistent responses to S-phase chemotherapeutics. Thus, modulating Cdk2 inhibitory phosphorylation may represent a unique approach to sensitize cancer cells with hyperactive Cdk2 to S-phase chemotherapy.
Materials and Methods
See SI Materials and Methods for more detailed information on materials and methods.
Drug Treatments.
Unless otherwise noted, Hydroxyurea (Sigma) was used at 2 mM for 16–18 h, APH (Sigma) was used at 2 μM for 16–18 h, roscovitine (Sigma) was used at 25 μM, and staurosporine was used at 0.2 μM.
Flow Cytometry.
For cell cycle analysis, cells were trypsinized and fixed in 70% (vol/vol) ethanol at 4 °C overnight. Cells were washed in PBS, and DNA was stained with propidium iodide. γH2AX and annexin V staining were performed according to manufacturer’s instructions (Millipore, BD Biosciences). All samples were analyzed on a Canto 1 (Becton Dickinson) flow cytometer.
Cell Cycle Analysis, Growth Assays, and Micronucleation.
In cell cycle progression studies, cells were incubated for 48–52 h in serum- and leucine-free media (MP Biomedicals) and released into media containing 40 ng/mL nocodazole to prevent entry into the next cell cycle. For S-phase arrest, cells were treated with HU or APH for 16–18 h and released into media containing 40 ng/mL nocodazole. For growth assays, cells were seeded on day 0 at 2,000 cells per well in a 96-well plate. The following day, either HU or APH was added at various concentrations. Drugs were removed after 24 h, and proliferation was assayed 3 d after release. Proliferation was assayed by using either Alamar Blue (Invitrogen) or Crystal Violet. Senesence-associated (SA)-β-gal and micronucleation assays were performed as described (35, 48).
Adeno-Associated Virus (AAV) Gene Targeting.
Gene targeting, including viral production, purification, vector cloning, Hct116 transfection, screening (PCR, Southern blot, and genomic sequencing), and Cre-mediated removal of the selectable marker was performed as described or by standard techniques (29, 30). Complete primer and targeting vector sequences are available upon request. A representative targeting strategy and clone screening by Southern blotting is shown in Fig. S1A.
PFGE.
PFGE was performed as described (49). Briefly, 5 × 105 cells were melted into 1% (wt/vol) agarose (InCert agarose; Lonza) and digested overnight at 50 °C in 0.5% (wt/vol) EDTA, 1% (wt/vol) N-laurylsarcosyl, and 1 mg/mL proteinase K. Plugs were washed four times in Tris-EDTA (TE), loaded into a 1% (wt/vol) chromosome grade agarose gel, and separated by PFGE for 24 h (CHEF system, Bio-Rad Laboratories; 14 °C, 4 V/cm2, 120° angle, 60–240 s switch time). DNA was visualized with ethidium bromide.
RNAi Experiments.
pBABE p53 shRNA and control vectors were described (35). p21 shRNA, cyclin E shRNA, cyclin A shRNA, and control lentiviral vectors were from Open Biosystems. Cdk2AF siRNA sequence: sense strand 5′-GCGCGUUCGGAGUUGUGUATT-3′, antisense strand 5′-UACACAACUCCGAACGCGCTT-3′. Mus81 siRNA sequence was described (16). siRNA transfections were performed by using Lipofectamine RNAiMAX as directed (Invitrogen). For siRNA kinome screen, cells were seeded at 2,000 cells per well in a 96-well plate and reverse transfected with 2.5 pmole siRNA (human kinome library; Qiagen) by using RNAiMAX. After 48 h, 0.5 mM HU was added for 24 h to arrest cells in S phase, and cells were released. Growth was measured by Alamar Blue 4 d after release.
Microfluidics-Assisted Replication Track Analysis.
Microchannel fabrication, DNA fiber stretching, and replication track analysis was done as described (37, 38). For more details, see SI Materials and Methods.
Supplementary Material
Acknowledgments
We thank Jon Grim, Brian Fissel, and Celine Jacquemont for assistance and advice. This work was supported by National Institutes of Health Grants CA084069 (to B.E.C.), CA102742 (to B.E.C.), 5P01 CA77852 (to R.J.M.), and CA009657-22 (to B.T.H.); and the American Cancer Society (B.T.H.).
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1302927110/-/DCSupplemental.
References
- 1.Morgan DO. Principles of CDK regulation. Nature. 1995;374(6518):131–134. doi: 10.1038/374131a0. [DOI] [PubMed] [Google Scholar]
- 2.Atherton-Fessler S, Hannig G, Piwnica-Worms H. Reversible tyrosine phosphorylation and cell cycle control. Semin Cell Biol. 1993;4(6):433–442. doi: 10.1006/scel.1993.1051. [DOI] [PubMed] [Google Scholar]
- 3.Berry LD, Gould KL. Regulation of Cdc2 activity by phosphorylation at T14/Y15. Prog Cell Cycle Res. 1996;2:99–105. doi: 10.1007/978-1-4615-5873-6_10. [DOI] [PubMed] [Google Scholar]
- 4.Coleman TR, Dunphy WG. Cdc2 regulatory factors. Curr Opin Cell Biol. 1994;6(6):877–882. doi: 10.1016/0955-0674(94)90060-4. [DOI] [PubMed] [Google Scholar]
- 5.Ferrell JE, Jr, et al. Simple, realistic models of complex biological processes: Positive feedback and bistability in a cell fate switch and a cell cycle oscillator. FEBS Lett. 2009;583(24):3999–4005. doi: 10.1016/j.febslet.2009.10.068. [DOI] [PubMed] [Google Scholar]
- 6.Bartek J, Lukas J. DNA damage checkpoints: From initiation to recovery or adaptation. Curr Opin Cell Biol. 2007;19(2):238–245. doi: 10.1016/j.ceb.2007.02.009. [DOI] [PubMed] [Google Scholar]
- 7.Sørensen CS, Syljuåsen RG. Safeguarding genome integrity: The checkpoint kinases ATR, CHK1 and WEE1 restrain CDK activity during normal DNA replication. Nucleic Acids Res. 2012;40(2):477–486. doi: 10.1093/nar/gkr697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hwang HC, Clurman BE. Cyclin E in normal and neoplastic cell cycles. Oncogene. 2005;24(17):2776–2786. doi: 10.1038/sj.onc.1208613. [DOI] [PubMed] [Google Scholar]
- 9.Sherr CJ. Mammalian G1 cyclins. Cell. 1993;73(6):1059–1065. doi: 10.1016/0092-8674(93)90636-5. [DOI] [PubMed] [Google Scholar]
- 10.Chow JP, et al. Differential contribution of inhibitory phosphorylation of CDC2 and CDK2 for unperturbed cell cycle control and DNA integrity checkpoints. J Biol Chem. 2003;278(42):40815–40828. doi: 10.1074/jbc.M306683200. [DOI] [PubMed] [Google Scholar]
- 11.Gabrielli BG, Lee MS, Walker DH, Piwnica-Worms H, Maller JL. Cdc25 regulates the phosphorylation and activity of the Xenopus cdk2 protein kinase complex. J Biol Chem. 1992;267(25):18040–18046. [PubMed] [Google Scholar]
- 12.Gu Y, Rosenblatt J, Morgan DO. Cell cycle regulation of CDK2 activity by phosphorylation of Thr160 and Tyr15. EMBO J. 1992;11(11):3995–4005. doi: 10.1002/j.1460-2075.1992.tb05493.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kiyokawa H, Ray D. In vivo roles of CDC25 phosphatases: Biological insight into the anti-cancer therapeutic targets. Anticancer Agents Med Chem. 2008;8(8):832–836. doi: 10.2174/187152008786847693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Beck H, et al. Regulators of cyclin-dependent kinases are crucial for maintaining genome integrity in S phase. J Cell Biol. 2010;188(5):629–638. doi: 10.1083/jcb.200905059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Beck H, et al. Cyclin-dependent kinase suppression by WEE1 kinase protects the genome through control of replication initiation and nucleotide consumption. Mol Cell Biol. 2012;32(20):4226–4236. doi: 10.1128/MCB.00412-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Domínguez-Kelly R, et al. Wee1 controls genomic stability during replication by regulating the Mus81-Eme1 endonuclease. J Cell Biol. 2011;194(4):567–579. doi: 10.1083/jcb.201101047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Falck J, Mailand N, Syljuåsen RG, Bartek J, Lukas J. The ATM-Chk2-Cdc25A checkpoint pathway guards against radioresistant DNA synthesis. Nature. 2001;410(6830):842–847. doi: 10.1038/35071124. [DOI] [PubMed] [Google Scholar]
- 18.Zhao H, Chen X, Gurian-West M, Roberts JM. Loss of cyclin-dependent kinase 2 (CDK2) inhibitory phosphorylation in a CDK2AF knock-in mouse causes misregulation of DNA replication and centrosome duplication. Mol Cell Biol. 2012;32(8):1421–1432. doi: 10.1128/MCB.06721-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Strohmaier H, et al. Human F-box protein hCdc4 targets cyclin E for proteolysis and is mutated in a breast cancer cell line. Nature. 2001;413(6853):316–322. doi: 10.1038/35095076. [DOI] [PubMed] [Google Scholar]
- 20.Koepp DM, et al. Phosphorylation-dependent ubiquitination of cyclin E by the SCFFbw7 ubiquitin ligase. Science. 2001;294(5540):173–177. doi: 10.1126/science.1065203. [DOI] [PubMed] [Google Scholar]
- 21.Moberg KH, Bell DW, Wahrer DC, Haber DA, Hariharan IK. Archipelago regulates Cyclin E levels in Drosophila and is mutated in human cancer cell lines. Nature. 2001;413(6853):311–316. doi: 10.1038/35095068. [DOI] [PubMed] [Google Scholar]
- 22.Ekholm-Reed S, et al. Mutation of hCDC4 leads to cell cycle deregulation of cyclin E in cancer. Cancer Res. 2004;64(3):795–800. doi: 10.1158/0008-5472.can-03-3417. [DOI] [PubMed] [Google Scholar]
- 23.Loeb KR, et al. A mouse model for cyclin E-dependent genetic instability and tumorigenesis. Cancer Cell. 2005;8(1):35–47. doi: 10.1016/j.ccr.2005.06.010. [DOI] [PubMed] [Google Scholar]
- 24.Minella AC, et al. p53 and p21 form an inducible barrier that protects cells against cyclin E-cdk2 deregulation. Curr Biol. 2002;12(21):1817–1827. doi: 10.1016/s0960-9822(02)01225-3. [DOI] [PubMed] [Google Scholar]
- 25.Minella AC, et al. Cyclin E phosphorylation regulates cell proliferation in hematopoietic and epithelial lineages in vivo. Genes Dev. 2008;22(12):1677–1689. doi: 10.1101/gad.1650208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Spruck CH, Won KA, Reed SI. Deregulated cyclin E induces chromosome instability. Nature. 1999;401(6750):297–300. doi: 10.1038/45836. [DOI] [PubMed] [Google Scholar]
- 27.Clurman BE, Sheaff RJ, Thress K, Groudine M, Roberts JM. Turnover of cyclin E by the ubiquitin-proteasome pathway is regulated by cdk2 binding and cyclin phosphorylation. Genes Dev. 1996;10(16):1979–1990. doi: 10.1101/gad.10.16.1979. [DOI] [PubMed] [Google Scholar]
- 28.Won KA, Reed SI. Activation of cyclin E/CDK2 is coupled to site-specific autophosphorylation and ubiquitin-dependent degradation of cyclin E. EMBO J. 1996;15(16):4182–4193. [PMC free article] [PubMed] [Google Scholar]
- 29.Grim JE, et al. Isoform- and cell cycle-dependent substrate degradation by the Fbw7 ubiquitin ligase. J Cell Biol. 2008;181(6):913–920. doi: 10.1083/jcb.200802076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Russell DW, Hirata RK. Human gene targeting by viral vectors. Nat Genet. 1998;18(4):325–330. doi: 10.1038/ng0498-325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bartek J, Lukas C, Lukas J. Checking on DNA damage in S phase. Nat Rev Mol Cell Biol. 2004;5(10):792–804. doi: 10.1038/nrm1493. [DOI] [PubMed] [Google Scholar]
- 32.Donzelli M, Draetta GF. Regulating mammalian checkpoints through Cdc25 inactivation. EMBO Rep. 2003;4(7):671–677. doi: 10.1038/sj.embor.embor887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hoffmann I, Draetta G, Karsenti E. Activation of the phosphatase activity of human cdc25A by a cdk2-cyclin E dependent phosphorylation at the G1/S transition. EMBO J. 1994;13(18):4302–4310. doi: 10.1002/j.1460-2075.1994.tb06750.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li C, Andrake M, Dunbrack R, Enders GH. A bifunctional regulatory element in human somatic Wee1 mediates cyclin A/Cdk2 binding and Crm1-dependent nuclear export. Mol Cell Biol. 2010;30(1):116–130. doi: 10.1128/MCB.01876-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Minella AC, Grim JE, Welcker M, Clurman BE. 2007. p53 and SCF(Fbw7) cooperatively restrain cyclin E-associated genome instability. Oncogene 26(48):6948–6953. [DOI] [PubMed]
- 36.Rajagopalan H, et al. Inactivation of hCDC4 can cause chromosomal instability. Nature. 2004;428(6978):77–81. doi: 10.1038/nature02313. [DOI] [PubMed] [Google Scholar]
- 37.Sidorova JM, Li N, Schwartz DC, Folch A, Monnat RJ., Jr Microfluidic-assisted analysis of replicating DNA molecules. Nat Protoc. 2009;4(6):849–861. doi: 10.1038/nprot.2009.54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sidorova JM, Kehrli K, Mao F, Monnat R., Jr Distinct functions of human RECQ helicases WRN and BLM in replication fork recovery and progression after hydroxyurea-induced stalling. DNA Repair (Amst) 2013;12(2):128–139. doi: 10.1016/j.dnarep.2012.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sabatinos SA, Green MD, Forsburg SL. Continued DNA synthesis in replication checkpoint mutants leads to fork collapse. Mol Cell Biol. 2012;32(24):4986–4997. doi: 10.1128/MCB.01060-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Poli J, et al. dNTP pools determine fork progression and origin usage under replication stress. EMBO J. 2012;31(4):883–894. doi: 10.1038/emboj.2011.470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Boutros R, Dozier C, Ducommun B. The when and wheres of CDC25 phosphatases. Curr Opin Cell Biol. 2006;18(2):185–191. doi: 10.1016/j.ceb.2006.02.003. [DOI] [PubMed] [Google Scholar]
- 42.Sørensen CS, et al. Chk1 regulates the S phase checkpoint by coupling the physiological turnover and ionizing radiation-induced accelerated proteolysis of Cdc25A. Cancer Cell. 2003;3(3):247–258. doi: 10.1016/s1535-6108(03)00048-5. [DOI] [PubMed] [Google Scholar]
- 43.Gagou ME, Zuazua-Villar P, Meuth M. Enhanced H2AX phosphorylation, DNA replication fork arrest, and cell death in the absence of Chk1. Mol Biol Cell. 2010;21(5):739–752. doi: 10.1091/mbc.E09-07-0618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Boutros R, Lobjois V, Ducommun B. CDC25 phosphatases in cancer cells: Key players? Good targets? Nat Rev Cancer. 2007;7(7):495–507. doi: 10.1038/nrc2169. [DOI] [PubMed] [Google Scholar]
- 45.Stathis A, Oza A. Targeting Wee1-like protein kinase to treat cancer. Drug News Perspect. 2010;23(7):425–429. doi: 10.1358/dnp.2010.23.7.1490760. [DOI] [PubMed] [Google Scholar]
- 46.Galaktionov K, et al. CDC25 phosphatases as potential human oncogenes. Science. 1995;269(5230):1575–1577. doi: 10.1126/science.7667636. [DOI] [PubMed] [Google Scholar]
- 47.Ray D, Kiyokawa H. CDC25A phosphatase: A rate-limiting oncogene that determines genomic stability. Cancer Res. 2008;68(5):1251–1253. doi: 10.1158/0008-5472.CAN-07-5983. [DOI] [PubMed] [Google Scholar]
- 48.Debacq-Chainiaux F, Erusalimsky JD, Campisi J, Toussaint O. Protocols to detect senescence-associated beta-galactosidase (SA-betagal) activity, a biomarker of senescent cells in culture and in vivo. Nat Protoc. 2009;4(12):1798–1806. doi: 10.1038/nprot.2009.191. [DOI] [PubMed] [Google Scholar]
- 49.Herschleb J, Ananiev G, Schwartz DC. Pulsed-field gel electrophoresis. Nat Protoc. 2007;2(3):677–684. doi: 10.1038/nprot.2007.94. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.