

Abstract
A widely accepted paradigm in cancer research holds that the development of cancers is rate-limited by the occurrence of oncogenic mutations. In particular, the exponential rise in the incidence of most cancers with age is thought to reflect the time required for cells to accumulate the multiple oncogenic mutations needed to confer the cancer phenotype. Here I will argue against the axiom that the occurrence of oncogenic mutations limits cancer incidence with age, based on several observations, including that the rate of mutation accumulation is maximal during ontogeny, oncogenic mutations are frequently detected in normal tissues, the evolution of complex multicellularity was not accompanied by reductions in mutation rates, and that many oncogenic mutations have been shown to impair stem cell activity. Moreover, while evidence that has been used to support the current paradigm includes increased cancer incidence in individuals with inherited DNA repair deficiencies or exposed to mutagens, the pleotropic effects of these contexts could enhance tumorigenesis at multiple levels. I will further argue that age-dependent alteration of selection for oncogenic mutations provides a more plausible explanation for increased cancer incidence in the elderly. While oncogenic mutations are clearly required for cancer evolution, together these observations counter the common view that age-dependence of cancers is largely explained by the time required to accumulate sufficient oncogenic mutations.
Keywords: mutation, evolution, aging
The field of cancer research is dominated by the view that oncogenesis is rate-limited by the incidence of oncogenic mutations, and that these mutations are typically advantageous when they occur in the right cell type. Such oncogenic mutations (including activation of proto-oncogenes and deactivation of tumor suppressor genes, whether by genetic or epigenetic mechanisms) are thought to provide cells with various “Hallmarks of Cancer”, including sustained proliferative signaling and resistance to growth suppressive and cell death signals (1). In particular, it is widely accepted that the exponential increase of cancer incidence with age reflects the time required for cells to accumulate sufficient numbers of genetic and epigenetic mutations to confer the cancer phenotype (2–8). This paradigm in part originates with the classic modeling studies of Armitage and Doll (9, 10), which showed that the incidence of cancers increases with around the 6th power of age, suggesting that the age-dependent accumulation of 6–7 oncogenic mutations is needed for cancer development. The logic is quite simple: Aging → Mutations (including Oncogenic Mutations) → Cancer.
Oncogenic mutations are clearly required for cancer evolution, and increases in genetic/epigenetic diversity in somatic cells associated with aging should contribute to cancer incidence. Increased rates of genomic instability in some cancers can also help promote tumor evolution (1). However, I will argue that the axiomatic attribution of the rising incidence of cancers with age primarily to the accumulation of oncogenic mutations is insufficiently justified, stimulating the following questions: Why is aging associated with increased cancer incidence? Is the association explained by the requirement to increase the accumulation of oncogenic mutations? Or are pre-existing oncogenic mutations largely the substrate upon which age-dependent alterations in selection act? This perspective will not address whether cancer evolution in general involves or requires accelerated mutation accumulation, unless this acceleration were due to aging.
I will describe evidence to challenge the axiom that the occurrence of oncogenic mutations limits cancer incidence with age. I will argue that the age-dependent accumulation of mutations plays a relatively minor role in the increased incidence of cancer with age. Instead, other aging-associated changes, such as alterations in tissues that influence the selection for oncogenic events, largely underlie the aging-association of cancers.
The rate of mutation accumulation over time is maximal during ontogeny (development to maturity)
While the commonly accepted idea is that accumulation of oncogenic mutations with age accounts for the age-dependence of cancer, many if not most mutations appear to accumulate during ontogeny, rather than during adulthood (11–13). The maintenance of self-renewing adult tissues may require relatively few stem cell divisions. Indeed, most telomere shortening in human cells occurs before birth (14). It is estimated that any given hematopoietic stem cell (HSC) will divide on average 5–10 times through the life of an adult mouse (from maturity to 2–3 years of age) (15), and yet one would surmise that the generation of each HSC in a young mouse required far larger numbers of cell divisions (counting from the one-celled zygote). Thus, it is not surprising that a substantial fraction of mutations and epigenetic changes would occur and accumulate during ontogeny (12, 13), followed by a more modest rate of accumulation during tissue maintenance post-maturity (although an accelerated accumulation of mutations/genomic rearrangements in late life has been observed (16, 17), and DNA repair mechanisms, at least as assessed in cell culture, may become impaired in old age (18)). At least as assessed using transgenic reporters in C57BL/6 mice, accumulation of mutations (including genome rearrangements) from maturity through old age is relatively modest (~2–3 fold in some tissues) (12). In particular, the accumulation of mutations in the spleen is negligible from maturity through old age (12, 19) (Figure 1), consistent with the very low division rates for HSC past maturity (15). Since leukocytes in the spleen are derived from HSC and are relatively short-lived, they should serve as adequate proxies for the analysis of mutation accumulation in HSC and hematopoietic progenitors. As the most common malignancies that develop in old C57BL/6 mice are lymphocytic (20), the paucity of aging-dependent accumulation of mutations in the hematopoietic compartment is at odds with the mutation-centric paradigm. That the incidence of most cancers rises late in life (Figure 1), with kinetics that are quite disconnected from the time-dependent accumulation of mutations (maximal during ontogeny) in the tissues from wince these cancers arise, argues that age-dependent acquisition of oncogenic mutations is not a rate-limiting step in tumorigenesis. Indeed, Frank has proposed that cancers that develop in old age may in fact depend on oncogenic mutations accumulated during ontogeny (13).
Figure 1. Comparison of the time-courses of mutation accumulation with onset of malignancies in the hematopoietic system in mice.
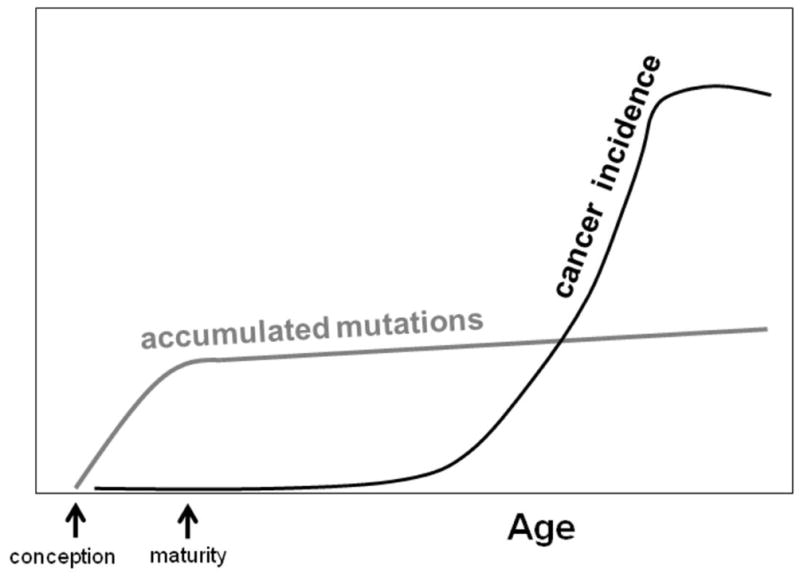
Stylized curves represent the numbers of mutations detected in spleens (gray) (12, 19) and lymphoma incidence (black) (20) in C57BL/6 mice.
Oncogenic mutations are frequently detected in non-diseased tissues
Oncogenic mutations are necessary, but not sufficient, for tumorigenesis. Many barriers to tumor progression exist and it is well known that multiple oncogenic mutations are required for the emergence of clinically-detectable cancers (21, 22). Still, if the incidence of oncogenic mutations were a rate-limiting step in tumorigenesis, one would not expect an abundance of oncogenic mutations in the absence of tumorigenesis. Nonetheless, cell clones with mutational or epigenetic inactivation of the PTEN or INK4A tumor suppressor genes are frequently found in histologically normal endometria and breast (respectively) of cancer free women (23, 24), far outpacing the incidence of the corresponding cancers. Furthermore, the presence of TEL-AML1 and AML1-ETO translocations in blood cells of newborns is ~100-fold greater than the risk of the associated leukemias (25). Perhaps most surprisingly, histologically advanced microscopic tumors are detected in many tissues of adult humans (22, 26), but which appear to be mostly held in check by unknown mechanisms. In addition, even though it is thought that the incidence of chronic myeloid leukemia (CML), which increases exponentially late in life, is limited by the occurrence of the initiating Bcr-Abl translocation (27), in frame Bcr-Abl fusions are detected in leukocytes of ~1 in 3 healthy individuals (28, 29), the vast majority of which will fortunately never develop this leukemia (despite persistence of the translocation in leukocytes for long enough to suggest an HSC origin (30)). Notably, CML in chronic phase is thought to be a simple leukemia (a myeloproliferative disorder), with Bcr-Abl as the only recurrent mutation (31). In mouse models, the expression of Bcr-Abl provides a much greater advantage to progenitor cells in an aged as compared to young hematopoietic system, leading to increased clonal expansion and leukemogenesis (32). Thus, at least in this mouse model, the occurrence of the same oncogenic mutation, Bcr-Abl, results in very different outcomes dependent on the age of the target tissue.
The evolution of multicellularity has not been accompanied by decreases in DNA mutation rates
If increased acquisition of oncogenic mutations is the primary driver of oncogenesis, organisms that are more likely to acquire oncogenic mutations should be more likely to get cancer, and larger/longer-lived animals would have been expected to have evolved mechanisms to limit mutation accumulation. Thus, it might seem surprising, but mutation rates are higher in larger and more complex organisms like mammals than they are in prokaryotes, single celled eukaryotes, and simpler multicellular organisms (11). Additionally, within mammals, somatic mutation rates are even higher than those for the germline. For example, the average somatic mutation rate for humans across four tissues (~1 × 10−9/site/cell division) is ~17-fold higher than the germline rate, and surprisingly, several-fold higher than rates for S. cerevisiae and E. coli (11). While some studies have shown that the efficiency of DNA excision repair among mammals is proportional to lifespan and/or body size (at least for fibroblasts exposed in vitro to ultraviolet light; UV) (33, 34), these differences have not been shown to coincide with similar changes in mutation accumulation in tissues in vivo and UV-induced excision repair may be less relevant for small nocturnal mammals (in fact, recognition of UV-induced cyclobutane pyrimidine dimers is suppressed in rodent cells (35)). Indeed, while the disparate methods used muddy comparisons (and highlight the need for direct measurements of mutations in mammals with age), rates of mutation accumulation in somatic tissues are similar between rodents and humans (11). So of all of the proposed evolved tumor suppressive mechanism that keep cancer rates sufficiently low in multicellular organisms long enough to promote reproductive success (36), improvements in DNA repair do not appear to have been harnessed during the evolution of bigger, more complex and longer-lived animals. DNA fidelity mechanisms were apparently already “good enough” to limit cancer through reproductive years, and the evolution of tumor suppressive mechanisms with increasing multicellularity did not require further refinement.
DNA repair deficiency, mutagens, and cancer: complicated relationships
The increased cancer incidence associated with inherited DNA repair deficiencies or exposure to DNA damaging agents is often cited as key support for the argument that time-dependent accumulation of oncogenic mutations is responsible for the rise of cancer rates with aging (e.g. (2, 4, 6, 37)). The logic seems simple: agents that increase mutation frequency also increase cancer incidence. Though easily understood, this rationale bypasses important characteristics of diseases associated with inherited DNA repair defects, particularly cancer-promoting characteristics that extend beyond increased frequencies of oncogenic mutations. For example, ATM (ataxia telangiectasia mutated) deficiency also reduces the fitness (see Box 1 for definitions) of HSC, increases reactive oxygen species, alters metabolism, promotes inflammation, and decreases immune function (37–40), all of which could contribute to cancer evolution at multiple levels. These pleotropic effects of ATM loss emanate both from impaired DNA repair as well as non-repair functions of ATM. Thus, for DNA repair deficiencies, it is difficult to assign the blame for increased cancer to a particular consequence of the genetic defect.
Box 1. Glossary.
Adaptive- increases fitness (e.g. a mutation that increases cellular fitness would be adaptive); note that whether a mutation is adaptive should be context dependent.
Adaptive Landscape- relationships between genotype and cellular fitness; in evolutionary biology, these landscapes describe how changes in genotype (and the corresponding phenotype) influence organismal fitness, but should also describe similar relationships for cells.
Cell fitness- a measure of the ability of a cell to pass its genotype on to future cell generations; cell fitness is not simply a measure of cell duplication rates; for example, certain stem cells need to limit cell cycling to maintain themselves within the stem cell niche (necessary to remain as stem cells)
Niche- for a stem cell, this refers to the environmental factors (other cells, matrix and soluble factors) that influence the properties of the cell
Moreover, an increase in mutation rates does not always confer increased cancer incidence. Heterozygous mutation of the DNA polymerase δ (at L604G and L604K) in mice increases mutation rates 4–5 fold in embryonic fibroblasts, with an even larger increase in the frequency of chromosomal aberrations (>17 fold), but without increasing the incidence of cancer (41). In particular, since the L604G/+ mice have normal lifespans and cancer incidence, increased mutation rates in these mice appear to have been uncoupled from any changes in both cancer and overall physiology (contrasting with ATM deficient mice). In contrast, heterozygous L604K mutation leads to a shorter lifespan, and cancers in L604K/+ mice show accelerated progression (but with similar tumor incidence to +/+ mice). One could conclude that L604K accelerates tumor development by either increasing genetic diversity or by altering selective pressures; the absence of a similar acceleration in the L604G/+ mice would argue for the latter, although the two explanations need not be mutually exclusive. Although in vivo mutation rates have not been determined for L604K/+ and L604G/+ mice, the increases in L604 DNA polymerase δ mutant mice are likely to eclipse the modest accumulation of mutations in mice from maturity to old age (12). Thus, increasing mutation rates is apparently not sufficient for increased tumorigenesis. Still, other loss-of-function mutations in DNA polymerases do increase cancer incidence in mice (42), indicating that there is either a threshold increase in mutations required to increase cancer rates over background or that other effects of these mutations on mouse physiology may promote the increased cancer incidence.
Analogous concerns can be raised for associations between exposure to DNA damaging agents and cancer, as these agents (and the resulting DNA damage) similarly cause pleotropic effects (reduced progenitor cell fitness, increased inflammation, increased cell turnover, decreased immune function, etc.) (43). Thus, the extent to which radiation, chemotherapy treatments, and other mutagenic exposures increase cancer rates by inducing oncogenic mutations cannot currently be determined.
Fitness, selection and cancer evolution: an alternative model
For organismal evolution, natural selection works on heritable diversity, and major periods of speciation (such as the Cambrian Explosion) were likely due to altered environmental selection pressures rather than increases in mutation rates. Analogously, numerous investigators have stressed the importance of the microenvironment in cancer development and the critical role of altered selection (21, 22, 44–49). Dramatic changes in tissue microenvironments occur with age, including stromal changes and increased inflammation (50, 51). These age-dependent changes should substantially alter adaptive landscapes (relationships between genotype and cellular fitness; Box 1), which describe how mutational changes can be adaptive, maladaptive or neutral in a context-dependent fashion. Alterations in adaptive landscapes in old age should promote selection for particular oncogenic mutations from within the standing genetic/epigenetic variation (Figure 2), whether it arose from endogenous (oxidative damage, replicative errors, etc.) or exogenous (exposure to environmental carcinogens) insults. Indeed, recent studies indicate that the frequency of cells with clonally-expanded genomic rearrangements increases substantially after 50–60 years of age in humans, correlating with cancer risk, which could reflect alterations in the adaptive landscape, increased rates of genomic alterations, and/or decreased stem cell polyclonality (52–54). Notably, in another study, 5 of the 6 detected clonally-expanded chromosomal abnormalities were present in both bladder and blood, suggesting an early embryonic origin of the events (55). Mutations that arise during ontogeny (or anytime after) but were neither adaptive nor maladaptive at the time, may be adaptive in the new landscape, thereby conferring a selective advantage and promoting clonal expansion (36, 48). Context-dependent selection leading to expansion of the oncogenically mutated clone would then greatly increase the likelihood of acquisition of secondary oncogenic mutations in cells that harbor an initiating lesion. Moreover, some of these oncogenic events selected for by the age or carcinogen altered adaptive landscapes could then contribute to increased genomic instability, providing more fuel for selection to act upon. While new mutations that accumulate with age should increase the cellular variation subject to selection, this alternative model does not depend on age-dependent accumulation of mutations to explain increased cancer incidence in old age.
Figure 2. Selection-centric model.

This model posits that aging is largely associated with cancer due to alterations in selection for oncogenic mutations. The weight of the arrow reflects the proposed contribution to cancer incidence.
To understand cancer evolution, we should consider why large and long-lived multicellular organisms like ourselves are so good at not getting cancer (36). For example, what tumor suppressive mechanism could allow for mammals as diverse as mice and blue whales to largely avoid cancer through their reproductive years (Peto’s Paradox)? As argued above, the commonly accepted view that cancer incidence is rate-limited by the occurrence of oncogenic mutations does not appear to be consistent with the common presence of oncogenic mutations in normal tissues, with the most rapid accumulation of mutations during ontogeny, and with the lack of reductions in somatic mutation rates during the evolution of complex multicellularity. We have proposed that cancer avoidance through reproductive years is dependent on the same basic principle that governs the avoidance of other hallmarks of aging: investments are made in tissue maintenance to the extent that provides the best return in terms of reproductive success. Thus, we have argued that the maintenance of tissue stem and progenitor cell fitness is inherently tumor suppressive, as high cellular fitness should disfavor selection for phenotype-altering somatic mutations (see (36, 48) for a full description of this “Adaptive Oncogenesis” model). Of course, other mechanisms, such as alterations in how telomeres are maintained (56, 57), could also contribute to similar tumor suppression through reproductive years for species with hugely different sizes and lifespans.
If we again consider HSC, given that hematopoietic malignancies are common in mice and that HSC are the best characterized stem cells, it is striking that mutations defined as oncogenic (activation of an oncogenic pathway, either by tumor suppressor gene deletion or oncogene expression) typically exhibit a common phenotype in HSC: loss of self-renewal (Table 1). For this table, I have assembled all published reports that I could find which describe oncogenic mutations engineered in mouse HSC under reasonably physiological contexts (i.e. in young unperturbed bone marrow at steady-state). It is notable that even mutations, such as in PTEN (58, 59), which increase proliferation (leading to initial expansion of short-term progenitors), impair HSC maintenance. In fact, a common effect of oncogenic mutations in HSC is to increase cell cycling (60), which likely contributes to loss of self-renewal: HSC maintenance necessitates an appropriate level of quiescence. Thus, we would expect that these mutations, should they occur in an individual HSC, would lead to clonal exhaustion by differentiation. Finally, it is notable that many of these mutations have been shown to be advantageous in vitro. For example, β-catenin activation increases HSC self-renewal and expansion in vitro (61). Animals did not evolve stem cells that would be well adapted to in vitro culture, and certain oncogenic events can be adaptive under such stressful conditions. The studies summarized in Table 1 provide support for the model that stem cells occupy a local fitness peak on the adaptive landscape, such that changes in phenotypic parameters will be rarely advantageous and typically disadvantageous (Figure 3).
Table 1. Oncogenic mutations typically impair HSC maintenance.
The effects of oncogenic mutations on HSC analyzed in genetically engineered mice. Data summarized for effects on HSC were, at least in large part, derived from analyses of unperturbed young mice (HSC at steady-state).
| Genotype | Pathway Deregulated | Effect on HSC | Corollary | References |
|---|---|---|---|---|
| Rb cKO | CDK/Rb/E2F | ↓self-renewal | ↑cell cycle | (74–76) |
| p21CIP1−/− | CDK/Rb/E2F | ↓self-renewal | ↑cell cycle | (77) |
| PTEN cKO | PI3K/AKT/mTOR | ↓self-renewal | ↑differentiation; ↑cell cycle | (58, 59) |
| p16INK4A−/− | CDK/Rb/E2F | ↓self-renewal | ↑cell cycle | (71) |
| ATM−/− | DDR/metabolism | ↓self-renewal | ↑ROS | (78) |
| p53−/− & cKO | p53 | no effect | (72, 73) | |
| GSK3 knockdown & GSK3b−/− | Wnt/β-catenin mTOR |
↓self-renewal; loss of HSC | ↑cell cycle | (79) |
| APC cKO | Wnt/β-catenin mTOR |
↓self-renewal | ↑differentiation; ↑ROS | (80) |
| TSC1−/− | mTOR | ↓self-renewal | ↑differentiation; ↑ROS | (81, 82) |
| LKB cKO | mTOR/AMPK | ↓self-renewal | ↑apoptosis ↑cell cycle |
(83) |
| EGR1−/− | various | ↓self-renewal; HSC exhaustion | ↑cell cycle; ↑mobilization | (84) |
| FBW7 cKO | Notch; Myc; Cyclin E | ↓self-renewal; ↓CRC | ↑cell cycle; ↑Myc; ↑apoptosis | (85, 86) |
| MEN1 cKO | Interacts with | ↓hematopoietic | ↓↓HSC function | (87) |
| (MENIN) | MLL | output; ↓CRC | under stress | |
| c-CBL−/− | Tyrosine kinase signaling | ↑HSC#; ↑CRC | ↑cell cycle; ↑STAT5 activation | (62) |
| Ikaros LOF mt | Ikaros | ↓self-renewal; loss of HSC | impaired differentiation | (88–90) |
| β-catenin activation | Wnt/β-catenin/mTOR | loss of HSC; ↓differentiation | ↑apoptosis ↑cell cycle |
(91–93) |
| Myc o/e | Myc | ↓self-renewal | ↑cell cycle | (94) |
| Rheb2 o/e | mTOR | ↓self-renewal | ↑cell cycle | (95) |
| Kras(G12D) | Ras/MAPK | ↑CRC; ↓↓HSC # | ↑cell cycle | (66) |
| Bcr-Abl (inducible) | Ras, AKT, STAT, others | ↓HSC #; ↑CRC | ↑cell cycle; ↑differentiation | (67) |
cKO, conditional knockout; o/e, transgenic overexpression; CDK, cyclin-dependent kinase; CRC, competitive repopulating capacity.
Figure 3. The Goldilocks Rule for stem cells.

A. Young healthy stem cells are proposed to possess parameters (cell cycle, differentiation, interactions with the niche, etc.) that are near optimal (“just right”) for maintenance as a stem cells. Stem cells are presumed to occupy a local fitness peak on the adaptive landscape; cancer cells in the same tissue could occupy a higher peak, but transitions to this peak would require passage through lower fitness states on the landscape (see (36)). Thus, acquisition of a single oncogenic mutation would typically be disadvantageous, by changing parameters from their optimum (see Table 1). B. For old or damaged stem cells, parameters are suboptimal or abnormal, and the stem cells no longer possess optimal or near optimal fitness. Changes in parameters could result from both cell-autonomous events (damage to the stem cells) or from non-cell autonomous changes (such as degradation of the niche or systemic changes). These changes in the stem cell pool can lead to selection for oncogenic events that are adaptive to this context.
While the Adaptive Oncogenesis model posits that oncogenic mutations should rarely be advantageous within young, fit stem cell pools, there are potential exceptions. First, c-CBL−/− mice exhibit increased numbers of HSC, and these HSC exhibit increased cycling and greater reconstitution potential in competitive bone marrow transplantation experiments (62) (Table 1). C-CBL is an E3 ligase that downregulates tyrosine kinase signaling. Gain-of-function mutations and translocation of c-CBL are implicated in several cancers including myeloid neoplasms (63, 64), and c-CBL−/− mice exhibit tissue hyperplasia (65). It will be interesting to determine if mutation of c-Cbl in an isolated HSC indeed proves advantageous in a young healthy mouse (as opposed to a mouse deficient in c-CBL in all tissues). Second, the induction of a KRASG12D mutation in mice leads to competitive expansion of the hematopoietic clones (including HSC) expressing activated K-Ras, despite a dramatic loss of functional HSC numbers (66). As noted by the authors, conditional activation of KRASG12D occurs in many (if not all) tissues, and thus K-RasG12D expression in non-hematopoietic tissues could alter the microenvironment for HSC (and thus the adaptive landscape), which could also explain the reductions in HSC numbers. Another possible exception is for Bcr-Abl. Reynaud et al showed that activation of Bcr-Abl expression in unperturbed mice results in reduced HSC numbers, apparently by increased differentiation to more committed myeloid progenitors (67), which nicely supports our model that young unperturbed HSC favor the status quo (the youthful phenotype). These results are also consistent with previous studies which indicate that Bcr-Abl promotes differentiation of human HSC, inhibiting self-renewal (68, 69), and that selection for Bcr-Abl is context dependent (32, 70). However, in the Reynaud et al study, following transplantation into irradiated recipient mice, Bcr-Abl provides a competitive advantage to HSC (67). Irradiation clearly alters the bone marrow microenvironment (and thus the stem cell niche), which could impact upon the adaptive landscape and promote selection for Bcr-Abl mutation. Of course, the alternative explanation is that some oncogenic mutations can be advantageous even in young healthy HSC pools, and it is the small size of this stem cell pool (together with other hurdles to tumorigenesis, such as the need for multiple oncogenic mutations) combined with the inability of these oncogenes to initiate cancer in more committed hematopoietic progenitors that sufficiently limits leukemias initiated by these oncogenes until older ages.
Just as high cellular fitness should prevent the fixation of phenotype-altering mutations, the converse should also be true: reductions in progenitor cell fitness with aging or other insults should increase selection for oncogenic mutations adaptive to the particular context (Figure 3). For example, while INK4A (encodes the p16 cyclin-dependent kinase inhibitor) mutation reduces the self-renewal of young HSC (Table 1), p16 loss actually increases the self-renewal of old HSC (which exhibit self-renewal defects) (71), and thus we would expect that p16 loss would be adaptive in old HSC pools. Similarly, while loss of p53 does not provide an advantage within young healthy hematopoietic pools, p53 mutation is potently selected for within HSC and more committed progenitor pools following irradiation of mice (72, 73). Finally, we have shown that Bcr-Abl is adaptive in old hematopoietic progenitor pools, but not young, by restoring kinase signaling pathways that are reduced in old progenitors (32). Thus, just as maintenance of fit stem cell pools should be tumor suppressive by disfavoring phenotype-altering mutations, reductions in the fitness of stem cell pools (such as during aging or following irradiation) should increase selection for particular oncogenic mutations adaptive to the altered context.
Conclusions
The current paradigm that the occurrence of oncogenic mutations with age is rate limiting for cancer development has provided a framework for a large body of cancer research, particularly for the field of carcinogenesis. I have raised questions to challenge this paradigm: if cancer were rate-limited by the occurrence of oncogenic mutations with age: Why would cancers increase exponentially late in life given that mutation accumulation rates are maximal during ontogeny? Would we expect the frequency of oncogenic mutations in tissues to far outpace the rates of corresponding cancers? Why have mutation rates not decreased during the evolution of larger and longer-lived species? Why would oncogenic mutations impair stem cell maintenance? In addition, I have argued that the cause-and-effect relationships between inherited DNA repair deficiencies (or mutagen exposure), oncogenic mutations and cancer incidence are far from established. There are important implications for a revised understanding of the relationships between aging, carcinogens and cancer incidence. From a practical standpoint, perhaps we should be more concerned about how aging, environmental exposures, and therapies impact on the overall tissue landscape, especially given that prevention of changes in adaptive landscapes is probably more feasible than the prevention of the occurrence of mutations. Whereas limiting the incidence of cancers through maintenance of healthier tissues might turn out to be a reasonable preventative approach, meaningful developments in this area will necessitate challenging the current dogma.
Acknowledgments
These studies were supported by grants from the National Institutes of Health (R01-CA157850) and the Leukemia Lymphoma Society. I thank Robert Sclafani, Michael Weil, Andriy Marusyk, Ruth Hershberg, Andrew Thorburn and members of my laboratory for their critical comments and suggestions.
References
- 1.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–74. doi: 10.1016/j.cell.2011.02.013. Epub 2011/03/08. [DOI] [PubMed] [Google Scholar]
- 2.Kennedy SR, Loeb LA, Herr AJ. Somatic mutations in aging, cancer and neurodegeneration. Mech Ageing Dev. 2011 doi: 10.1016/j.mad.2011.10.009. Epub 2011/11/15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Vogelstein B, Kinzler KW. Cancer genes and the pathways they control. Nat Med. 2004;10(8):789–99. doi: 10.1038/nm1087. Epub 2004/08/03. [DOI] [PubMed] [Google Scholar]
- 4.Weinberg RA. The Biology of Cancer. Chapter 11. New York: Garland Science; 2007. [Google Scholar]
- 5.Serrano M, Blasco MA. Cancer and ageing: convergent and divergent mechanisms. Nat Rev Mol Cell Biol. 2007;8(9):715–22. doi: 10.1038/nrm2242. Epub 2007/08/25. [DOI] [PubMed] [Google Scholar]
- 6.Hoeijmakers JH. DNA damage, aging, and cancer. N Engl J Med. 2009;361(15):1475–85. doi: 10.1056/NEJMra0804615. Epub 2009/10/09. [DOI] [PubMed] [Google Scholar]
- 7.Peto R, Roe FJ, Lee PN, Levy L, Clack J. Cancer and ageing in mice and men. Br J Cancer. 1975;32(4):411–26. doi: 10.1038/bjc.1975.242. Epub 1975/10/01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nowell PC. The clonal evolution of tumor cell populations. Science. 1976;194(4260):23–8. doi: 10.1126/science.959840. [DOI] [PubMed] [Google Scholar]
- 9.Armitage P, Doll R. The age distribution of cancer and a multi-stage theory of carcinogenesis. Br J Cancer. 1954;8(1):1–12. doi: 10.1038/bjc.1954.1. Epub 1954/03/01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Armitage P, Doll R. A two-stage theory of carcinogenesis in relation to the age distribution of human cancer. Br J Cancer. 1957;11(2):161–9. doi: 10.1038/bjc.1957.22. Epub 1957/06/01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lynch M. Evolution of the mutation rate. Trends Genet. 2010;26(8):345–52. doi: 10.1016/j.tig.2010.05.003. Epub 2010/07/03. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vijg J, Busuttil RA, Bahar R, Dolle ME. Aging and genome maintenance. Ann N Y Acad Sci. 2005;1055:35–47. doi: 10.1196/annals.1323.007. [DOI] [PubMed] [Google Scholar]
- 13.Frank SA. Evolution in health and medicine Sackler colloquium: Somatic evolutionary genomics: mutations during development cause highly variable genetic mosaicism with risk of cancer and neurodegeneration. Proc Natl Acad Sci U S A. 2010;107(Suppl 1):1725–30. doi: 10.1073/pnas.0909343106. Epub 2009/10/07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Weng NP, Hathcock KS, Hodes RJ. Regulation of telomere length and telomerase in T and B cells: a mechanism for maintaining replicative potential. Immunity. 1998;9(2):151–7. doi: 10.1016/s1074-7613(00)80597-x. Epub 1998/09/05. [DOI] [PubMed] [Google Scholar]
- 15.Wilson A, Laurenti E, Oser G, van der Wath RC, Blanco-Bose W, Jaworski M, et al. Hematopoietic stem cells reversibly switch from dormancy to self-renewal during homeostasis and repair. Cell. 2008;135(6):1118–29. doi: 10.1016/j.cell.2008.10.048. Epub 2008/12/09. [DOI] [PubMed] [Google Scholar]
- 16.Li W, Vijg J. Measuring genome instability in aging - a mini-review. Gerontology. 2012;58(2):129–38. doi: 10.1159/000334368. Epub 2011/12/14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dolle ME, Giese H, Hopkins CL, Martus HJ, Hausdorff JM, Vijg J. Rapid accumulation of genome rearrangements in liver but not in brain of old mice. Nat Genet. 1997;17(4):431–4. doi: 10.1038/ng1297-431. [DOI] [PubMed] [Google Scholar]
- 18.Freitas AA, de Magalhaes JP. A review and appraisal of the DNA damage theory of ageing. Mutat Res. 2011;728(1–2):12–22. doi: 10.1016/j.mrrev.2011.05.001. Epub 2011/05/24. [DOI] [PubMed] [Google Scholar]
- 19.Giese H, Snyder WK, van Oostrom C, van Steeg H, Dolle ME, Vijg J. Age-related mutation accumulation at a lacZ reporter locus in normal and tumor tissues of Trp53-deficient mice. Mutat Res. 2002;514(1–2):153–63. doi: 10.1016/s1383-5718(01)00329-1. Epub 2002/01/30. [DOI] [PubMed] [Google Scholar]
- 20.Frith CH, Ward JM, Chandra M. The morphology, immunohistochemistry, and incidence of hematopoietic neoplasms in mice and rats. Toxicol Pathol. 1993;21(2):206–18. doi: 10.1177/019262339302100213. Epub 1993/01/01. [DOI] [PubMed] [Google Scholar]
- 21.Gatenby RA, Gillies RJ. A microenvironmental model of carcinogenesis. Nat Rev Cancer. 2008;8(1):56–61. doi: 10.1038/nrc2255. [DOI] [PubMed] [Google Scholar]
- 22.Greaves M, Maley CC. Clonal evolution in cancer. Nature. 2012;481(7381):306–13. doi: 10.1038/nature10762. Epub 2012/01/20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mutter GL, Ince TA, Baak JP, Kust GA, Zhou XP, Eng C. Molecular identification of latent precancers in histologically normal endometrium. Cancer Res. 2001;61(11):4311–4. [PubMed] [Google Scholar]
- 24.Crawford YG, Gauthier ML, Joubel A, Mantei K, Kozakiewicz K, Afshari CA, et al. Histologically normal human mammary epithelia with silenced p16(INK4a) overexpress COX-2, promoting a premalignant program. Cancer Cell. 2004;5(3):263–73. doi: 10.1016/s1535-6108(04)00023-6. [DOI] [PubMed] [Google Scholar]
- 25.Greaves MF, Wiemels J. Origins of Chromosome Translocations in Childhood Leukemia. Nat Rev Cancer. 2003;3:1–10. doi: 10.1038/nrc1164. [DOI] [PubMed] [Google Scholar]
- 26.Naumov GN, Akslen LA, Folkman J. Role of angiogenesis in human tumor dormancy: animal models of the angiogenic switch. Cell Cycle. 2006;5(16):1779–87. doi: 10.4161/cc.5.16.3018. Epub 2006/08/26. [DOI] [PubMed] [Google Scholar]
- 27.Vickers M. Estimation of the number of mutations necessary to cause chronic myeloid leukaemia from epidemiological data. British journal of haematology. 1996;94(1):1–4. doi: 10.1046/j.1365-2141.1996.d01-1751.x. [DOI] [PubMed] [Google Scholar]
- 28.Bose S, Deininger M, Gora-Tybor J, Goldman JM, Melo JV. The presence of typical and atypical BCR-ABL fusion genes in leukocytes of normal individuals: biologic significance and implications for the assessment of minimal residual disease. Blood. 1998;92(9):3362–7. [PubMed] [Google Scholar]
- 29.Biernaux C, Loos M, Sels A, Huez G, Stryckmans P. Detection of major bcr-abl gene expression at a very low level in blood cells of some healthy individuals. Blood. 1995;86(8):3118–22. [PubMed] [Google Scholar]
- 30.Matioli GT. BCR-ABL insufficiency for the transformation of human stem cells into CML. Med Hypotheses. 2002;59(5):588–9. doi: 10.1016/s0306-9877(02)00220-7. [DOI] [PubMed] [Google Scholar]
- 31.Mullighan CG, Miller CB, Radtke I, Phillips LA, Dalton J, Ma J, et al. BCR-ABL1 lymphoblastic leukaemia is characterized by the deletion of Ikaros. Nature. 2008 doi: 10.1038/nature06866. [DOI] [PubMed] [Google Scholar]
- 32.Henry CJ, Marusyk A, Zaberezhnyy V, Adane B, DeGregori J. Declining lymphoid progenitor fitness promotes aging-associated leukemogenesis. Proc Natl Acad Sci U S A. 2010;107(50):21713–8. doi: 10.1073/pnas.1005486107. Epub 2010/11/26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cortopassi GA, Wang E. There is substantial agreement among interspecies estimates of DNA repair activity. Mech Ageing Dev. 1996;91(3):211–8. doi: 10.1016/s0047-6374(96)01788-5. Epub 1996/11/13. [DOI] [PubMed] [Google Scholar]
- 34.Promislow DE. DNA repair and the evolution of longevity: a critical analysis. J Theor Biol. 1994;170(3):291–300. doi: 10.1006/jtbi.1994.1190. Epub 1994/10/07. [DOI] [PubMed] [Google Scholar]
- 35.Tang JY, Hwang BJ, Ford JM, Hanawalt PC, Chu G. Xeroderma pigmentosum p48 gene enhances global genomic repair and suppresses UV-induced mutagenesis. Mol Cell. 2000;5(4):737–44. doi: 10.1016/s1097-2765(00)80252-x. Epub 2000/07/06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.DeGregori J. Evolved tumor suppression: why are we so good at not getting cancer? Cancer Res. 2011;71(11):3739–44. doi: 10.1158/0008-5472.CAN-11-0342. Epub 2011/05/26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Thompson LH, Schild D. Recombinational DNA repair and human disease. Mutat Res. 2002;509(1–2):49–78. doi: 10.1016/s0027-5107(02)00224-5. [DOI] [PubMed] [Google Scholar]
- 38.Bagley J, Cortes ML, Breakefield XO, Iacomini J. Bone marrow transplantation restores immune system function and prevents lymphoma in Atm-deficient mice. Blood. 2004;104(2):572–8. doi: 10.1182/blood-2003-12-4226. [DOI] [PubMed] [Google Scholar]
- 39.Bensimon A, Aebersold R, Shiloh Y. Beyond ATM: the protein kinase landscape of the DNA damage response. FEBS Lett. 2011;585(11):1625–39. doi: 10.1016/j.febslet.2011.05.013. Epub 2011/05/17. [DOI] [PubMed] [Google Scholar]
- 40.Westbrook AM, Schiestl RH. Atm-deficient mice exhibit increased sensitivity to dextran sulfate sodium-induced colitis characterized by elevated DNA damage and persistent immune activation. Cancer Res. 2010;70(5):1875–84. doi: 10.1158/0008-5472.CAN-09-2584. Epub 2010/02/25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Venkatesan RN, Treuting PM, Fuller ED, Goldsby RE, Norwood TH, Gooley TA, et al. Mutation at the polymerase active site of mouse DNA polymerase delta increases genomic instability and accelerates tumorigenesis. Mol Cell Biol. 2007;27(21):7669–82. doi: 10.1128/MCB.00002-07. Epub 2007/09/06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Preston BD, Albertson TM, Herr AJ. DNA replication fidelity and cancer. Semin Cancer Biol. 2010;20(5):281–93. doi: 10.1016/j.semcancer.2010.10.009. Epub 2010/10/19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fleenor CJ, Marusyk A, DeGregori J. Ionizing radiation and hematopoietic malignancies: altering the adaptive landscape. Cell Cycle. 2010;9(15):3005–11. doi: 10.4161/cc.9.15.12311. Epub 2010/08/03. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Laconi E, Doratiotto S, Vineis P. The microenvironments of multistage carcinogenesis. Semin Cancer Biol. 2008;18:322–9. doi: 10.1016/j.semcancer.2008.03.019. [DOI] [PubMed] [Google Scholar]
- 45.Bagby GC, Fleischman AG. The stem cell fitness landscape and pathways of molecular leukemogenesis. Front Biosci (Schol Ed) 2011;3:487–500. doi: 10.2741/s167. Epub 2011/01/05. [DOI] [PubMed] [Google Scholar]
- 46.Bissell MJ, Hines WC. Why don’t we get more cancer? A proposed role of the microenvironment in restraining cancer progression. Nat Med. 2011;17(3):320–9. doi: 10.1038/nm.2328. Epub 2011/03/09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Blagosklonny MV. Carcinogenesis, cancer therapy and chemoprevention. Cell Death Differ. 2005;12(6):592–602. doi: 10.1038/sj.cdd.4401610. [DOI] [PubMed] [Google Scholar]
- 48.Marusyk A, DeGregori J. Declining cellular fitness with age promotes cancer initiation by selecting for adaptive oncogenic mutations. Biochim Biophys Acta. 2008;1785(1):1–11. doi: 10.1016/j.bbcan.2007.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sieber OM, Tomlinson SR, Tomlinson IP. Tissue, cell and stage specificity of (epi)mutations in cancers. Nat Rev Cancer. 2005;5(8):649–55. doi: 10.1038/nrc1674. [DOI] [PubMed] [Google Scholar]
- 50.Henry CJ, Marusyk A, DeGregori J. Aging-associated changes in hematopoiesis and leukemogenesis: what’s the connection? Aging (Albany NY) 2011;3(6):643–56. doi: 10.18632/aging.100351. Epub 2011/07/19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rando TA. Stem cells, ageing and the quest for immortality. Nature. 2006;441(7097):1080–6. doi: 10.1038/nature04958. [DOI] [PubMed] [Google Scholar]
- 52.Laurie CC, Laurie CA, Rice K, Doheny KF, Zelnick LR, McHugh CP, et al. Detectable clonal mosaicism from birth to old age and its relationship to cancer. Nat Genet. 2012;44(6):642–50. doi: 10.1038/ng.2271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Jacobs KB, Yeager M, Zhou W, Wacholder S, Wang Z, Rodriguez-Santiago B, et al. Detectable clonal mosaicism and its relationship to aging and cancer. Nat Genet. 2012;44(6):651–8. doi: 10.1038/ng.2270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Forsberg LA, Rasi C, Razzaghian HR, Pakalapati G, Waite L, Thilbeault KS, et al. Age-related somatic structural changes in the nuclear genome of human blood cells. Am J Hum Genet. 2012;90(2):217–28. doi: 10.1016/j.ajhg.2011.12.009. Epub 2012/02/07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rodriguez-Santiago B, Malats N, Rothman N, Armengol L, Garcia-Closas M, Kogevinas M, et al. Mosaic uniparental disomies and aneuploidies as large structural variants of the human genome. Am J Hum Genet. 2010;87(1):129–38. doi: 10.1016/j.ajhg.2010.06.002. Epub 2010/07/06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gomes NM, Ryder OA, Houck ML, Charter SJ, Walker W, Forsyth NR, et al. Comparative biology of mammalian telomeres: hypotheses on ancestral states and the roles of telomeres in longevity determination. Aging Cell. 2011;10(5):761–8. doi: 10.1111/j.1474-9726.2011.00718.x. Epub 2011/04/27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gorbunova V, Seluanov A. Coevolution of telomerase activity and body mass in mammals: from mice to beavers. Mech Ageing Dev. 2009;130(1–2):3–9. doi: 10.1016/j.mad.2008.02.008. Epub 2008/04/05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yilmaz OH, Valdez R, Theisen BK, Guo W, Ferguson DO, Wu H, et al. Pten dependence distinguishes haematopoietic stem cells from leukaemia-initiating cells. Nature. 2006;441(7092):475–82. doi: 10.1038/nature04703. [DOI] [PubMed] [Google Scholar]
- 59.Zhang J, Grindley JC, Yin T, Jayasinghe S, He XC, Ross JT, et al. PTEN maintains haematopoietic stem cells and acts in lineage choice and leukaemia prevention. Nature. 2006;441(7092):518–22. doi: 10.1038/nature04747. [DOI] [PubMed] [Google Scholar]
- 60.Wilson A, Laurenti E, Trumpp A. Balancing dormant and self-renewing hematopoietic stem cells. Current opinion in genetics & development. 2009;19(5):461–8. doi: 10.1016/j.gde.2009.08.005. Epub 2009/10/09. [DOI] [PubMed] [Google Scholar]
- 61.Reya T, Duncan AW, Ailles L, Domen J, Scherer DC, Willert K, et al. A role for Wnt signalling in self-renewal of haematopoietic stem cells. Nature. 2003;423(6938):409–14. doi: 10.1038/nature01593. Epub 2003/04/30. [DOI] [PubMed] [Google Scholar]
- 62.Rathinam C, Thien CB, Langdon WY, Gu H, Flavell RA. The E3 ubiquitin ligase c-Cbl restricts development and functions of hematopoietic stem cells. Genes Dev. 2008;22(8):992–7. doi: 10.1101/gad.1651408. Epub 2008/04/17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ogawa S, Shih LY, Suzuki T, Otsu M, Nakauchi H, Koeffler HP, et al. Deregulated intracellular signaling by mutated c-CBL in myeloid neoplasms. Clin Cancer Res. 2010;16(15):3825–31. doi: 10.1158/1078-0432.CCR-09-2341. Epub 2010/06/16. [DOI] [PubMed] [Google Scholar]
- 64.Rathinam C, Thien CB, Flavell RA, Langdon WY. Myeloid leukemia development in c-Cbl RING finger mutant mice is dependent on FLT3 signaling. Cancer Cell. 2010;18(4):341–52. doi: 10.1016/j.ccr.2010.09.008. Epub 2010/10/19. [DOI] [PubMed] [Google Scholar]
- 65.Murphy MA, Schnall RG, Venter DJ, Barnett L, Bertoncello I, Thien CB, et al. Tissue hyperplasia and enhanced T-cell signalling via ZAP-70 in c-Cbl-deficient mice. Mol Cell Biol. 1998;18(8):4872–82. doi: 10.1128/mcb.18.8.4872. Epub 1998/07/22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Sabnis AJ, Cheung LS, Dail M, Kang HC, Santaguida M, Hermiston ML, et al. Oncogenic Kras initiates leukemia in hematopoietic stem cells. PLoS Biol. 2009;7(3):e59. doi: 10.1371/journal.pbio.1000059. Epub 2009/03/20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Reynaud D, Pietras E, Barry-Holson K, Mir A, Binnewies M, Jeanne M, et al. IL-6 controls leukemic multipotent progenitor cell fate and contributes to chronic myelogenous leukemia development. Cancer Cell. 2011;20(5):661–73. doi: 10.1016/j.ccr.2011.10.012. Epub 2011/11/19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Holyoake TL, Jiang X, Drummond MW, Eaves AC, Eaves CJ. Elucidating critical mechanisms of deregulated stem cell turnover in the chronic phase of chronic myeloid leukemia. Leukemia. 2002;16(4):549–58. doi: 10.1038/sj.leu.2402444. [DOI] [PubMed] [Google Scholar]
- 69.Schemionek M, Elling C, Steidl U, Baumer N, Hamilton A, Spieker T, et al. BCR-ABL enhances differentiation of long-term repopulating hematopoietic stem cells. Blood. 2010;115(16):3185–95. doi: 10.1182/blood-2009-04-215376. Epub 2010/01/08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Bilousova G, Marusyk A, Porter CC, Cardiff RD, DeGregori J. Impaired DNA Replication within Progenitor Cell Pools Promotes Leukemogenesis. PLoS Biology. 2005;3(12):e401. doi: 10.1371/journal.pbio.0030401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Janzen V, Forkert R, Fleming HE, Saito Y, Waring MT, Dombkowski DM, et al. Stem-cell ageing modified by the cyclin-dependent kinase inhibitor p16INK4a. Nature. 2006;443(7110):421–6. doi: 10.1038/nature05159. [DOI] [PubMed] [Google Scholar]
- 72.Bondar T, Medzhitov R. p53-mediated hematopoietic stem and progenitor cell competition. Cell Stem Cell. 2010;6(4):309–22. doi: 10.1016/j.stem.2010.03.002. Epub 2010/04/07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Marusyk A, Porter CC, Zaberezhnyy V, DeGregori J. Irradiation selects for p53-deficient hematopoietic progenitors. PLoS Biol. 2010;8(3):e1000324. doi: 10.1371/journal.pbio.1000324. Epub 2010/03/09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Daria D, Filippi MD, Knudsen ES, Faccio R, Li Z, Kalfa T, et al. The retinoblastoma tumor suppressor is a critical intrinsic regulator for hematopoietic stem and progenitor cells under stress. Blood. 2008;111(4):1894–902. doi: 10.1182/blood-2007-02-071746. Epub 2007/12/01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Walkley CR, Shea JM, Sims NA, Purton LE, Orkin SH. Rb regulates interactions between hematopoietic stem cells and their bone marrow microenvironment. Cell. 2007;129(6):1081–95. doi: 10.1016/j.cell.2007.03.055. Epub 2007/06/19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Spike BT, Dirlam A, Dibling BC, Marvin J, Williams BO, Jacks T, et al. The Rb tumor suppressor is required for stress erythropoiesis. EMBO J. 2004;23(21):4319–29. doi: 10.1038/sj.emboj.7600432. Epub 2004/10/01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Cheng T, Rodrigues N, Shen H, Yang Y, Dombkowski D, Sykes M, et al. Hematopoietic stem cell quiescence maintained by p21cip1/waf1. Science. 2000;287(5459):1804–8. doi: 10.1126/science.287.5459.1804. [DOI] [PubMed] [Google Scholar]
- 78.Ito K, Hirao A, Arai F, Matsuoka S, Takubo K, Hamaguchi I, et al. Regulation of oxidative stress by ATM is required for self-renewal of haematopoietic stem cells. Nature. 2004;431(7011):997–1002. doi: 10.1038/nature02989. Epub 2004/10/22. [DOI] [PubMed] [Google Scholar]
- 79.Huang J, Zhang Y, Bersenev A, O’Brien WT, Tong W, Emerson SG, et al. Pivotal role for glycogen synthase kinase-3 in hematopoietic stem cell homeostasis in mice. J Clin Invest. 2009;119(12):3519–29. doi: 10.1172/JCI40572. Epub 2009/12/05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Qian Z, Chen L, Fernald AA, Williams BO, Le Beau MM. A critical role for Apc in hematopoietic stem and progenitor cell survival. J Exp Med. 2008;205(9):2163–75. doi: 10.1084/jem.20080578. Epub 2008/08/30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Chen C, Liu Y, Liu R, Ikenoue T, Guan KL, Zheng P. TSC-mTOR maintains quiescence and function of hematopoietic stem cells by repressing mitochondrial biogenesis and reactive oxygen species. J Exp Med. 2008;205(10):2397–408. doi: 10.1084/jem.20081297. Epub 2008/09/24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Gan B, Sahin E, Jiang S, Sanchez-Aguilera A, Scott KL, Chin L, et al. mTORC1-dependent and -independent regulation of stem cell renewal, differentiation, and mobilization. Proc Natl Acad Sci U S A. 2008;105(49):19384–9. doi: 10.1073/pnas.0810584105. Epub 2008/12/05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Gan B, Hu J, Jiang S, Liu Y, Sahin E, Zhuang L, et al. Lkb1 regulates quiescence and metabolic homeostasis of haematopoietic stem cells. Nature. 2010;468(7324):701–4. doi: 10.1038/nature09595. Epub 2010/12/03. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Min IM, Pietramaggiori G, Kim FS, Passegue E, Stevenson KE, Wagers AJ. The transcription factor EGR1 controls both the proliferation and localization of hematopoietic stem cells. Cell Stem Cell. 2008;2(4):380–91. doi: 10.1016/j.stem.2008.01.015. Epub 2008/04/10. [DOI] [PubMed] [Google Scholar]
- 85.Thompson BJ, Jankovic V, Gao J, Buonamici S, Vest A, Lee JM, et al. Control of hematopoietic stem cell quiescence by the E3 ubiquitin ligase Fbw7. J Exp Med. 2008;205(6):1395–408. doi: 10.1084/jem.20080277. Epub 2008/05/14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Matsuoka S, Oike Y, Onoyama I, Iwama A, Arai F, Takubo K, et al. Fbxw7 acts as a critical fail-safe against premature loss of hematopoietic stem cells and development of T-ALL. Genes Dev. 2008;22(8):986–91. doi: 10.1101/gad.1621808. Epub 2008/03/28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Maillard I, Chen YX, Friedman A, Yang Y, Tubbs AT, Shestova O, et al. Menin regulates the function of hematopoietic stem cells and lymphoid progenitors. Blood. 2009;113(8):1661–9. doi: 10.1182/blood-2009-01-135012. Epub 2009/02/21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Papathanasiou P, Attema JL, Karsunky H, Hosen N, Sontani Y, Hoyne GF, et al. Self-renewal of the long-term reconstituting subset of hematopoietic stem cells is regulated by Ikaros. Stem Cells. 2009;27(12):3082–92. doi: 10.1002/stem.232. Epub 2009/10/10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Nichogiannopoulou A, Trevisan M, Neben S, Friedrich C, Georgopoulos K. Defects in hemopoietic stem cell activity in Ikaros mutant mice. J Exp Med. 1999;190(9):1201–14. doi: 10.1084/jem.190.9.1201. Epub 1999/11/02. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Papathanasiou P, Perkins AC, Cobb BS, Ferrini R, Sridharan R, Hoyne GF, et al. Widespread failure of hematolymphoid differentiation caused by a recessive niche-filling allele of the Ikaros transcription factor. Immunity. 2003;19(1):131–44. doi: 10.1016/s1074-7613(03)00168-7. Epub 2003/07/23. [DOI] [PubMed] [Google Scholar]
- 91.Perry JM, He XC, Sugimura R, Grindley JC, Haug JS, Ding S, et al. Cooperation between both Wnt/{beta}-catenin and PTEN/PI3K/Akt signaling promotes primitive hematopoietic stem cell self-renewal and expansion. Genes Dev. 2011;25(18):1928–42. doi: 10.1101/gad.17421911. Epub 2011/09/06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Kirstetter P, Anderson K, Porse BT, Jacobsen SE, Nerlov C. Activation of the canonical Wnt pathway leads to loss of hematopoietic stem cell repopulation and multilineage differentiation block. Nat Immunol. 2006;7(10):1048–56. doi: 10.1038/ni1381. Epub 2006/09/05. [DOI] [PubMed] [Google Scholar]
- 93.Scheller M, Huelsken J, Rosenbauer F, Taketo MM, Birchmeier W, Tenen DG, et al. Hematopoietic stem cell and multilineage defects generated by constitutive beta-catenin activation. Nat Immunol. 2006;7(10):1037–47. doi: 10.1038/ni1387. Epub 2006/09/05. [DOI] [PubMed] [Google Scholar]
- 94.Wilson A, Murphy MJ, Oskarsson T, Kaloulis K, Bettess MD, Oser GM, et al. c-Myc controls the balance between hematopoietic stem cell self-renewal and differentiation. Genes Dev. 2004;18(22):2747–63. doi: 10.1101/gad.313104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Campbell TB, Basu S, Hangoc G, Tao W, Broxmeyer HE. Overexpression of Rheb2 enhances mouse hematopoietic progenitor cell growth while impairing stem cell repopulation. Blood. 2009;114(16):3392–401. doi: 10.1182/blood-2008-12-195214. Epub 2009/08/20. [DOI] [PMC free article] [PubMed] [Google Scholar]