

Abstract
The “small molecule universe” (SMU), the set of all synthetically feasible organic molecules of 500 Daltons molecular weight or less, is estimated to contain over 1060 structures, making exhaustive searches for structures of interest impractical. Here, we describe the construction of a “representative universal library” spanning the SMU that samples the full extent of feasible small molecule chemistries. This library was generated using the newly developed Algorithm for Chemical Space Exploration with Stochastic Search (ACSESS). ACSESS makes two important contributions to chemical space exploration: it allows the systematic search of the unexplored regions of the small molecule universe, and it facilitates the mining of chemical libraries that do not yet exist, providing a near-infinite source of diverse novel compounds.
Keywords: chemical space, small molecule universe, chemical diversity
INTRODUCTION
Many grand challenges in science and biomedicine require molecular and materials discovery.1–4 Yet, the fraction of “chemical space” that has been explored over human history is infinitesimal – less than one part in 1050.5 The vast unexplored molecular frontier suggests that there is reason for optimism in the face of grand scientific tasks.
Current experimental and theoretical tools are poorly matched to the scale and scope of the molecular discovery undertaking. Enumerating all compounds or materials in the vastness of molecular space is impossible, and assessing their properties is even more unimaginable; even synthetically accessible small organic compounds number over 1060.5 Further, current compound libraries are notably lacking in diversity, meaning that much of available small molecule chemistry has yet to be explored.5–7 Chemical libraries that capture the much broader diversity of the entire chemical universe promise to be a more empowering starting point for molecular discovery.8–11
Synthetic methods for expanding the diversity of compound collections, collectively known as diversity-oriented synthesis, arose as a reaction to the relatively non-diverse libraries generated by combinatorial synthesis and used in high-throughput screening.3,8,12 Computational approaches to aid in chemical space exploration can be very broadly classified into molecular optimization techniques – those that aim to identify or design compounds with optimal activity – and enumerative techniques, which aim to explore the full extent of a given chemical space. Optimization techniques include both search techniques based on stochastic and evolutionary algorithm frameworks13–17, directed combinatorial library design18, and optimization within continuous alchemical spaces.19–21 Other methods aim to construct molecules that have high similarity to a set of known compounds as a basis for lead discovery.22,23
In contrast to these focused design techniques, others have sought to enumerate all structures and to explore the full range of chemistries available within a given chemical space. Reymond has reported enumeration of all possible organic compounds (within a set of rules for synthetic feasibility) of 13 or fewer heavy atoms.24–26 This “GDB13” database contains nearly 1 billion compounds, most of which are not found in any other compound library. Compound mining in GDB13 has already led to successes in the drug discovery process.27,28 Oprea has also made considerable progress in quantifying chemical topology, having enumerated all possible ring topologies up to eight rings.6,29 Both of these studies yield valuable information about the diversity of the small molecule chemical space.
Although the number of compounds in the small molecule universe (SMU) is far too great to be enumerated, we show here that this astronomically large collection can be characterized in a way similar to enumerative techniques, but that only requires consideration of a far smaller set of chemical structures. Well-established chemoinformatics techniques allow the construction of “representative sublibraries” – maximally diverse collections of compounds that contain as much diversity as the parent library expressed in a much smaller number of compounds.30–33 Combining these techniques with concepts from chemical evolutionary algorithms, as described below, allows the mapping of humongous chemical spaces such as the SMU.
A schematic overview of the Algorithm for Chemical Space Exploration with Stochastic Search (ACSESS) is shown in Figure 1. By combining stochastic chemical structure mutations with methods for maximizing molecular diversity, ACSESS efficiently produces representative sublibraries of a vast chemical space. This procedure fundamentally differs from existing chemical genetic algorithms; it is designed to explore rigorously the full diversity available in targeted chemical spaces, including astronomically large ones, such as the space of druglike molecules in the SMU (vide infra), or functional chemical regions that contain molecules with specific desirable properties (see supporting information). We term the compound libraries thus generated “representative universal libraries” (RUL), collections of compounds that represent the full extent of chemical diversity within a much larger set of molecular structures.
Figure 1. The ACSESS procedure.

allows the construction of a representative universal library in an arbitrary chemical space. A, a library of initial molecules is expanded using chemical mutations and crossover; compounds outside the target chemical space are discarded; and a maximally diverse subset of the remaining molecules is selected. This process is repeated until the diversity of the set converges. B, chemical structure modifications, which include addition or deletion of terminal atoms (1), bond order modifications (2), addition or deletion of in-chain atoms (3), removal or addition of cyclic bonds (4), and modifications of atom type (5). C, an example of a chemical space trajectory. The final compound occupies unexplored chemical space in the SMU.
“Chemical space” is defined here as an M-dimensional Cartesian space in which compounds are located by a set of M physiochemical and/or chemoinformatic descriptors. We focus on chemical spaces defined by selected properties – the SMU, for instance, contains all stable compounds of 500 Da or less. A representative universal library contains chemical compounds that span the full extent of accessible descriptor values in the M-dimensional space. While the choice of the M descriptors and diversity measures may depend on specific applications, this approach remains generally applicable. Unlike previous techniques for selecting a maximally diverse sub-library, ACSESS is unique in that the parent collection does not need to be enumerated, allowing systematic exploration of uncharted and astronomically large chemical spaces.
METHODS
The ACSESS Algorithm
To map an arbitrary chemical space, ACSESS is seeded with a set of compounds; often, a very small library suffices to initialize the algorithm (a library of consisting benzene and cyclohexane, for example, was used to seed all work shown here). This library is enlarged and diversified over multiple computational generations. In each generation, the library is modified as follows (Figure 1A): 1) the library is expanded by creating new structures using “chemical mutations”, 2) compounds not in the chemical space of interest are removed (including those assessed as not being synthetically feasible or lacking the property of interest), and 3) the size of the library is reduced by selecting a maximally diverse subset of compounds. The qualitative features of the algorithm are discussed below; a complete description is provided in the supplementary information.
Note that the ACSESS algorithm requires concrete choices of chemical descriptor, diversity function, and target chemical space. As described below, we have chosen descriptors, chemical space filters, and diversity definitions that are A) relatively general and transferable, and B) computationally efficient, allowing construction of a large compound library and exploration of a large compound space. For more focused problems, other descriptors, diversity definitions, filters, or even chemical mutation types can be used as “drop-in” replacements for those described here.
1) Reproduction and mutation
ACSESS begins a generation by generating novel chemical structures from the previous generation. First, a set of new compounds is produced by “crossover” mutation (Scheme S1): two “parent” compounds are copied from the library, and each is split into two fragments by cutting a random acyclic bond. Two of the resulting fragments, one from each parent, are then bonded together, and the resulting structure is added back into the library.
After generating crossover mutants, further novel compounds are generated by copying random individual structures from the existing library, stochastically modifying them, and adding the new, modified structures to the library. These “chemical mutations”, as shown in Figure 1B, consist of addition/removal of an atom, either a terminal atom (1) or “within” an existing bond (3); creation/removal of a ring bond (4), modification of atom type (5, e.g., changing a carbon atom to a nitrogen atom), and modification of bond order (2). The mutation process and the probabilities of each mutation type are shown in Scheme S2. Because the descriptor set used in this study depends only on molecular connectivity (vide infra), stereochemical information was not tracked in these calculations. However, for descriptor sets or property calculations that depend upon absolute or relative configuration, stereochemical mutations can be included as well. For such systems, configurations may be inverted, and cis-trans diastereomers may be isomerized.
2) Filters
After new molecular structures are generated using the above chemical mutations, those that fall outside the chemical space of interest must be discarded. In this study, we focus specifically on stable, synthetically accessible drug-like molecules in the SMU; compounds were therefore screened using a combination of chemical subgroup filters (eliminating compounds that contain reactive or hydrolytically labile moieties such as strained allenes, cumulenes, hemiacetals, aminals, orthesters, etc.), steric strain filters based on generated 3-dimensional conformations (for example, removing compounds with non-tetrahedral sp3 carbons), and simple physiochemical filters (XlogP, Lipinski and Veber rules, among others).24 A complete list of filters is given in the supplementary information. Because these calculations did not track stereochemical information, the software used to generate 3D geometries was used to generate any energetically reasonable configuration for each structure. More robust ab initio stability filters would be an appealing future alternative to the heuristic ones employed here.34
3) Maximally diverse subset selection
At the final stage of each ACSESS generation, only a maximally diverse subset of the remaining molecular structures is retained – all other compounds are removed from the library. These structures are used to seed the next generation of the ACSESS procedure. Because the new library is chosen from both the new “child” compounds from the current generation and the “parent” compounds from the previous generation, the diversity of the library must necessarily improve or at least remain constant after each generation.
Many quantitative definitions of diversity exist, as do methods for selecting small maximally diverse libraries from larger libraries.30 One common definition of “diversity” is as the average nearest-neighbor chemical space distance within a set of compounds. Given this definition of diversity, a maximally diverse collection can be selected using the “maximin” algorithm, which creates a representative subset by choosing compounds from a larger library one-by-one, such that each new structure has the largest minimum distance to existing compounds in the subset.32
A cell-based definition of diversity can be used if the principal components of the chemical space are known.35 For cell diversity, chemical space is divided into a discrete, multidimensional grid, and diversity is then simply defined as the number of cells that contain at least one chemical structure. A maximally diverse set of compounds can be selected simply by choosing a single structure from each cell.
Chemical space descriptors
Any diversity analysis is highly dependent on the descriptor set chosen, which defines the chemical space coordinates of the structures. A large number of chemical descriptor sets exist,36 ranging from simple counts of topological properties37 to measures of 3-dimensional shape.38
For the mapping presented here, chemical space coordinates were calculated using Moreau-Broto autocorrelation descriptors.39 This well-established set of descriptors encodes structural information from an arbitrary chemical structure into a fixed-length vector,40 and has been successful in diverse tasks such as defining biologically relevant similarities in large compound sets41 and correlating structural diversity with biological activity.42 Autocorrelation descriptors describe topological correlations between atomic properties on a molecule:
| (1) |
where
| (2) |
dij is the number of bonds separating atoms i and j, and pi is the value of atomic property p on atom i. Here, the properties p include the atomic number, Gasteiger-Marsili partial charge,43 atomic polarizability,44 topological steric index,45 and unity (i.e., for all i); values of d from 0 to 7 were used, providing a 40-dimensional chemical space. Note that these descriptors are based solely on molecular topology and do not reflect stereochemistry or 3-dimensional structure.
Principal component analysis of the SMU
First, a small RUL of 2,000 compounds was constructed using the maximin method to select maximally diverse subsets (see SI). The 40 autocorrelation vectors of these 2,000 molecules were mean-centered and normalized to have unit variance, and principal component analysis (PCA) was performed. Loadings for the first 10 principal components of the SMU are shown in Table S1.
Construction of a representative universal library of the small molecule universe
Next, cell-based diversity was used to construct a large, synthetically optimized representative universal library of the small molecule universe (SMU-RUL). A partitioning scheme was developed based on the largest 10 principal components of the SMU (PCs). These PCs collectively account for 98.5% of the SMU’s chemical space variance. Each PC was then partitioned into bins, with the number of bins proportional each PC’s standard deviation, yielding a 20×15×12×11×9×8×8×6×6×4 grid that partitions the SMU chemical space into 3.3×109 cells. ACSESS was then used to construct the 8.9×106-structure SMU-RUL, with maximally diverse subsets selected by choosing one compound per grid cell. In cases where more than one compound was present in a cell, the compound with the highest estimated synthetic accessibility score was selected.
Synthetic accessibility scores
The SAScore algorithm estimates a structure’s synthetic accessibility based on both its topological complexity and how frequently its substructures appear in large chemical databases.46 SAScores reported here are based on a substructure analysis of the full ZINC database, and higher SAScores indicate a more facile synthesis. The distribution of SAScores for compounds in the PubChem library is shown in Figure 2E.
Figure 2. Comparison to existing libraries.
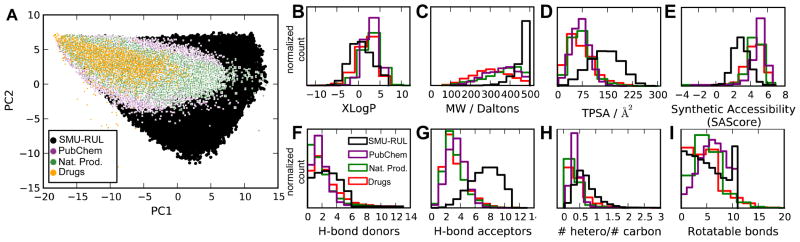
The SMU-RUL (black), ZINC natural product library (green), ZINC drug library (orange), and druglike compounds in PubChem (purple) are shown. A, compound locations along the first two principal components of the SMU-RUL library. B–I, histograms of physiochemical properties for the four libraries; y-axes correspond to normalized compound counts within each library. The properties include B, estimated LogP (XLogP)54; C, molecular weight (MW); D, topologically estimated polar surface area (TPSA)55; E, synthetic accessibility score (SAScore)46; F–G, number of hydrogen-bond donors and acceptors; H, ratio of non-carbon heavy atoms to carbon atoms; and I, number of rotatable bonds. Compared to the PubChem database, molecules in the SMU-RUL are, on average, more polar and have a larger molecular weight. Lower synthetic accessibility scores (E) for SMU-RUL compounds are expected because of their novelty and dissimilarity to known compounds.
Software
ACSESS was implemented in Python 2.7 using OpenEye chemoinformatics toolkits.47 For screening of 3-dimensional geometries, conformers were generated using the OpenEye OMEGA program.48
RESULTS
Proof of principle: the GDB13 chemical space
The GDB13 database enumerates all possible compounds of 13 heavy atoms or fewer (within a set of synthetic criteria used as inspiration for the present work), and is currently the largest available database of chemical structures.24 We first used GDB13 to test the ability of the ACSESS method to capture the diversity of a large molecular ensemble. An RUL of 10,000 compounds in GDB13 (GDB13-RUL) was constructed. Compounds were filtered for synthetic feasibility using the same criteria as in GDB13; the diversity of the set converged after 1,000 generations. The 10,000-member GDB13-RUL was as diverse as the fully enumerated GDB13 library, but required a factor of 104 fewer compounds to be processed computationally than the GDB13 enumeration. Additionally, principal component analysis (and other mappings) of the GDB13-RUL produced diversity metrics similar to those of the fully enumerated library.
These results indicate two important properties of the ACSESS method. First, at convergence of the diversity measure, the RUL captured the full diversity of its parent space. Second, ACSESS generated the GDB13 RUL by enumerating far fewer compounds.
A representative library of the SMU
ACSESS was employed to build a representative library of the entire small molecule universe (SMU-RUL) consisting of 8.9×106 structures (database S1), with local optimization for synthetic accessibility. Chemical structures were restricted to 150–500 Da with estimated logP < 7.0, and were filtered for reactivity, stability, and druglikeness. Chemical space coordinates were computed using Moreau-Broto autocorrelation descriptors.36 A total of 3.6×109 structures were screened.
Structures in the SMU-RUL represent a widely spaced mesh over the complete SMU chemical space as defined above. It is important to note, however, that the specific set of compounds in the SMU-RUL is not unique. Instead, each SMU-RUL compound indicates the existence of minimally one, and, on average, ~1053 related structures (given the estimate of 1060 possible SMU structures4) in a particular region of chemical space. Convergence of the diversity measure, while not indicating that a global optimum has been reached, shows that we have obtained a set of structures that is representative of the accessible chemical space.
Comparison to existing databases
The chemical space coverage of the SMU-RUL was compared to that of three existing databases: PubChem, a database of over 3×107 pure chemical compounds49; ZINC natural products, a database of 2×105 natural products and metabolites relevant to drug discovery; and ZINC drugs, a database of over 7,000 approved drugs.50 For comparison to the SMURUL, databases were filtered according to druglikeness and atom content using a subset of the SMU-RUL filters.
Although 99.9% of the generated SMU-RUL structures obey Lipinski’s rules for drug-likeness,51 only 11,000 are present in the PubChem database. The scaffolds in the SMU-RUL (defined as the set of atoms that are in or link the molecule’s ring systems29,49) are also highly novel. Of the 5.1×106 unique scaffold topologies in the SMU-RUL, only 23,000 are found among the 3.2×106 scaffold topologies in the PubChem database. Interestingly, the SMURUL also contains two known drugs, acetanilide and phenytoin.
Figure 2A shows the chemical space occupied by SMURUL compounds along its first two principal components, as well as the positions of compounds from the ZINC and PubChem databases. Even in this 2-dimensional projection of the 40-dimensional chemical space, the SMU-RUL covers a much larger region of chemical space than existing chemical libraries. Figure 2B–I shows the distribution of eight physiochemical properties in the four sample libraries. Most strikingly, the SMU-RUL, on the whole, contains heavier, more polar and synthetically more challenging members (given current methodologies) than existing compound libraries (Figure 2B–G).
The property distributions in Figure 2 show that much of the available SMU diversity is concentrated at higher molecular weights and more polar structures than currently known compounds. This is not surprising, given the autocorrelation descriptors used to describe diversity here; larger compounds can support a larger range of chemical functions, which allows descriptor values to be explored. The relatively low synthetic accessibility scores of the SMU-RUL structures are also expected. The relatively low SAScores indicate that in many regions of chemical space there were few compounds with similar substructure to known compounds.
Self-organizing map of the SMU
A self-organizing map (SOM) was constructed from the SMU-RUL to visualize the high-dimensional SMU chemical space (Figure 3). SOMs have a rich history in chemical diversity analyses. 52 An SOM consists of a lattice of “neurons”, each associated with a chemical space coordinate.22,23 The SOM is randomly presented with “cue” coordinates from the training set (here, the SMU-RUL). For each cue, the neuron with coordinates closest to the cue is said to “fire,” and its and its neighbors’ coordinates are adjusted in the direction of the cue. Iteration of this procedure creates a low-dimensional representation of the high dimensional chemical space.
Figure 3. Map of the small molecule universe.

A 300×300 toroidal self-organizing map map was created using normalized autocorrelation descriptors of SMU-RUL compounds. For clarity, the map is divided into 36 labeled sections (AI, BII, etc.), each containing a 50×50 grid of neurons. A, number of PubChem compounds assigned to a neuron; white indicates neurons which are unoccupied by any PubChem compounds (84% of total). The PubChem compounds are highly clustered to a relatively small region of chemical space; 98% are assigned to only 2% of the neurons. The black circle in region EI encompasses the positions of all GDB13 compounds. B–D, molecular properties; each neuron is colored by the median value of its SMU-RUL compounds.
A toroidal 300×300 SOM was trained using the autocorrelation chemical space coordinates of the SMU-RUL. Each SMU-RUL compound was then assigned to its closest neuron. The compounds were spread relatively evenly throughout the map, with an average of 98.5±25.3 (and at least 16) chemical structures assigned to each neuron. A small region of the map (region EI in Figure 3B) corresponds to relatively low molecular weight structures with 15–20 heavy atoms, while others correspond to higher molecular weights nearer to the 500 Da limit. Figure 3D shows well-defined regions containing either large, fused ring systems or simpler monocyclic and fused bicyclic structures. Variations of other topological and physiochemical properties over the map are shown in Figure 3CF and Figure S3A.
The autocorrelation vectors of all PubChem library compounds were computed and assigned to neurons on the SOM (Figure 3A). The PubChem compounds were concentrated in a very restricted area compared to the SMU-RUL, with 98% of PubChem compounds assigned to 2% of the neurons. The most significant PubChem compound cluster is centered on region EI and is characterized by compounds with low molecular weights and few rings. The cluster can be further divided into two regions, one corresponding to rigid structures without rotatable bonds, and the other to more flexible molecules. A smaller cluster in region DIV corresponds to compounds with higher molecular weights and more complex scaffolds.
Large regions of chemical space populated by SMURUL structures are unrepresented in PubChem (white spaces in Figure 3A). Note that the inverse is not true; a SOM constructed using compounds from both PubChem and the SMU-RUL shows that SMU-RUL structures occupy all of the space occupied by PubChem compounds (Figure S3B). The unexplored regions of chemical space were, like the SMU-RUL in general, almost entirely druglike based on Lipinski’s rules. Examples of SMU-RUL structures from unexplored portions of chemical space (Chart 1 and Figure S4) include complex ring structures (AIII, BVI), many simple ring systems (AV), bridged macrocycles (CIII), and high heteroatom content (CI–CII).
Chart 1. SMU-RUL compounds from unexplored chemical space.

Each compound shown here was selected from a SOM map neuron unoccupied by any PubChem compounds, and was among the most synthetically accessible compounds assigned to the neuron. Letters/numerals refer to the regions shown in figure 3. The stereochemical assignments shown reflect the generated 3D conformations, which are shown as ball-and-stick models in the SI.
CONCLUSIONS
The stochastic exploration described here is a computationally efficient tool for accessing the astronomical number of feasible organic structures. As a comparison of stochastic and enumerative approaches, all 970×106 compounds from the enumerated GDB13 library were assigned to the SMU-RUL SOM. 98% of GDB13 compounds were assigned to just 10 neurons in the low-molecular weight portion of the map, and the GDB13 compounds overall occupy a total of only 61 adjacent neurons, 0.07% of the total (Figure 3A). The combinatorial explosion of new molecules available at higher molecular weights is simply not accessible in the smaller chemical spaces amenable to enumeration.
Importantly, in our stochastic exploration, large gaps were observed in the currently known compound collections. There has never been an attempt made to explore the full range of chemical diversity, either by nature or by man. Nature uses readily available building blocks and biosynthetic tools to develop structural motifs, and arguably has employed repetitive patterns and quantum leaps in molecular weight (biopolymers) to address the diversity-intense aspects of data storage, immune defense, scaffolding, etc. Laboratory synthesis relies on a nucleation-based building-block approach, using iterative bond formations and a limited pool of available reagents. In the absence of obvious incentives otherwise, laboratory synthesis thus emphasizes simplicity and uses small functional group-specific tools to carve out niches around known biologically active scaffolds.
The ACSESS algorithm makes two important contributions to chemical space exploration, both of which are immediately available for further experimentation. The gaps identified in the known chemical universe may now be explored systematically. ACSESS further allows the mining of chemical libraries that do not yet exist, providing a near-infinite source of novel compounds. For instance, we have used ACSESS to search a chemical space of unprecedented size to create a library of compounds with high similarity to bretazenil, a benzodiazepine anxiolytic drug discovered in 1988. Similarity here was defined using the Tanimoto coefficient of the PubChem-format fingerprints.53 The resulting library represents both a universal library of structural isomers of the target drug and a collection of novel, unpatented candidates for future development. Because of ACSESS’s efficiency, more computationally intensive metrics than structural similarity can be employed, affording opportunities for molecular discovery in fields well beyond biology and medicine.
Supplementary Material
Acknowledgments
We thank Professor Sean Xie for helpful discussion and the NIH for support of the UPCMLD (P50-GM067082).
ABBREVIATIONS
- SMU
small molecule universe
- ACSESS
algorithm for chemical space exploration with stochastic search
- GDB13
generated database of 13 atoms or less
- RUL
representative universal library
- PCA
principal component analysis
- SOM
self-organizing map
Footnotes
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
ASSOCIATED CONTENT
Supporting Information. SMU-RUL compounds in SMILES format; ACSESS source code; detailed methods and chemical space definitions; computational details of library construction; SOM and PCA analysis of SMU-RUL; construction of bretazenil isomer library; GDB13 proofs-of-principle. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Beyond the Molecular Frontier: Challenges for Chemistry and Chemical Engineering. The National Academies Press; Washington, D.C: 2003. [PubMed] [Google Scholar]
- 2.Sauer WHB, Schwarz MK. J Chem Inf Comp Sci. 2003;43:987. doi: 10.1021/ci025599w. [DOI] [PubMed] [Google Scholar]
- 3.Schreiber SL. Nature. 2009;457:153. doi: 10.1038/457153a. [DOI] [PubMed] [Google Scholar]
- 4.Dandapani S, Marcaurelle LA. Nature Chem Bio. 2010;6:861. doi: 10.1038/nchembio.479. [DOI] [PubMed] [Google Scholar]
- 5.Bohacek RS, McMartin C, Guida WC. Med Res Rev. 1996;16:3. doi: 10.1002/(SICI)1098-1128(199601)16:1<3::AID-MED1>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- 6.Wester MJ, Pollock SN, Coutsias EA, Allu TK, Muresan S, Oprea TI. J Chem Inf Model. 2008;48:1311. doi: 10.1021/ci700342h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Triggle DJ. Biochem Pharmacol. 2009;78:217. doi: 10.1016/j.bcp.2009.02.015. [DOI] [PubMed] [Google Scholar]
- 8.Tan DS. Nat Chem Biol. 2005;1:74. doi: 10.1038/nchembio0705-74. [DOI] [PubMed] [Google Scholar]
- 9.Thomas GL, Wyatt EE, Spring DR. Curr Opin Drug Discov Dev. 2006;9:700. [PubMed] [Google Scholar]
- 10.Hajduk PJ, Galloway WRJD, Spring DR. Nature. 2011;470:42. doi: 10.1038/470042a. [DOI] [PubMed] [Google Scholar]
- 11.Brown LE, Cheng KCC, Wei WG, Yuan P, Dai P, Trilles R, Ni F, Yuan J, MacArthur R, Guha R, Johnson RL, Su XZ, Dominguez MM, Snyder JK, Beeler AB, Schaus SE, Inglese J, Porco JA., Jr Proc Natl Acad Sci U S A. 2011;108:6775. doi: 10.1073/pnas.1017666108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dow M, Fisher M, James T, Marchetti F, Nelson A. Org Biomol Chem. 2012;10:17. doi: 10.1039/c1ob06098h. [DOI] [PubMed] [Google Scholar]
- 13.Nicolaou CA, Brown N, Pattichis CS. Curr Opin Drug Discov Dev. 2007;10:316. [PubMed] [Google Scholar]
- 14.Schneider G, Hartenfeller M, Reutlinger M, Tanrikulu Y, Proschak E, Schneider P. Trends Biotechnol. 2009;27:18. doi: 10.1016/j.tibtech.2008.09.005. [DOI] [PubMed] [Google Scholar]
- 15.Besnard J, Ruda GF, Setola V, Abecassis K, Rodriguiz RM, Huang X-P, Norval S, Sassano MF, Shin AI, Webster LA, Simeons FRC, Stojanovski L, Prat A, Seidah NG, Constam DB, Bickerton GR, Read KD, Wetsel WC, Gilbert IH, Roth BL, Hopkins AL. Nature. 2012;492:215. doi: 10.1038/nature11691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zablocki J. J Am Chem Soc. 2007;129:12586. [Google Scholar]
- 17.Gillet VJ. Struct Bond. 2004;110:133. [Google Scholar]
- 18.Gillet VJ, Willett P, Fleming PJ, Green DVS. J Mol Graph Model. 2002;20:491. doi: 10.1016/s1093-3263(01)00150-4. [DOI] [PubMed] [Google Scholar]
- 19.Hu XQ, Beratan DN, Yang W. J Chem Phys. 2008;129:064102. doi: 10.1063/1.2958255. [DOI] [PubMed] [Google Scholar]
- 20.Balamurugan D, Yang W, Beratan DN. J Chem Phys. 2008;129:174105. doi: 10.1063/1.2987711. [DOI] [PubMed] [Google Scholar]
- 21.Wang M, Hu XQ, Beratan DN, Yang W. J Am Chem Soc. 2006;128:3228. doi: 10.1021/ja0572046. [DOI] [PubMed] [Google Scholar]
- 22.Brown N, McKay B, Gasteiger J. J Comput Aided Mol Des. 2004;18:761. doi: 10.1007/s10822-004-6986-2. [DOI] [PubMed] [Google Scholar]
- 23.van Deursen R, Reymond J-L. Chem Med Chem. 2007;2:636. doi: 10.1002/cmdc.200700021. [DOI] [PubMed] [Google Scholar]
- 24.Blum LC, Reymond J-L. J Am Chem Soc. 2009;131:8732. doi: 10.1021/ja902302h. [DOI] [PubMed] [Google Scholar]
- 25.Fink T, Bruggesser H, Reymond JL. Angew Chem Int Ed. 2005;44:1504. doi: 10.1002/anie.200462457. [DOI] [PubMed] [Google Scholar]
- 26.Fink T, Reymond JL. J Chem Inf Model. 2007;47:342. doi: 10.1021/ci600423u. [DOI] [PubMed] [Google Scholar]
- 27.Luethi E, Nguyen KT, Bürzle M, Blum LC, Suzuki Y, Hediger M, Reymond JL. J Med Chem. 2010;53:7236. doi: 10.1021/jm100959g. [DOI] [PubMed] [Google Scholar]
- 28.Nguyen KT, Syed S, Urwyler S, Bertrand S, Bertrand D, Reymond JL. ChemMedChem. 2008;3:1520. doi: 10.1002/cmdc.200800198. [DOI] [PubMed] [Google Scholar]
- 29.Pollock SN, Coutsias EA, Wester MJ, Oprea TI. J Chem Inf Model. 2008;48:1304. doi: 10.1021/ci7003412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Farnum MA, Desjarlais RL, Agrafiotis DK. In: In Handbook of chemoinformatics : from data to knowledge. Gasteiger J, editor. Vol. 4. Wiley-VCH; Weinheim: 2003. p. 1640. [Google Scholar]
- 31.Gillet VJ, Willett P, Bradshaw J, Green DVS. J Chem Inf Comput Sci. 1999;39:169. [Google Scholar]
- 32.Agrafiotis DK. J Chem Inf Comp Sci. 1997;37:841. [Google Scholar]
- 33.Gillet V. In: In Molecular Diversity in Drug Design. Dean P, Lewis R, editors. Springer Netherlands; 2002. p. 43. [Google Scholar]
- 34.Hoffmann R, Schleyer PvR, Schaefer HF., III Angew Chem Int Ed. 2008;47:7164. doi: 10.1002/anie.200801206. [DOI] [PubMed] [Google Scholar]
- 35.Xue L, Stahura FL, Bajorath J. In: Methods in Molecular Biology. Bajorath J, editor. Vol. 275. Humana Press; 2004. p. 279. [DOI] [PubMed] [Google Scholar]
- 36.Todeschini R, Consonni V. Molecular Descriptors for Chemoinformatics. 2. Wiley-VCH; Weinheim: 2009. [Google Scholar]
- 37.Nguyen KT, Blum LC, van Deursen R, Reymond J-L. ChemMedChem. 2009;4:1803. doi: 10.1002/cmdc.200900317. [DOI] [PubMed] [Google Scholar]
- 38.Arteca GA. In Reviews in Computational Chemistry. Vol. 9. John Wiley & Sons, Inc; 2007. p. 191. [Google Scholar]
- 39.Moreau G, Broto P. Nouv J Chim. 1980;4:359. [Google Scholar]
- 40.Gasteiger J. In Handbook of Chemoinformatics. Wiley-VCH Verlag GmbH; 2003. p. 1034. [Google Scholar]
- 41.Bauknecht H, Zell A, Bayer H, Levi P, Wagener M, Sadowski J, Gasteiger J. J Chem Inf Comp Sci. 1996;36:1205. doi: 10.1021/ci960346m. [DOI] [PubMed] [Google Scholar]
- 42.Matter H. J Med Chem. 1997;40:1219. doi: 10.1021/jm960352+. [DOI] [PubMed] [Google Scholar]
- 43.Gasteiger J, Marsili M. Tetrahedron Lett. 1978;19:3181. [Google Scholar]
- 44.Miller KJ, Savchik J. J Am Chem Soc. 1979;101:7206. [Google Scholar]
- 45.Cao C, Liu L. J Chem Inf Comp Sci. 2004;44:678. [Google Scholar]
- 46.Ertl P, Schuffenhauer A. J Chemoinf. 2008;1:8. [Google Scholar]
- 47.1.7.7. OpenEye Scientific Software, Inc; 2012. [Google Scholar]
- 48.Hawkins PCD, Skillman AG, Warren GL, Ellingson BA, Stahl MT. J Chem Inf Model. 2010;50:572. doi: 10.1021/ci100031x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bemis GW, Murcko MA. J Med Chem. 1996;39:2887. doi: 10.1021/jm9602928. [DOI] [PubMed] [Google Scholar]
- 50.Irwin JJ, Sterling T, Mysinger MM, Bolstad ES, Coleman RG. J Chem Inf Model. 2012;52:1757. doi: 10.1021/ci3001277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Adv Drug Deliv Rev. 1997;23:3. doi: 10.1016/s0169-409x(00)00129-0. [DOI] [PubMed] [Google Scholar]
- 52.Sadowski J, Wagener M, Gasteiger J. Angew Chem Int Ed. 1996;34:2674. [Google Scholar]
- 53.ftp://ftp.ncbi.nlm.nih.gov/pubchem/specifications/pubchem_fingerprints.txt.
- 54.Wang R, Ying F, Lai LJ. Chem Info Comput Sci. 1997;37:615. [Google Scholar]
- 55.Ertl P, Rohde B, Selzer P. J Med Chem. 2000;43:3714. doi: 10.1021/jm000942e. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.