

Abstract
We have devised protein-folding sensors that link protein stability to TEM-1 β-lactamase activity. Addition of osmolytes and other compounds with chemical chaperone activity to the growth media of bacteria containing these sensors increases β-lactamase activity up to 207-fold in a dose-dependent manner. This enables the rapid detection and sensitive quantification of compounds that enhance in vivo protein stability.
Keywords: in vivo, folding sensor, β-lactamase, osmolytes, protein stability
INTRODUCTION
Proteins are often frustratingly unstable, creating major problems in their biochemical and structural analyses [1]. Despite the presence of molecular chaperones that assist protein folding in vivo, as much as 30% of newly synthesized polypeptides form misfolded conformers in the cell [2; 3; 4]. Not surprisingly, a large number of disease states have been associated with protein misfolding. These include cystic fibrosis, Alzheimer’s disease, and Huntington’s disease [5]. A number of naturally occurring low molecular weight compounds, such as glycerol, sorbitol, and L- proline, are known to stabilize proteins in vivo [6]. These compounds were first identified as natural osmolytes in living tissues. They act to maintain cell volume and stabilize macromolecules under conditions of environmental stress like high osmotic and salt pressure [7; 8; 9]. Subsequent studies showed that these compounds stabilize proteins in vitro and assist in their refolding [10]. Intense efforts have also resulted in the discovery of compounds termed pharmacological chaperones that specifically stabilize disease related mutant proteins [11; 12]. The identification of chemical compounds or conditions that help stabilize proteins typically involves an expensive and time-consuming trial and error process. For all these reasons, a simple assay that allows for the rapid identification of stabilizing compounds would be very useful.
We present here a method that allows us to readily assess the influence of chemical compounds on the in vivo stability of proteins. Our approach is based on a sandwich fusion in which an unstable test protein is inserted into β-lactamase [1]. The in vivo stability of the test protein determines β-lactamase enzymatic activity, which can be measured by a simple colorimetric assay [13]. This results in a simple, sensitive, and quantitative assay that can be used to screen for chemicals that enhance protein stability in vivo.
In our sandwich fusion constructs that serve as protein stability indicators, a test protein is inserted between amino acids 196 and 197 of β-lactamase [1]. If the inserted protein is stable, the N- and C-terminal portions of β-lactamase will remain close enough together to form a functional entity that confers enzymatic activity. Conversely, unstable protein inserts will be targeted for proteasomal degradation. This results in the separation of the two halves of β-lactamase and reduced enzymatic activity (Fig. 1). This system provides a sensitive and convenient way of linking the in vivo stability of test proteins that may lack an easily assayable function to β-lactamase activity for which a simple colorimetric assay is available.
Figure 1.
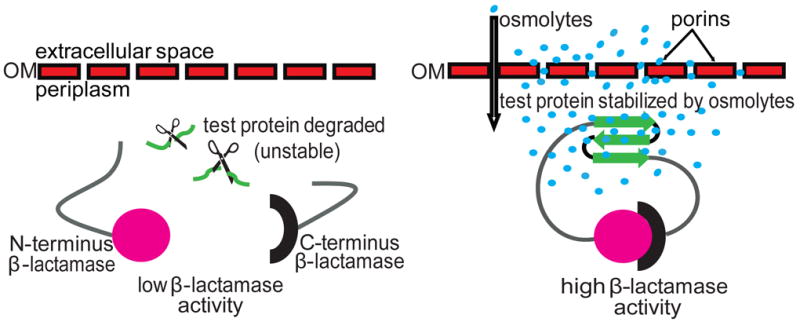
Schematic representation of a sandwich fusion system for assessing the influence of osmolytes on protein stability in vivo. An unstable test protein (green) is inserted into β-lactamase in the form of a sandwich fusion. (a) In the absence of osmolytes, the inserted protein is subject to degradation by periplasmic proteases, symbolized by scissors. This results in a separation of the two β-lactamase fragments and hence decreased β-lactamase activity. (b) Osmolytes (blue circles) diffuse into the periplasm through the holes generated in the outer membrane (OM) by endogenous porins. The presence of osmolytes favors a stable fold of the inserted protein. This allows an association of the N- (magenta circle) and C- (black semi-circle) fragments of β-lactamase, resulting in high enzymatic activity.
Due to the presence of outer membrane porins, the periplasm of gram-negative bacteria is permeable to molecules smaller than ~600 Da [14; 15]. We reasoned that the periplasmic location of our protein folding biosensors allows for a simple enzymatic readout for the presence of osmolytes and chemical and pharmacological chaperones in the media that positively affect the folding of unstable proteins.
MATERIALS AND METHODS
Chemicals and antibodies
Nitrocefin was purchased from Calbiochem (La Jolla, CA). 10 mg/ml stock solutions of nitrocefin were prepared in DMSO and stored at −20 °C. A 500 μg/ml working solution of nitrocefin was prepared in 100 mM sodium phosphate buffer, pH 7.0 prior to each experiment. Since nitrocefin is extremely light sensitive in solution, steps involving nitrocefin handing were performed in the dark and tubes were wrapped in aluminum foil. B-PER bacterial protein extraction reagent was purchased from Thermo Scientific (Rockford, IL). EDTA-free Protease inhibitor cocktail was purchased from Roche (Indianapolis, IN). EDTA, NaCl, urea, and glycerol were purchased from MP Biomedical LLC (Solon, OH). All other chemicals were purchased from Sigma Aldrich (St. Louis, MO). All additives tested as osmolytes were of analytical grade and filter sterilized after dissolving in LB. Polyclonal anti-β-lactamase antibody was obtained from Millipore (Billerica, MA). Polyclonal GroEL and DnaJ, and monoclonal DnaK antibody were purchased from Stressgen (Ann Arbor, MI).
Bacterial strains and expression vectors
NEB 10-beta cells were used for cloning. After cloning, plasmids were transformed into MG1655 ΔampCΔhsdR or NEB 10-beta strains for subsequent experiments. Wild type (WT) β-lactamase was expressed from pBR322. Plasmids for the expression of β-lactamase-link (pBR322*link) or β-lactamase -Im7-β-lactamase (pBR322* -Im7) were constructed as previously described [1]. Human muscle acylphosphatase 2 (hAcP2) was expressed from the plasmid pMB1 (pMB1- AcyP2). pMB1 was constructed by cloning together the origin of replication and antibiotic resistance marker of pBR322 with the arabinose inducible control elements of pBAD33. To achieve this, the β-lactamase gene with the linker encoding region was amplified from pBR322*link and cloned into pBAD33 using EcorI and XbaI sites. Whole plasmid PCR was then performed to introduce additional restriction sites for cloning Acp2 without altering the amino acid sequence of β-lactamase or the linker region.
A variant of AcP2 where the cysteine at position 21 has been mutated to serine was used for assays. This variant is used to avoid folding kinetics complexities and has essentially the same properties as WT AcP2 [16].
Bacterial growth
Bacterial growth was performed in 5 ml tubes in LB (Lysogeny Broth) medium. In all experiments, plasmids were maintained by growth in tetracycline (12 μg/ml). To prepare cultures for the β-lactamase assay, each strain containing the desired plasmids was inoculated from plates into 5 ml tubes and grown overnight in LB in a rotary drum incubator at 600 rpm at 37 °C. For strains expressing Im7 fusions 10 μl aliquots of overnight cultures were used to inoculate 5 ml LB medium containing the appropriate additives and grown overnight at 37 °C. For hAcP2, 100 μl of overnight cultures were used to inoculate 5 ml LB and grown for 2.5 hours and then induced with 0.5 % arabinose and grown for an additional 4 hours.
Bacterial harvest and lysis
2 or 4 ml of the bacterial cell cultures, for the Im7 and hAcP2 expressing strains respectively were harvested by centrifuging at 13,000 rpm for 10 min at 4 °C and the supernatant discarded. The pellets were resuspended in 1 ml (or 0.5 ml for hAcP2) ice cold B-PER lysis buffer supplemented with one tablet per 10 ml of Complete mini® protease inhibitor cocktail EDTA free and 20 mM EDTA and incubated for 1 hr with shaking at 1000 rpm at 4 °C to ensure efficient lysis. The lysate was then centrifuged at 13,000 rpm for 20 min at 4 °C to separate soluble proteins from the insoluble fraction. The lysate supernatant was transferred to new 96-well polypropylene blocks kept on ice and used for subsequent β-lactamase activity assays.
β-lactamase colorimetric assay
To quantify β-lactamase activity, 10 μl of nitrocefin (100 μM) was added to each well of a clear-bottom 96-well microtiter plate (Costar, Corning, NY). 90 μl of cell lysate, appropriately diluted in lysis buffer to ensure that the linear stage of hydrolysis reaction is monitored, was added to each well containing nitrocefin. Nitrocefin alone with buffer was used a blank. The increase in absorbance at 486 nm over time was recorded with a BioTek Synergy 2 plate reader (Winooski, VT). Nitrocefin hydrolysis was monitored for 30 min and rates of hydrolysis were calculated from plots of the linear range of increasing absorbance. The obtained hydrolysis rates were normalized to the cell OD measured at 600 nm of the respective sample.
Western blot analysis
Whole cell extracts and Western blot analysis were performed as previously described [1]. Briefly, 1 ml of overnight culture was harvested and pelleted as described above. The cell pellet was resuspended with the appropriate amounts of 1 × phosphate buffered saline (137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 1.76 mM KH2PO4, pH 7.0) to obtain an OD at 600 nm of 5. 100 μl of the resuspension was transferred into a new 1 ml tube and mixed with 25 μl of reducing loading dye, leading to a final cell OD at 600 nm of 4. The mixture was incubated at 100 °C for 10 min to lyse the cells and centrifuged at 13, 000 rpm for 1 minute. The resulting total cell extract was separated on a 7% Tris-acetate precast gel (Invitrogen, Carlsbad, CA) and probed with antibodies.
Quantitative real-time polymerase chain reaction
Overnight cultures were diluted 1:100 in LB and grown until early-log phase under tetracycline selection. 3 ml of these cultures were harvested and 6 ml of RNA Protect Reagent was added to the cultures to stabilize the RNA. RNA was extracted using Qiagen’s RNeasy mini kit (Valentia, CA). The total mRNA extracted was diluted to 0.75 μg/μl for cDNA synthesis. cDNA was synthesized using random hexamer primers to prime total mRNA by incubation at 65 °C for 5 min followed by incubation on ice for 5 min. Reverse transcription was carried out by incubation of the total mRNA-random hexamer mix at 25 °C for 10 min, 50 °C for 55 min, followed by incubation at 80 °C for 10 min to inactivate the reverse transcriptase. Following reverse transcription, the mRNA was digested with RNase H. Quantitative real-time polymerase chain reaction was performed with gene-specific primers using SYBR green (Invitrogen) according to the manufacturer’s instructions.
Presentation of data
The experiments presented in the figures were performed at least three times (unless stated otherwise). The data are shown as mean ± standard deviation from representative experiments.
RESULTS AND DISCUSSION
We have previously developed fusion biosensors to assess protein stability in vivo [1]. Using these biosensors, we have shown that there is a very good relationship between the thermodynamic stability of mutant variants of the immunity protein 7 (Im7) test protein and the β-lactamase activity of strains containing tripartite sandwich fusions made between these test protein variants and β-lactamase [1]. This fusion approach enables one to link the in vivo stability of Im7, which is difficult to assay for, with the simple enzymatic assay for β-lactamase. We wondered if our previously developed protein stability biosensors could be adapted to use β-lactamase activity to detect the presence of compounds that when added to the media affected the stability and therefore the steady-state level of the biosensor. Examples of such compounds are osmolytes or chemical chaperones. β-lactamase is active only when expressed in the periplasmic space and inactive when expressed in the cytosol [17]. Thus, protein stability biosensors based on monitoring β-lactamase activity are ideal for detecting changes in the periplasmic folding environment. We have extensively utilized Im7 as a test protein because of the ready availability of mutants with well-defined levels of stability and the well-studied nature of Im7’s folding pathway [18]. We have previously shown that very unstable mutants of the test protein Im7, when inserted into our stability biosensors, show an up to 4 fold enhancement β-lactamase levels upon overproduction of the periplasmic chaperone Spy [19]. More stable mutants of Im7 and wild type Im7 show smaller or insignificant fold increases in β-lactamase levels upon the overproduction of this chaperone (unpublished data). Given that unstable insert proteins provide the possibility of substantial stabilization via the action of chemical chaperones and osmolytes [1; 18], we reasoned that using unstable mutants of Im7 would allow us to very sensitively detect changes in the folding environment due to chemical chaperone or osmolyte additions to the media. As our first insert protein, we tested Im7 I22V, which is known to be substantially unfolded[18], into β-lactamase via flexible glycine-serine linkers. This resulted in the protein stability indicators fusion β-lactamase -Im7 I22V-β-lactamase.
Insertion of Im7 I22V into β-lactamase led to a 500-fold decrease in enzymatic activity in cell extracts compared to cell extracts of strains expressing WT β-lactamase in the absence of any additive (Fig. 2a), reflecting the very poor stability of the inserted test protein. We then tested the effect of a number of additives that are known to act as protein stabilizing agents or osmolytes in vitro and in vivo [6; 7; 10; 20] on the β-lactamase activity of this protein stability indicator. Cultivation of cells expressing β-lactamase -Im7 I22V- β-lactamase in the presence of the osmolytes trimethylamine N-oxide (TMAO), betaine, arabitol, glycine, sucrose, L-proline, sorbitol and glycerol substantially restored β-lactamase activity to 4–39 % of that seen for WT β-lactamase in the absence of any insert in LB (Fig. 2a). These results correspond to 22- to 207-fold increases in the β-lactamase activities of the sandwich fusion β-lactamase -Im7 I22V- β-lactamase in the presence of additives compared to the cultivation of the strain in the absence of these additives in normal LB media (Fig. 2b). The largest increases (157- and 207-fold) were observed for 1 M sorbitol and 2 M glycerol, which correspond to 29 % and 39 % restoration of WT β-lactamase activity in the absence of additives, respectively (Fig 2 a and b). We examined the four most potent osmolytes (glycerol, sorbitol, L-proline, and sucrose) further to determine whether the increase in β-lactamase activity was dependent on the concentration of the osmolyte added to the medium. We found activity to be very nicely dose-dependent for all four compounds, suggesting that a higher concentration of these osmolytes leads to increased stabilization of the test protein (Fig. 3).
Figure 2.
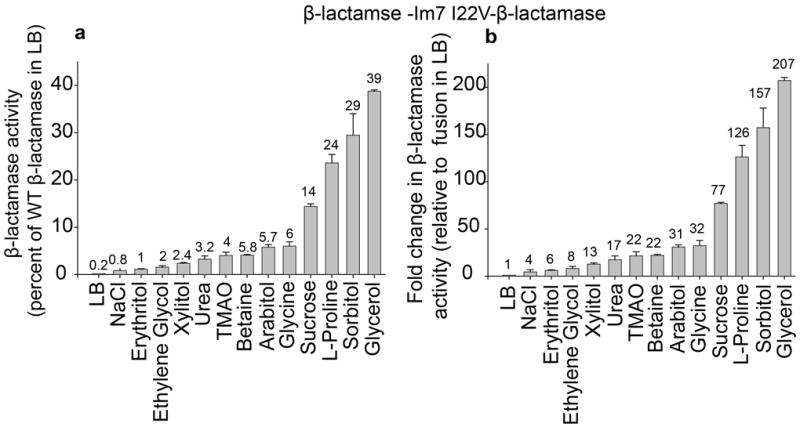
Influence of selected osmolytes on the β-lactamase activity of tripartite fusions containing the destabilized Im7 variant Im7 I22V. (a) β-lactamase activity (determined by measuring absorbance at 486 nm over time and adjusted to the cell OD of the cell extract) in whole cell extracts of Escherichia coli MG1655 ΔampCΔhsdR expressing the tripartite fusion β-lactamase -Im7 I22V-β-lactamase in the presence or absence of protein stabilizing agents in the cultivation medium. Enzymatic activity is expressed as % of the activity of cells expressing WT β-lactamase in LB (100%). (b) Fold change in β-lactamase activity in the presence of osmolytes in the cultivation medium compared to activity in LB of the same strain. 0.25 M NaCl was used as a negative control. The additives were all present at 0.5 M except for glycerol, which was used at 2 M, sorbitol, which was used at 1 M, urea, which was used at 200 mM and glycine, which was used at 0.1 M. Mean values ± standard deviations are shown for independent triplicate experiments.
Figure 3.
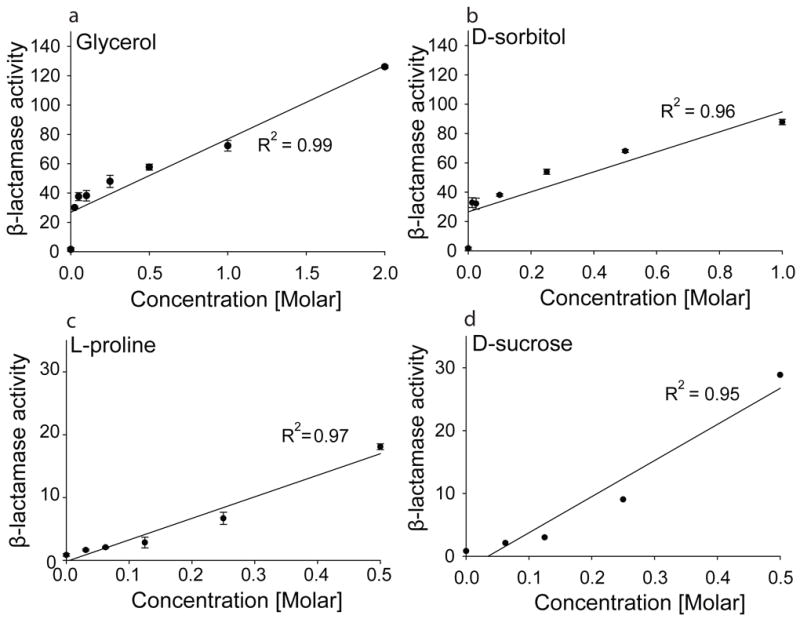
Addition of the osmolytes glycerol, D-sorbitol, L-proline, and D-sucrose to the cultivation medium restores β-lactamase activity in a dose-dependent manner in extracts of E. coli MG1655 ΔampCΔhsdR expressing β-lactamase -Im7 I22V-β-lactamase. Absolute enzymatic activity is expressed as the rate of hydrolysis of nitrocefin (determined by measuring absorbance at 486 nm over time), adjusted to the cell OD of the cell extract. Mean values ± standard deviations are shown for independent triplicate experiments except for sucrose in which data are from one experiment
The increase in the enzymatic activity of the reporter protein (β-lactamase) in the presence of osmolytes correlates well with an increase in steady-state protein levels of WT β-lactamase itself, as observed by western blotting using anti-β-lactamase antibody (Figs. S1 and S2 and S3). This suggests that the increase in β-lactamase activity for the β-lactamase -Im7 I22V- β-lactamase fusion is partly due to a stabilization of the reporter protein itself. However, since the fusion protein shows a much larger increase in β-lactamase activity and steady-state protein level in the presence of chemical chaperones and osmolytes compared to the increase observed for WT β-lactamase or a fusion containing the short 30 amino acid linker containing the multiple cloning sites for guest protein insertions (for Instance Im7) (Figs. S1 A and S3), the osmolytes seem to preferentially stabilize the insert protein rather than the reporter itself (Sucrose, for example showed a 16.5 fold increase in steady state protein level for strains expressing β-lactamase -Im7 I22V-β-lactamase versus only a 1.9 fold increase in steady-state level for strains expressing WT β-lactamase). Such preferential stabilization of Im7 I22V over β-lactamase by chemical chaperones is consistent with the relative stabilities of the two proteins that have a free energy of unfolding of -16 kJ/mol and -41.86 kJ/mol for Im7 I22V and WT β-lactamase respectively. These results indicate that osmolytes work at least in part by increasing the levels of the sandwich fusion in the bacterial periplasm. These increased levels could be the result of the osmolytes either enhancing the in vivo folding yield of the fusion protein or enhancing their in vivo proteolytic resistance or a synergistic combination of these effects. Although it is possible that osmolytes can also change the specific activity of enzymes by directly interacting with them [21], it is unlikely that these potential changes would be detected in our system since the β-lactamase assays were done after removing the osmolytes.
Even though molar quantities of osmolyte are generally used in vitro, our in vivo assay is sensitive enough to detect the effects of even millimolar quantities. For example, the addition of 25 mM glycerol or sorbitol led to 30- and 33-fold enhancements in β-lactamase activity respectively, for the Im7 I22V fusion (Fig. 3). That these compounds are effective at such low concentrations in vivo is surprising. Although the precise mechanism of protein stabilization by osmolyte and chemical chaperones is still unclear [3; 6; 20; 22; 23] many models propose that they act indirectly by inducing molecular crowding or by changing the structure and dynamics of water. These models suggest that the osmolytes must be present in high concentrations to be active. However, a direct interaction with the polypeptide backbone has also been proposed; this model might allow these compounds to be effective at lower concentrations as we observed [22]. For instance TMAO, a protecting osmolyte, appears to act by promoting H-bonding, an interaction that exists even within unfolded polypeptide chains such as in glycine serine linkers [24; 25; 26]. Our finding that these additives are active at lower concentrations in vivo is interesting and potentially important because it may make their clinical use more feasible.
One result that was initially surprising is that we saw a slight increase of 3.2 fold in β-lactamase activity in the presence of 0.2 M urea. Upon searching the literature, we found that a previous study has shown that the two most common denaturants, urea and guanidinium hydrochloride, can act as protein stabilizers when used in sub-denaturing concentrations [27]. The authors of this study suggest that these denaturants can act as chemical cross-linkers that help establish hydrogen bonds and van der Waal’s interactions between main chain and side-chain atoms of proteins leading to limited freedom of motion. This effect has been termed “protein stiffening” [27].
As described above, Im7 I22V was selected as a test protein because it is a very unstable Im7 variant destabilized by 8.9 kJ/mol relative to wild type Im7. We thought this high degree of instability would provide the potential for a high degree of stabilization through the action of osmolytes and chemical chaperones and reasoned that tripartite fusions between β-lactamase and Im7 I22V might make for a sensitive chemical chaperone biosensor. The large enhancement of β-lactamase enzymatic activity upon the addition of osmolytes and chemical chaperones shows that this choice was appropriate and enabled our development of a very sensitive sensor. Most of our experiments were performed using the β-lactamase -Im7 I22V-β-lactamase protein stability indicator. To determine if other similarly destabilized mutants of Im7 were also useful as osmolyte/chemical chaperone biosensors, we performed a number of experiments with an alternate protein stability indicator sandwich fusion, β-lactamase -Im7 L34A-β-lactamase. This biosensor utilizes a different destabilized Im7 mutant (Im7 L34A), which is destabilized by 7.7 kJ/mol relative WT Im7 and similar to the degree of destabilization shown by Im7 I22V. The chemical chaperone biosensor constructed using the Im7L34A variant gave very similar results to the one made with Im7 I22V both in terms of the fold increase of β-lactamase activity and β-lactamase steady-state protein level (Figs. S1 C and S4). To see if other destabilized test proteins could also be used to construct chemical chaperone biosensors we studied the response of tripartite fusions between β-lactamase and two destabilized mutants of hAcP2, C21S/M61A and C21S/L65V. These biosensors still responded to the chemical chaperones glycerol, sorbitol and trehalose, albeit to a much lesser degree than β-lactamase -Im7 I22V-β-lactamase or β-lactamase -Im7 L34A-β-lactamase (Fig S5).
We also tested the effects of osmolytes on a β-lactamase variant that did not contain an Im7 insert, but instead contained a 59-residue glycine-serine linker (β-lactamase -link long-β-lactamase). Cells expressing this construct that were cultivated in LB exhibited very low β-lactamase activities compared to WT β-lactamase. However, the activities were comparable to those measured in extracts from cells expressing tripartite fusions containing the destabilized Im7 mutants β-lactamase -Im7 I22V-β-lactamase or β-lactamase -Im7L34A-β-lactamase (Fig. S4a). This suggested that the insertion of this 59 residue linker into β-lactamase substantially destabilized the protein. Adding osmolytes to the media increased the β-lactamase activity of β-lactamase - link long (59 aa)-β-lactamase 19- to 77-fold (Fig. S4b), suggesting that these osmolytes not only help stabilize proteins, but can also stabilize proteins that contain flexible, unfolded structures like the long glycine-serine linker construct. Previously, we showed that β-lactamase activity in vivo could be used to quantitatively monitor the thermodynamic stability of various proteins inserted into β-lactamase [1]. Our results here raise the possibility that many kinds of polypeptides inserted into β-lactamase, including unstable mutants of Im7, hAcP2 and even a simple 59-residue glycine-serine linker, can be used as osmolyte and chemical chaperone detectors.
We also considered the possibility that the osmolytes were exerting their effect on β-lactamase activity indirectly, either by inducing expression of the fusions at the transcriptional level or by causing the induction of host encoded molecular chaperones[6; 28; 29], which then acted to stabilize the β-lactamase fusions. We therefore performed quantitative real-time polymerase chain reactions on total mRNA extracted from cells expressing WT β-lactamase, β-lactamase -link- β-lactamase or β-lactamase -Im7 I22V-β-lactamase cultured in the presence or absence of 2 M glycerol, 1 M sorbitol, or 0.5 M sucrose. We found no increase in mRNA encoding the fusion proteins; rather, a small osmolyte-dependent decrease was observed (Fig. S6). Similarly, quantitative real-time polymerase chain reactions on mRNA levels of periplasmic chaperones or proteins that influence chaperone expression, including DegP, Spy, RpoH, SurA, Skp, and CpxP showed no significant differences in the presence or absence of the osmolyte (Fig. S7). Further, Western blots with antisera against the molecular chaperones DnaJ, DnaK, and GroEL showed only small osmolyte-dependent increases and decreases that were neither of sufficient magnitude or direction to explain the increased levels of the tripartite fusions observed (Fig. S8).
Because of the relatively insensitive and laborious nature of previously available assays for osmolytes and chemical chaperones, most studies have tested the activity of only one or two of these compounds [28; 30], making it difficult to evaluate the relative efficiency of different osmolytes. Mishra et al ’s., in vitro study [10], however, used the same conditions and compared the ability of 5 different chemical additives (glycerol, sorbitol, xylitol, ethylene glycol, and erythritol) to enhance citrate synthase (CS) refolding yield at 25 °C. Consistent with our in vivo results, they found that glycerol and sorbitol were the most effective as osmolytes, with 2 M and 7 M glycerol enhancing CS refolding yield 1.5- and 2.25-fold, respectively. Sorbitol at 1 M was also effective, enhancing CS refolding yield 1.5-fold [10]. Like us, they also observed that ethylene glycol and erythritol were less effective as osmolytes than glycerol or sorbitol. Erythritol, which we found to be a very weak osmolyte, actually decreased refolding yield in their studies. Similar to our results, they found 0.5 M xylitol (the concentration we used) to be relatively ineffective. Thus, the rank order of effectiveness of the osmolytes in vitro was very similar to what we observe in vivo. However, our assay appears to be much more sensitive than the citrate synthase refolding assay, has a much greater dynamic range, and scales much better to osmolyte concentration (see Figs. 2 and 3).
In particular, we note that the increases that Mishra et al observe in citrate synthase refolding yield (up to 2.3 fold) are very much smaller than the up to 207 fold increases that we observe in stability biosensor activities. Further, Mishra et al observed a very poor correlation between osmolyte concentration and refolding yield [10]. Higher concentrations of osmolytes in general were found to actually decrease citrate synthase refolding yields and, as noted, some osmolytes like erythritol even decreased folding yields at all concentrations tested. Consistent with our observations, Mishra et al observed that addition of 3M glycerol increased the midpoint of guanidine hydrochloride denaturation of citrate synthase from ~1.3M to ~2M. However, measurement of midpoints of guanidine denaturation is laborious and required large amounts of purified protein. In contrast, our in vivo folding biosensor based assay for osmolytes not only shows much larger fold increases in the observed signal but also shows an excellent and positive correlation between osmolyte concentrations and β-lactamase activity (Fig. 3). It is clear that the relatively small effects that osmolytes have on in vitro refolding makes these types of assays much less suitable to screen for chemical chaperone activity of compounds compared to our in vivo biosensor screen.
CONCLUSION
Taken together, these results suggest that the increased β-lactamase activity and the increased steady-state protein levels observed in the presence of osmolytes are most likely attributable to the direct stabilizing effect osmolytes have on the unstable protein constructs. Our method relies on a simple colorimetric assay and should therefore be useful in screening for compounds that either generally enhance protein stability or enhance the stability of specific proteins. Because there is a thermodynamic link between ligand binding and protein stability [31], this assay may also be useful in screening for compounds that bind tightly to and therefore either stabilize or inhibit specific proteins, including those involved in human disease [32]. This capability may enable the rapid discovery of pharmacological chaperones or protein inhibitors of pharmacological interest and may facilitate the identification of compounds that stabilize proteins for crystallography.
Supplementary Material
Steady-state protein levels of β-lactamase and β-lactamase-Im7 fusion proteins in the absence or presence of different osmolytes in the cultivation medium. Whole cell extracts of cells expressing the corresponding constructs and cultivated in the presence or absence of osmolytes were separated on a 7% Tris-glycine SDS-PAGE gel. The proteins were then transferred to a polyvinylidene difluoride membrane and probed with an anti-β-lactamase antibody. Values beneath the respective Western blot bands represent the fold increase in protein level compared to cells cultivated in LB. Mean values ± standard deviations are shown for independent duplicate experiments.
Correlation of β-lactamase activity with steady-state β-lactamase protein levels. Fold increase in β-lactamase activity was plotted against fold increase in steady-state protein level for WT β-lactamase (a), β-lactamase - link (30 aa) - β-lactamase (b), and our tripartite fusion proteins (c and d). Graphs show linear correlations between the two variables. Fold changes are expressed as changes in activity or steady-state protein level in comparison to each strain in LB, expressed as 1.
β-lactamase activity in whole cell extracts of E. coli MG1655 ΔampCΔhsdR expressing either WT β-lactamase or β-lactamase- link (30 aa) - β-lactamase was measured in the presence or absence of osmolytes in the cultivation medium. Activity is expressed as fold change relative to activity in LB of the same strain. 0.25 M NaCl was used as a negative control. Mean values ± standard deviations are shown for independent triplicate experiments.
Effect of various osmolytes on β-lactamase activity in extracts of E. coli MG1655 ΔampCΔhsdR cells expressing β-lactamase -link (59 aa) -β-lactamase, β-lactamase -Im7 I22V-β-lactamase and β-lactamase -Im7 L34A-β-lactamase. (a) Enzymatic activity expressed as % of the activity of cells expressing WT β-lactamase in LB (100%). (b) Fold increase in β-lactamase activity in the presence of osmolytes in the cultivation medium relative to activity in LB of the same strain. In the absence of osmolytes, insertion of a 59-residue linker (black bars) results in very low enzymatic activity, similar to activities observed for β-lactamase-Im7 I22V-β-lactamase or β-lactamase -Im7 L34A-β-lactamase. In the presence of osmolytes like glycerol, sorbitol, and sucrose, the activity of cells expressing β-lactamase –link (59 aa)- β-lactamase is essentially restored to WT β-lactamase levels, whereas the activity of cells expressing β-lactamase Im7 I22V-β-lactamase or β-lactamase -Im7 L34A-β-lactamase is only restored to up to 20% or 35%, respectively, of WT β-lactamase levels (a). This trend is also reflected in the fold increases in β-lactamase activity upon cultivation in osmolytes compared to cultivation in LB (b). 0.25 M NaCl was used as a negative control. Mean values ± standard deviations are shown for independent triplicate experiments. The somewhat overall lower fold increases in β-lactamase activity observed for these experiments relative to those reported in Figure 2 reflects the observation that the β-lactamase activity of the β-lactamase -Im7 I22V-β-lactamase fusion in the absence of any media additive is extremely low and somewhat variable. This greatly affects the fold increases observed (b), but has a relatively small influence on the activities expressed as a % of WT in LB (a).
Effect of osmolytes on β-lactamase activity in extracts of E. coli NEB 10-beta cells expressing β-lactamase AcP2 C21S/M61A-β-lactamase and β-lactamase-AcP2 C21S/L65V-β-lactamase. Fold increase in β-lactamase activity in the presence of osmolytes in the cultivation medium relative to activity in LB of the same strain is shown. Mean values ± standard deviations are shown for independent triplicate experiments. The somewhat overall lower fold increases in β-lactamase activity observed for AcP2 relative to those reported for Im7 fusions indicates that some sensors are better in reporting for protein folding and activity in the presence or absence of media additive.
mRNA levels in the presence and absence of osmolytes. The presence of osmolytes in the culture media does not increase the mRNA levels of the fusion constructs indicating that osmolytes act on the protein level to stabilize the fusions rather than upregualting the expression of the proteins at the mRNA level. A small osmolyte induced decrease is observed for glycerol and sorbitol.
mRNA levels of periplasmic chaperones. Cells expressing the constructs were cultured in LB and harvested at mid-log phase. Total mRNA was extracted using Qiagen’s RNeasy mini kit. After cDNA synthesis (see Supporting Information Methods), quantitative real-time PCR was carried out using SYBR Orange according to the manufacturer’s instructions. Data were normalized for the mRNA level of DnaQ and fold increases were calculated with reference to mRNA levels in LB.
Steady-state protein levels of three molecular chaperones (DnaJ, DnaK, and GroEL) in response to the absence or presence of osmolytes. The crude extracts of whole cell lysates of WT β-lactamase, β-lactamase -link (30 aa) - β-lactamase, and tripartite fusions expressed in the presence or in the absence of the osmolytes were separated on a 7% Tris-glycine SDS-PAGE gel, the proteins transferred to polyvinylidene difluoride membrane, and probed with anti-DnaK antibody (a), anti-GroEL antibody (b), or anti-DnaJ antibody (c).
Acknowledgments
Funding Sources
This work was funded by the Howard Hughes Medical Institute.
Footnotes
Author Contributions
All authors have given approval to the final version of the manuscript.
Supporting Information
Supplementary figures can be found in the Supporting Information. This material is available free of charge via the Internet at http://www.sciencedirect.com.
The authors declare no competing financial interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Tsinatkeab T. Hailu, Email: tsenat@umich.edu.
Linda Foit, Email: lfoit@umich.edu.
James C. A. Bardwell, Email: jbardwel@umich.edu.
References
- 1.Foit L, Morgan GJ, Kern MJ, Steimer LR, von Hacht AA, Titchmarsh J, Warriner SL, Radford SE, Bardwell JC. Optimizing protein stability in vivo. Mol Cell. 2009;36:861–71. doi: 10.1016/j.molcel.2009.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hartl FU, Hayer-Hartl M. Converging concepts of protein folding in vitro and in vivo. Nat Struct Mol Biol. 2009;16:574–81. doi: 10.1038/nsmb.1591. [DOI] [PubMed] [Google Scholar]
- 3.Liu FF, Ji L, Zhang L, Dong XY, Sun Y. Molecular basis for polyol-induced protein stability revealed by molecular dynamics simulations. J Chem Phys. 2010;132:225103. doi: 10.1063/1.3453713. [DOI] [PubMed] [Google Scholar]
- 4.Schubert U, Anton LC, Gibbs J, Norbury CC, Yewdell JW, Bennink JR. Rapid degradation of a large fraction of newly synthesized proteins by proteasomes. Nature. 2000;404:770–4. doi: 10.1038/35008096. [DOI] [PubMed] [Google Scholar]
- 5.Voisine C, Pedersen JS, Morimoto RI. Chaperone networks: tipping the balance in protein folding diseases. Neurobiol Dis. 2010;40:12–20. doi: 10.1016/j.nbd.2010.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Arakawa T, Ejima D, Kita Y, Tsumoto K. Small molecule pharmacological chaperones: From thermodynamic stabilization to pharmaceutical drugs. Biochim Biophys Acta. 2006;1764:1677–87. doi: 10.1016/j.bbapap.2006.08.012. [DOI] [PubMed] [Google Scholar]
- 7.Papp E, Csermely P. Chemical chaperones: mechanisms of action and potential use. Handb Exp Pharmacol. 2006:405–16. doi: 10.1007/3-540-29717-0_16. [DOI] [PubMed] [Google Scholar]
- 8.Yancey PH. Organic osmolytes as compatible, metabolic and counteracting cytoprotectants in high osmolarity and other stresses. J Exp Biol. 2005;208:2819–30. doi: 10.1242/jeb.01730. [DOI] [PubMed] [Google Scholar]
- 9.Yancey PH, Blake WR, Conley J. Unusual organic osmolytes in deep-sea animals: adaptations to hydrostatic pressure and other perturbants. Comp Biochem Physiol A Mol Integr Physiol. 2002;133:667–76. doi: 10.1016/s1095-6433(02)00182-4. [DOI] [PubMed] [Google Scholar]
- 10.Mishra R, Seckler R, Bhat R. Efficient refolding of aggregation-prone citrate synthase by polyol osmolytes: how well are protein folding and stability aspects coupled? J Biol Chem. 2005;280:15553–60. doi: 10.1074/jbc.M410947200. [DOI] [PubMed] [Google Scholar]
- 11.Zhao JH, Liu HL, Lin HY, Huang CH, Fang HW, Chen SS, Ho Y, Tsai WB, Chen WY. Chemical chaperone and inhibitor discovery: potential treatments for protein conformational diseases. Perspect Medicin Chem. 2008;1:39–48. doi: 10.4137/pmc.s212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zheng W, Padia J, Urban DJ, Jadhav A, Goker-Alpan O, Simeonov A, Goldin E, Auld D, LaMarca ME, Inglese J, Austin CP, Sidransky E. Three classes of glucocerebrosidase inhibitors identified by quantitative high-throughput screening are chaperone leads for Gaucher disease. Proc Natl Acad Sci U S A. 2007;104:13192–7. doi: 10.1073/pnas.0705637104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Galarneau A, Primeau M, Trudeau LE, Michnick SW. Beta-lactamase protein fragment complementation assays as in vivo and in vitro sensors of protein protein interactions. Nat Biotechnol. 2002;20:619–22. doi: 10.1038/nbt0602-619. [DOI] [PubMed] [Google Scholar]
- 14.Csonka LN, Hanson AD. Prokaryotic osmoregulation: genetics and physiology. Annu Rev Microbiol. 1991;45:569–606. doi: 10.1146/annurev.mi.45.100191.003033. [DOI] [PubMed] [Google Scholar]
- 15.Nikaido H. Molecular basis of bacterial outer membrane permeability revisited. Microbiol Mol Biol Rev. 2003;67:593–656. doi: 10.1128/MMBR.67.4.593-656.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chiti F, Taddei N, Webster P, Hamada D, Fiaschi T, Ramponi G, Dobson CM. Acceleration of the folding of acylphosphatase by stabilization of local secondary structure. Nat Struct Biol. 1999;6:380–7. doi: 10.1038/7616. [DOI] [PubMed] [Google Scholar]
- 17.Tan R, Jiang X, Jackson A, Jin P, Yang J, Lee E, Duggan B, Stuve LL, Fu GK. E. coli selection of human genes encoding secreted and membrane proteins based on cDNA fusions to a leaderless beta-lactamase reporter. Genome Res. 2003;13:1938–43. doi: 10.1101/gr.1000903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Capaldi AP, Kleanthous C, Radford SE. Im7 folding mechanism: misfolding on a path to the native state. Nat Struct Biol. 2002;9:209–16. doi: 10.1038/nsb757. [DOI] [PubMed] [Google Scholar]
- 19.Quan S, Koldewey P, Tapley T, Kirsch N, Ruane KM, Pfizenmaier J, Shi R, Hofmann S, Foit L, Ren G, Jakob U, Xu Z, Cygler M, Bardwell JC. Genetic selection designed to stabilize proteins uncovers a chaperone called Spy. Nat Struct Mol Biol. 2011;18:262–9. doi: 10.1038/nsmb.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vagenende V, Yap MG, Trout BL. Mechanisms of protein stabilization and prevention of protein aggregation by glycerol. Biochemistry. 2009;48:11084–96. doi: 10.1021/bi900649t. [DOI] [PubMed] [Google Scholar]
- 21.Leandro P, Lechner MC, Tavares de Almeida I, Konecki D. Glycerol increases the yield and activity of human phenylalanine hydroxylase mutant enzymes produced in a prokaryotic expression system. Mol Genet Metab. 2001;73:173–8. doi: 10.1006/mgme.2001.3172. [DOI] [PubMed] [Google Scholar]
- 22.Street TO, Bolen DW, Rose GD. A molecular mechanism for osmolyte-induced protein stability. Proc Natl Acad Sci U S A. 2006;103:13997–4002. doi: 10.1073/pnas.0606236103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang Y, Cremer PS. Chemistry of Hofmeister anions and osmolytes. Annu Rev Phys Chem. 2010;61:63–83. doi: 10.1146/annurev.physchem.59.032607.093635. [DOI] [PubMed] [Google Scholar]
- 24.Holthauzen LM, Rosgen J, Bolen DW. Hydrogen bonding progressively strengthens upon transfer of the protein urea-denatured state to water and protecting osmolytes. Biochemistry. 2010;49:1310–8. doi: 10.1021/bi9015499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Moglich A, Joder K, Kiefhaber T. End-to-end distance distributions and intrachain diffusion constants in unfolded polypeptide chains indicate intramolecular hydrogen bond formation. Proc Natl Acad Sci U S A. 2006;103:12394–9. doi: 10.1073/pnas.0604748103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nick Pace C, Huyghues-Despointes BM, Fu H, Takano K, Scholtz JM, Grimsley GR. Urea denatured state ensembles contain extensive secondary structure that is increased in hydrophobic proteins. Protein Sci. 2010;19:929–43. doi: 10.1002/pro.370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kumar R, Prabhu NP, Yadaiah M, Bhuyan AK. Protein stiffening and entropic stabilization in the subdenaturing limit of guanidine hydrochloride. Biophys J. 2004;87:2656–62. doi: 10.1529/biophysj.104.044701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Majtan T, Liu L, Carpenter JF, Kraus JP. Rescue of cystathionine beta-synthase (CBS) mutants with chemical chaperones: purification and characterization of eight CBS mutant enzymes. J Biol Chem. 2010;285:15866–73. doi: 10.1074/jbc.M110.107722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ignatova Z, Krishnan B, Bombardier JP, Marcelino AM, Hong J, Gierasch LM. From the test tube to the cell: exploring the folding and aggregation of a beta-clam protein. Biopolymers. 2007;88:157–63. doi: 10.1002/bip.20665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Paul S, Punam S, Chaudhuri TK. Chaperone-assisted refolding of Escherichia coli maltodextrin glucosidase. FEBS J. 2007;274:6000–10. doi: 10.1111/j.1742-4658.2007.06122.x. [DOI] [PubMed] [Google Scholar]
- 31.Shriver JW, Edmondson SP. Ligand-binding interactions and stability. Methods Mol Biol. 2009;490:135–64. doi: 10.1007/978-1-59745-367-7_6. [DOI] [PubMed] [Google Scholar]
- 32.Cohen FE, Kelly JW. Therapeutic approaches to protein-misfolding diseases. Nature. 2003;426:905–9. doi: 10.1038/nature02265. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Steady-state protein levels of β-lactamase and β-lactamase-Im7 fusion proteins in the absence or presence of different osmolytes in the cultivation medium. Whole cell extracts of cells expressing the corresponding constructs and cultivated in the presence or absence of osmolytes were separated on a 7% Tris-glycine SDS-PAGE gel. The proteins were then transferred to a polyvinylidene difluoride membrane and probed with an anti-β-lactamase antibody. Values beneath the respective Western blot bands represent the fold increase in protein level compared to cells cultivated in LB. Mean values ± standard deviations are shown for independent duplicate experiments.
Correlation of β-lactamase activity with steady-state β-lactamase protein levels. Fold increase in β-lactamase activity was plotted against fold increase in steady-state protein level for WT β-lactamase (a), β-lactamase - link (30 aa) - β-lactamase (b), and our tripartite fusion proteins (c and d). Graphs show linear correlations between the two variables. Fold changes are expressed as changes in activity or steady-state protein level in comparison to each strain in LB, expressed as 1.
β-lactamase activity in whole cell extracts of E. coli MG1655 ΔampCΔhsdR expressing either WT β-lactamase or β-lactamase- link (30 aa) - β-lactamase was measured in the presence or absence of osmolytes in the cultivation medium. Activity is expressed as fold change relative to activity in LB of the same strain. 0.25 M NaCl was used as a negative control. Mean values ± standard deviations are shown for independent triplicate experiments.
Effect of various osmolytes on β-lactamase activity in extracts of E. coli MG1655 ΔampCΔhsdR cells expressing β-lactamase -link (59 aa) -β-lactamase, β-lactamase -Im7 I22V-β-lactamase and β-lactamase -Im7 L34A-β-lactamase. (a) Enzymatic activity expressed as % of the activity of cells expressing WT β-lactamase in LB (100%). (b) Fold increase in β-lactamase activity in the presence of osmolytes in the cultivation medium relative to activity in LB of the same strain. In the absence of osmolytes, insertion of a 59-residue linker (black bars) results in very low enzymatic activity, similar to activities observed for β-lactamase-Im7 I22V-β-lactamase or β-lactamase -Im7 L34A-β-lactamase. In the presence of osmolytes like glycerol, sorbitol, and sucrose, the activity of cells expressing β-lactamase –link (59 aa)- β-lactamase is essentially restored to WT β-lactamase levels, whereas the activity of cells expressing β-lactamase Im7 I22V-β-lactamase or β-lactamase -Im7 L34A-β-lactamase is only restored to up to 20% or 35%, respectively, of WT β-lactamase levels (a). This trend is also reflected in the fold increases in β-lactamase activity upon cultivation in osmolytes compared to cultivation in LB (b). 0.25 M NaCl was used as a negative control. Mean values ± standard deviations are shown for independent triplicate experiments. The somewhat overall lower fold increases in β-lactamase activity observed for these experiments relative to those reported in Figure 2 reflects the observation that the β-lactamase activity of the β-lactamase -Im7 I22V-β-lactamase fusion in the absence of any media additive is extremely low and somewhat variable. This greatly affects the fold increases observed (b), but has a relatively small influence on the activities expressed as a % of WT in LB (a).
Effect of osmolytes on β-lactamase activity in extracts of E. coli NEB 10-beta cells expressing β-lactamase AcP2 C21S/M61A-β-lactamase and β-lactamase-AcP2 C21S/L65V-β-lactamase. Fold increase in β-lactamase activity in the presence of osmolytes in the cultivation medium relative to activity in LB of the same strain is shown. Mean values ± standard deviations are shown for independent triplicate experiments. The somewhat overall lower fold increases in β-lactamase activity observed for AcP2 relative to those reported for Im7 fusions indicates that some sensors are better in reporting for protein folding and activity in the presence or absence of media additive.
mRNA levels in the presence and absence of osmolytes. The presence of osmolytes in the culture media does not increase the mRNA levels of the fusion constructs indicating that osmolytes act on the protein level to stabilize the fusions rather than upregualting the expression of the proteins at the mRNA level. A small osmolyte induced decrease is observed for glycerol and sorbitol.
mRNA levels of periplasmic chaperones. Cells expressing the constructs were cultured in LB and harvested at mid-log phase. Total mRNA was extracted using Qiagen’s RNeasy mini kit. After cDNA synthesis (see Supporting Information Methods), quantitative real-time PCR was carried out using SYBR Orange according to the manufacturer’s instructions. Data were normalized for the mRNA level of DnaQ and fold increases were calculated with reference to mRNA levels in LB.
Steady-state protein levels of three molecular chaperones (DnaJ, DnaK, and GroEL) in response to the absence or presence of osmolytes. The crude extracts of whole cell lysates of WT β-lactamase, β-lactamase -link (30 aa) - β-lactamase, and tripartite fusions expressed in the presence or in the absence of the osmolytes were separated on a 7% Tris-glycine SDS-PAGE gel, the proteins transferred to polyvinylidene difluoride membrane, and probed with anti-DnaK antibody (a), anti-GroEL antibody (b), or anti-DnaJ antibody (c).