

Abstract
Background:
Most of the heritable risk of glioma is presently unaccounted for by mutations in known genes. In addition to rare inactivating germline mutations in TP53 causing glioma in the context of the Li-Fraumeni syndrome, polymorphic variation in TP53 may also contribute to the risk of developing glioma.
Methods:
To comprehensively evaluate the impact of variation in TP53 on risk, we analysed 23 tagSNPs and imputed 2377 unobserved genotypes in four series totaling 4147 glioma cases and 7435 controls.
Results:
The strongest validated association signal was shown by the imputed single-nucleotide polymorphism (SNP) rs78378222 (P=6.86 × 10−24, minor allele frequency ∼0.013). Confirmatory genotyping confirmed the high quality of the imputation. The association between rs78378222 and risk was seen for both glioblastoma multiforme (GBM) and non-GBM tumours. We comprehensively examined the relationship between rs78378222 and overall survival in two of the case series totaling 1699 individuals. Despite employing statistical tests sensitive to the detection of differences in early survival, no association was shown.
Conclusion:
Our data provided strong validation of rs78378222 as a risk factor for glioma but do not support the tenet that the polymorphism being a clinically useful prognostic marker. Acquired TP53 inactivation is a common feature of glioma. As rs78378222 changes the polyadenylation signal of TP53 leading to impaired 3′-end processing of TP53 mRNA, the SNP has strong plausibility for being directly functional contributing to the aetiological basis of glioma.
Keywords: inherited susceptibility, glioma, risk
Gliomas account for ∼40% of all primary brain tumours and are responsible for around 13 000 deaths in the United States of America each year (Bondy et al, 2008). To date, the only established environmental risk factor for glioma is exposure to ionising radiation (Bondy et al, 2008).
The two-fold increased risk of glioma seen in relatives of glioma patients provides evidence for inherited genetic susceptibility to the disease and a number of rare inherited cancer predisposition disorders such as Li-Fraumeni and Turcot syndromes, and neurofibromatosis are recognised to be associated with increased risk (Bondy et al, 2008). Recently, genome-wide association studies (GWAS) have demonstrated that common genetic variation also contributes to the development of glioma identifying risk single-nucleotide polymorphisms (SNPs) at six genetic loci – 5p15.33 (TERT), 7p11.2 (EGFR, two independent regions), 8q24.21 (CCDC26), 9p21.3 (CDKN2A/CDKN2B), 11q23.3 (PHLDB1) and 20q13.33 (RTEL1; Shete et al, 2009; Wrensch et al, 2009; Sanson et al, 2011). Although these GWAS SNPs have provided insight into genetic susceptibility to glioma based on the population frequency of risk alleles, collectively these SNPs account for only around10% of the 1.8-fold increased risk of all glioma seen in the relatives of patients (Hemminki et al, 2009), hence there remains considerable ‘missing heritability'. In addition to common genetic variation impacting on glioma risk, it is likely that rare disease-causing variants also contribute to disease heritability. Such variants are generally not directly identifiable through GWAS because of incomplete SNP tagging.
A TP53 mutation is among the most frequent and earliest detectable genetic alteration in glioma (Ohgaki and Kleihues, 2011). In addition to the rare inactivating germline mutations in TP53 that cause the Li-Fraumeni syndrome, it is entirely plausible that variants conferring modest risks also contribute to the risk of glioma. A number of studies have variously reported associations between specific TP53 polymorphisms and glioma risk (Rajaraman et al, 2007; Idbaih et al, 2008; Jha et al, 2011; Stacey et al, 2011; Egan et al, 2012; Shi et al, 2012), but many observations have been based on small case–control series with limited replication of findings. Most recently, rs78378222 that maps 3′ to TP53 has been implicated as a risk factor influencing glioma risk from an analysis of two case–control series (P=1.0 × 10−5; Stacey et al, 2011) and possibly patient prognosis (Egan et al, 2012).
To comprehensively evaluate the role of polymorphic variation at the TP53 locus on glioma risk, we analysed SNP genotype data in four series totalling 4147 glioma patients and 7435 controls, and imputed unobserved SNPs in cases and controls.
Materials and Methods
Subjects
We used data previously generated on four non-overlapping case–control series of Northern European ancestry, which had been the subject of previous GWAS (Shete et al, 2009; Sanson et al, 2011). Briefly, the UK GWAS was based on 636 cases (401 male; mean age 46 years) ascertained through the INTERPHONE Study (Cardis et al, 2007; Shete et al, 2009). Individuals from the 1958 Birth Cohort served as a source of controls. The US GWAS was based on 1281 cases (786 male; mean age 47 years) ascertained through the MD Anderson Cancer Center, Texas, between 1990 and 2008. Individuals from the Cancer Genetic Markers of Susceptibility (CGEMS) studies served as controls (Hunter et al, 2007; Yeager et al, 2007; Shete et al, 2009). The French GWAS study comprised of 1495 incident patients with glioma ascertained through the Service de Neurologie Mazarin, Groupe Hospitalier Pitié-Salpêtrière, Paris (Sanson et al, 2011). The controls were ascertained from the SU.VI.MAX (SUpplementation en VItamines et MinerauxAntioXydants) study of 12 735 healthy subjects (women aged 35–60 years; men aged 45–60 years; Hercberg et al, 2004). The German GWAS comprised of 880 patients who underwent surgery for a glioma at the Department of Neurosurgery, University of Bonn Medical Center, between 1996 and 2008 (Sanson et al, 2011); samples being collected while under care of Neurosurgeons. Control subjects were taken from three population studies: KORA (Co-operative Health Research in the Region of Augsburg; n=488; Holle et al, 2005), POPGEN (Population Genetic Cohort; n=678; Wichmann et al, 2005) and from the Heinz Nixdorf Recall study (n=380; Krawczak et al, 2006).
Collection of blood samples and clinical information from subjects was undertaken with informed consent and relevant ethical review board approval in accordance with the tenets of the Declaration of Helsinki.
Genotyping
Full details of the genotyping of cases and quality control using Illumina Infinium HD Human610-Quad BeadChips (Illumina, San Diego, CA, USA) are detailed in previously published work (Shete et al, 2009; Sanson et al, 2011). Briefly, duplicate samples were used to check genotyping quality. SNPs and samples with <95% SNPs called were eliminated from the analyses. Genotype frequencies at each SNP were tested for deviation from the Hardy–Weinberg equilibrium and rejected at P<10−4. We have previously confirmed an absence of systematic genetic differences between cases and controls and shown no significant evidence of population stratification in these sample sets. Genotyping of rs78378222 and rs3585075 was conducted by competitive allele-specific PCR KASPar chemistry (KBiosciences) or Taqman technology implemented on an ABI7900HT platform (Applied Biosystems, Foster City, USA); details of primers and probes used are available on request. Genotyping quality control was further evaluated through inclusion of duplicate DNA samples in SNP assays, together with direct sequencing of a subset of samples to confirm genotyping accuracy (details available on request).
Statistical analysis
Analyses were primarily undertaken using R (v2.6), STATA v.10 (State College) and PLINK (v1.06) software. The association between each SNP and risk of glioma was assessed by the Cochran-Armitage trend test. Odds ratios and associated 95% confidence intervals (CI) were calculated by unconditional logistic regression. Prediction of the untyped SNPs was carried out using IMPUTEv2 based on data from the 1000 Genomes Project (Phase 1 interim June 2011). Imputed data were analysed using SNPTEST v2 to account for uncertainties in SNP prediction. Association meta-analyses only included markers with proper_info scores >0.4 and MAFs>0.01. Meta-analyses were carried out with META using the genotype probabilities from IMPUTEv2, where a SNP was not directly typed. To test for the presence of additional independent risk alleles in each region, we carried out conditional logistic regression analysis that included SNPs with evidence of association in the meta-analysis at P<5.0 × 10−4. We calculated Cochran's Q statistic to test for heterogeneity and the I2 statistic to quantify the proportion of the total variation that was caused by heterogeneity (Higgins and Thompson, 2002). I2 values ⩾75% are considered to indicate substantial heterogeneity. To address heterogeneity between case–control series, we derived pooled ORs under a random effects model.
The familial relative risk of glioma attributable to a variant was calculated using the formula (Houlston and Ford, 1996):
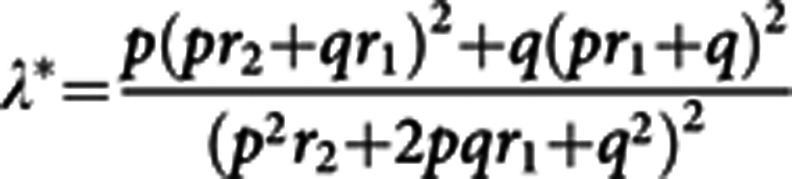 |
where p is the population frequency of the minor allele, q=1—p, and r1 and r2 are the relative risks (approximated by odds ratios) for heterozygotes and the rarer homozygotes relative to the more common homozygotes, respectively. From λ*, it is possible to quantify the influence of the locus on the overall familial risk of glioma in first-degree relatives of glioma patients. Assuming a multiplicative interaction between risk alleles, the proportion of the overall familial risk attributable to the locus is given by log (λ*)/log (λ0), where λ0 the overall familial risk of glioma, shown in epidemiological studies is 1.8 (Hemminki et al, 2009).
Measurement of LD between SNPs were based on Data Release 27, phase 3 (Feb 2009 on NCBI B36 assembly, HapMap dbSNP126), viewed using Haploview software (v4.2) and plotted using SNP annotation and proxy search (SNAP). LD blocks were defined according to HapMap recombination rate (centimorgans per megabase), as determined using the Oxford recombination hotspots (Myers et al, 2005) and the previously set distribution of CIs (Gabriel et al, 2002).
Complete survival data, together with clinical (age at diagnosis, sex, preoperative Karnofsky performance index (KPI)), degree of resection, chemotherapy and radiotherapy) covariate data were available for 1699 patients (75%) in the French and German case series, with a median follow-up interval of 8.9 years for patients without an event. Overall survival (OS) of patients was the end point of the analysis. Survival time was calculated from the date of diagnosis of glioma to the date of death. Patients who were not deceased were censored at the date of last contact. Mean follow-up time was computed among censored observations only. Kaplan–Meier survival curves according to genotype were generated and the homogeneity of the survival curves between genotypes was evaluated using the log-rank, and Wilcoxon and Fleming–Harrington tests (Klein (1997)). The log-rank test is usually the preferred test, but the other tests were conducted to show the influence of the polymorphic variation at different times of follow-up in order to detect any difference in early and late stages of disease. Cox regression analysis was used to estimate hazard ratios and their 95% CI, whereas adjusting for age, gender, treatment and grade.
Tumour genotyping
Tumour samples were available from a subset of the French patients ascertained through the Service de Neurologie Mazarin, Groupe Hospitalier Pitié-Salpêtrière, Paris. Tumours were snap-frozen in liquid nitrogen and DNA was extracted using the QIAmp DNA minikit, according to the manufacturer's recommendations (Qiagen, USA). DNA was analysed for large-scale copy-number variation by CGH-array as previously described (Simon et al, 2010). In the cases not analysed by CGH-array, 9p, 10q, 1p and 19q status were assigned using PCR microsatellites, and EGFR amplification and CDKN2A-p16-INK4a homozygous deletion by quantitative PCR. IDH1 codon R132 status was determined by sequencing. A search for mutations in exons 2-11 of TP53 mutations was conducted using previously reported primers and methods (Idbaih et al, 2007).
Results
We studied four non-overlapping glioma case–control series of Northern European ancestry providing data on 4147 cases and 7435 controls. We considered a ∼200-kb region from 100 kb 3′ to 100 kb 5′ of TP53 (rs11870250 at 7 471 783 bps to rs191403470 at 7 678 716 bps). Genotypes for 23 SNPs mapping to this region were available from each of the four GWAS data sets. Confining our analysis to the relationship between 17p13.1 variation and glioma risk, 8 of the 23 SNPs provided evidence for an association at P<0.05 (Figure 1). The strongest association was provided by rs1619016, which maps at 7 550 553 bps (OR=1.16, per allele P=4.93 × 10−4; Phet=0.17, I2=41% Figure 1).
Figure 1.

Case–control association plot for glioma and genomic structure of the chromosome 17p13.1 region (A) All glioma, (B) GBM, (C) non-GBM tumours. Overview of single-point SNP association results of both genotyped (triangles) and imputed (circles) SNPs are shown with recombination rates within the region. The region shown is chr17:7,471,719-7,678,811 (hg18 Built 36). −log10 P values (y axis) of the SNPs are shown according to their chromosomal positions (x axis). The top genotyped SNP in each combined analysis is a large CIRCLE and is rs78378222. The colour intensity of each symbol reflects the extent of LD with the top genotyped SNP, from white (r2=0) to dark gray (r2=1.0). Genetic recombination rates (cM/Mb), estimated using HapMap CEU samples, are shown with a light blue line. Also shown are the relative positions of genes and transcripts mapping to the region of association.
Using these data, we sought to establish whether we could identify SNPs better correlated with the risk of glioma through imputation of untyped SNPs referencing the 1000 Genomes project. Of a total of 6 403 imputed SNPs, 2,377 passed post-imputation QC filtering. Thirty-one of these SNPs provided superior evidence for an association with glioma risk than that provided by rs1619016 (Figure 1).
The strongest signals were shown by rs78378222, which maps 3′ to TP53, and rs35850753, which maps 5′ to TP53 (P<10−24). These associations are many orders of magnitude stronger than the association with rs1619016. Imputed information statistics for rs78378222 and rs3585075 SNPs were 0.66 and 0.61, respectively. To validate imputation of rs78378222 and rs3585075, we directly typed these SNPs in available DNAs from three of the four case–control series. Specifically, from the French GWAS we genotyped in all 1423 cases and 1827 controls, 816 of the German cases, all 631 UK cases and 700 UK controls. In each of these data sets, the concordance between imputed and directly typed SNP genotype was 99.0% for rs78378222 and 98.5% for rs3585075 (r2=0.90 and 0.89 for correlation between directly typed and imputed SNP genotypes, respectively).
The association between rs78378222 and glioma risk was shown in each of the four case–control studies (Figure 2), with the pooled OR under a fixed-effects model being 3.74 (95% CI: 2.89–4.83; P=6.86 × 10−24), however, there was significance between-study heterogeneity (Phet=0.045, I2=63%). Under a random effects model, the pooled OR was 3.65 (95% CI: 2.38–5.59; P=2.83 × 10−9). Between-study heterogeneity was largely ascribable to the German case–control series, which showed the weakest association. Omitting this study, the pooled OR was 4.45 (95% CI: 3.33–5.94; P=3.72 × 10−24; Phet=0.50, I2=0%).
Figure 2.

Association between glioma risk and rs78378222, for all glioma, GBM and non-GBM. Summary of association results for rs78378222. (A–C) Forest plots of rs78378222 per allele odds ratio (OR) for (A) all glioma, (B) GBM and (C) non-GBM. The x axis corresponds to the OR. Horizontal lines represent 95% confidence intervals. Each box represents the OR point estimate and its area is proportional to the statistical weight of the study. The diamonds (and broken lines) represent the summary odds ratios obtained from fixed-effect pooled analysis with 95% confidence intervals given by their widths. The unbroken vertical line is at the null value (OR=1.0). For all glioma, the MAFs for each study are: French cases: 0.024, French controls: 0.008, German cases: 0.023, German controls: 0.015, USA cases: 0.032, USA controls: 0.012, UK cases: 0.030, UK controls: 0.014.
rs78378222 is in strong LD with rs3585075 (r2=0.94). Conditional analysis is consistent with the 17p13.1 association being attributable to rs78378222 variation per se. Furthermore, the risk allele of rs78378222 has been shown to be directly functional leading to impaired 3′-end processing of TP53 mRNA, thereby presumably impacting on cellular availability of TP53 (Stacey et al, 2011). Logistic regression analysis of SNPs with evidence of association at P<5.0 × 10−4 did not identify any additional independent risk alleles in the region with variation at rs78378222 genotype sufficient to capture the variation impacting on glioma risk.
Gliomas are heterogeneous and different tumour subtypes defined in part by malignancy grade (e.g., pilocytic astrocytoma WHO grade I, diffuse ‘low grade' glioma WHO grade II, anaplastic glioma WHO grade III and glioblastoma multiforme (GBM; WHO grade IV)) can be distinguished. Accumulating data have established that the subtypes of glioma have different molecular profiles possibly resulting from different etiologic pathways. In view of this using data from all studies, we first examined the risk for GBM and non-GBM tumours associated with rs78378222 (Figure 2). Respective ORs for GBM and non-GBM tumours under a fixed-effects model were 5.59 (P=4.90 × 10−17) and 5.00 (P=1.47 × 10−20); under a random effects model respective ORs were 5.36 (P=2.32 × 10−6) and 5.09 (P=8.64 × 10−6; Figure 2).
We then examined the association by subtype in more detail using histology data of WHO grade. Although there was an evidence of between-study heterogeneity, this analysis showed that the association was seen in all glioma grades (Figure 3). Subsequent to this, using data from the French series of directly genotyped cases and controls, we examined the association between glioma risk and rs78378222 stratified by tumour histology and molecular characteristics of tumours (Tables 1 and 2). This analysis indicated that the association was not driven by a relationship with specific cell lineage, with strong associations being shown for tumours of both oligo- and astrocyte origin (Table 1). Within non-GBM tumours, the strongest associations were shown for IDH mutation, EGFR non-amplification, 10q non-deleted and 1p/19q co-deleted. Within GBM tumours, the strongest associations were shown for tumours that were 1p-19q non-codeleted and 10q non-deleted (Table 2). For 231 of the cases, TP53 mutational status in tumours was available. Although the frequency of the risk allele for rs78378222 was higher in TP53-mutated tumours, the difference was not significant, 0.045 vs 0.028, respectively (P=0.49).
Figure 3.

Association between glioma risk and rs78378222, for grade II, III and IV glioma. Summary of association results for rs78378222. (A–C) Forest plots of rs78378222 per allele odds ratio (OR) for (A) grade II (B) Grade III and (C) Grade IV glioma. The x axis corresponds to the OR. Horizontal lines represent 95% confidence intervals. Each box represents the OR point estimate and its area is proportional to the statistical weight of the study. The diamonds (and broken lines) represent the summary odds ratios obtained from fixed-effect pooled analysis with 95% confidence intervals given by their widths. The unbroken vertical line is at the null value (OR=1.0).
Table 1. Glioma risk and rs78378222 by morphology in the French case–control series.
| Grade | Histology | N | OR | 95% CI | P-value |
|---|---|---|---|---|---|
| |
Low-grade astrocytoma |
81 |
2.8 |
1.0–8.2 |
0.05 |
| II |
Oligoastrocytoma |
97 |
1.7 |
0.5–5.8 |
0.36 |
| |
Oligodendroglioma |
218 |
2.9 |
1.4–5.8 |
1.8 × 10−3 |
| |
Anaplastic astrocytoma |
48 |
4.9 |
1.7–14.6 |
1.3 × 10−3 |
| III |
Anaplastic oligoastrocytoma |
100 |
2.3 |
0.8–6.5 |
0.12 |
| |
Anaplastic oligodendroglioma |
179 |
1.6 |
0.6–4.0 |
0.36 |
| IV | GBM | 424 | 2.8 | 1.6–5.0 | 1.0 × 10−4 |
Abbreviations: CI=confidence interval; GBM=glioblastoma multiforme; OR=odds ratio.The analysis is based directly on genotyped data for 1147 cases and 1827 controls. Per allele odds ratios are shown with the risk allele for all tests being the C-allele with the reference being AA homozygotes.
Table 2. Glioma risk and rs78378222 stratified by morphology type: IDH1 mutation, EGFR amplification, CDKN2A-p16-INKa homozygous deletion, 9p and 10q loss, 1p-19q co-deletion status in the French case–control series.
| Non-GBM | GBM | |||||||
|---|---|---|---|---|---|---|---|---|
|
Tumour alteration |
N |
OR |
95% CI |
P-value |
N |
OR |
95% CI |
P-value |
|
IDH1+ |
272 |
3.0 |
1.6–5.6 |
5.0 × 10−4 |
24 |
7.8 |
2.2–27.3 |
2.0 × 10−4 |
|
IDH1− |
204 |
1.4 |
0.5–3.5 |
0.52 |
238 |
2.6 |
1.3–5.3 |
4.7 × 10−3 |
|
EGFR amplified |
35 |
1.6 |
0.2–12.0 |
0.65 |
81 |
1.4 |
0.3–5.8 |
0.66 |
|
EGFR nonamplified |
438 |
2.5 |
1.4–4.4 |
1.5 × 10−3 |
185 |
3.8 |
1.9–7.4 |
4.1 × 10−5 |
| p16 Deleted |
73 |
3.2 |
1.1–9.1 |
0.03 |
97 |
3.6 |
1.5–8.8 |
2.9 × 10−3 |
| p16 Nondeleted |
393 |
2.2 |
1.2–4.0 |
0.01 |
164 |
2.8 |
1.3–6.1 |
8.0 × 10−3 |
| 9p Deleted |
140 |
2.0 |
0.8–5.2 |
0.14 |
128 |
3.2 |
1.4–7.3 |
4.7 × 10−3 |
| 9p Nondeleted |
320 |
2.5 |
1.3–4.7 |
3.8 × 10−3 |
135 |
3.0 |
1.3–6.9 |
7.3 × 10−3 |
| 10q Deleted |
140 |
2.4 |
1.0–5.9 |
0.04 |
203 |
2.0 |
0.9–4.5 |
0.1 |
| 10q Nondeleted |
316 |
2.3 |
1.2–4.5 |
9.0 × 10−3 |
62 |
6.9 |
2.9–16.3 |
3.4 × 10−7 |
| 1p-19q Codeleted |
146 |
1.9 |
0.7–5.0 |
0.17 |
28 |
NA |
NA |
0.47 |
| 1p-19q Noncodeleted | 316 | 2.7 | 1.5–5.0 | 1.1 × 10−3 | 235 | 3.4 | 1.8–6.5 | 1.0 × 10−4 |
Abbreviations: CI=confidence interval; GBM=glioblastoma multiforme; OR=odds ratio.The analysis is based on directly genotyped data for 739 cases and 1,827 controls. Per allele odds ratios are shown with the risk allele for all tests being the C-allele with the reference being AA homozygotes.
To explore the possibility that rs78378222 genotype may influence tumour progression, we examined the relationship between directed typed genotypes and patient outcome in the French (n=1106) and German (n=593) case series. Survival analysis by each grade and adjusting for age at diagnosis, sex, chemotherapy and radiotherapy provided no evidence for an independent relationship between rs78378222 genotype and OS in either of patient cohort (Figure 4) with log-rank tests all yielding P-values >0.1. Similarly, Wilcoxon and Fleming–Harrington tests were also all non-significant (P>0.1). After inclusion of KPI data, all results remained non-significant (data not shown).
Figure 4.

Kaplan–Meier curves for glioma patients according to rs78378222 genotype. Survival curves for the common homozygotes (AA) are shown as a solid line. The dashed line depicts the survival curve for the heterozygotes (AC) and rare homozygotes (CC). (A) French grade II cases (HR=0.95, 95% CI: 0.40–2.28); (B) French grade III cases (HR=1.51, 95% CI: 0.74–3.09); (C) French grade IV cases (HR=1.24, 95% CI: 0.70–2.18); (D) German grade II cases (HR=3.11, 95% CI: 0.27–35.88); (E) German grade III cases (HR=NA); (F) German grade IV cases (HR=1.23, 95% CI: 0.68–2.21).
Discussion
Our findings provide strong evidence that the rs78378222 variant has a role in determining the risk of developing glioma, thereby confirming the recent observation made by Stacey et al (2011). Moreover, the strength of the association we have demonstrated provides unambiguous evidence for rs78378222 being a risk factor for glioma.
Glioma is increasingly being viewed as a highly heterogeneous cancer. Primary and secondary forms of GBM are recognised; with secondary GBM developing through progression from low-grade diffuse astrocytomas or anaplastic astrocytomas. Although usually indistinguishable histologically, distinct molecular pathways characterise the primary and secondary forms. Notably, IDH1 mutations are commonly detectable in low-grade glioma and secondary GBM, but are rare in primary GBMs (Ohgaki and Kleihues, 2007; Nobusawa et al, 2009). Furthermore, there is increased evidence that each of the non-GBM forms of glioma are typified by certain molecular characteristics, which may well reflect different genetic predispositions. In this study, we investigated the impact of genetic variation on risk of glioma by tumour subtype. Although the effect of rs78378222 on risk is in part generic, there was evidence for stronger associations being displayed by tumours in which EGFR was not deleted.
Major strengths of our study are its overall and individual study size; that these data have been systematically ascertained and that by making use of GWAS data bias from population stratification, confounding has been avoided. This type of bias is especially relevant in studies of low frequency variants. Furthermore, we were able to demonstrate high concordance between imputed and directly typed SNPs.
We found no evidence that rs78378222 genotype is an independent predictor of prognosis for glioma in either of the two case series studied. Stratification by histological subtype did not impact on the significance of findings. Our observations are therefore in contrast with those recently reported by Egan et al (2012), supporting a strong correlation between rs78378222 genotype and OS in an analysis of 566 glioma patients. Even though bias from non-uniform treatment is a potential confounder in studies of some cancers, the management of glioma is limited, as there are only a restricted number of chemotherapeutic agents and prognosis is uniformly poor. Even accepting that there may be differences in clinical management of glioma between centres, no association between genotype and outcome was seen in either the German or the French case series. It is therefore unlikely that any spurious influence as a consequence of study design will have impacted significantly on a failure to demonstrate a relationship between genotype and OS. It is unlikely a priori that a single SNP, albeit one with functional effects, will impart substantial differences in glioma prognosis independently and it is not uncommon for the first published studies to report over-inflated estimates of effects, which subsequently larger studies cannot replicate. A caveat to such an assertion is that carriers of rs78378222 are rare and hence, the power of any one study to demonstrate an association with outcome is inherently limited (i.e., <40% to detect a 1.2-fold difference on surviourship in the French and German case series, stipulating a P-value of 0.05).
rs78378222 localises within the 3′ untranslated region of TP53 with the risk variant changing the AATAAA polyadenylation signal to AATACA (Stacey et al, 2011). This nucleotide substitution has been shown to lead to impaired 3′-end processing of TP53 mRNA, thereby presumably impacting on cellular availability of TP53. Given the role of inactivating TP53 mutations in the development of glial tumours, an association between rs78378222 and glioma risk mediated through loss of TP53 function is therefore eminently plausible.
Although the allele frequency of rs78378222 is only around 1% in the European population, the associated genotype risks translate to the variant accounting for ∼6% of the familial risk of glioma. This single uncommon SNP therefore accounts for a greater percentage of the familial risk than the collective impact of all previously identified common risk SNPs for glioma.
In summary, our data provide robust evidence for rs78378222 being a determinant of glioma risk. Furthermore, our study demonstrates the use of imputation of untyped genotypes as a powerful means of exploiting existing GWAS data sets to identify low-frequency disease-causing variants.
URLs
The R suite can be found at: http://www.r-project.org/. Detailed information on the tagSNP panel can be found at: http://www.illumina.com/
dbSNP: http://www.ncbi.nlm.nih.gov/projects/SNP/
HAPMAP: http://www.hapmap.org/
1958 Birth Cohort: http://www.cls.ioe.ac.uk/page.aspx?&sitesectionid=724&sitesectiontitle=Welcome+to+the+1958+National+Child+Development+Study
CGEMS: http://cgems.cancer.gov/
SNPTEST: https://mathgen.stats.ox.ac.uk/genetics_software/snptest/snptest.html
KORA: http://epi.helmholtz-muenchen.de/kora-gen/index_e.php
POPGEN: http://www.popgen.de/
Acknowledgments
We are grateful to all the patients and individuals for their participation and we also thank the clinicians and other hospital staff, cancer registries and study staff in respective centres who contributed to the blood sample and data collection. For the UK GWA study, we acknowledge the participation of the clinicians and other hospital staff, cancer registries, study staff and funders who contributed to the blood sample and data collection for this study as listed in Hepworth et al (2006). MD Anderson acknowledges the work on the USA GWA study of Phyllis Adatto, Fabian Morice, Hui Zhang, Victor Levin, Alfred WK Yung, Mark Gilbert, Raymond Sawaya, Vinay Puduvalli, Charles Conrad, Fredrick Lang and Jeffrey Weinberg from the Brain and Spine Center. For the German GWA study, we are indebted to B Harzheim (Bonn) and Dr A Müller-Erkwoh (Bonn) for help with the acquisition of clinical data and R Mahlberg (Bonn) who provided technical support. The UK GWA study made use of control genotyping data generated by the Wellcome Trust Case–Control Consortium. A full list of the investigators who contributed to the generation of the data is available from http://www.wtccc.org.uk. The US GWA study made use of control genotypes from the CGEMS prostate and breast cancer studies. A full list of the investigators who contributed to the generation of the data is available from http://cgems.cancer.gov/. French controls were taken from the SU.VI.MAX study. The German GWA study made use of genotyping data from three population control sources: KORA-gen, The Heinz-Nixdorf RECALL study and POPGEN. We are extremely grateful to all investigators who contributed to the generation of these data sets. In the United Kingdom, funding was provided by Cancer Research UK (C1298/A8362 supported by the Bobby Moore Fund), the Wellcome Trust and the DJ Fielding Medical Research Trust. The UK INTERPHONE study was supported by the European Union Fifth Framework Program ‘Quality of life and Management of Living Resources' (QLK4-CT-1999-01563) and the International Union against Cancer (UICC). The UICC received funds from the Mobile Manufacturers' Forum and GSM Association. Provision of funds via the UICC was governed by agreements that guaranteed INTERPHONE's scientific independence (http://www.iarc.fr/ENG/Units/RCAd.html). The UK centres were also supported by the Mobile Telecommunications and Health Research (MTHR) Programme and the Northern UK Centre was supported by the Health and Safety Executive, Department of Health and Safety Executive and the UK Network Operators. The views expressed in this paper are not necessarily those of the funders. In the United States, funding was provided by NIH grants 5R01(CA119215&5R01 CA070917). Support was also obtained from the American Brain Tumor Association and the National Brain Tumor Society. In France, funding was provided by the Délégation à la Recherche Clinique (MUL03012), the Association pour la Recherche sur les Tumeurs Cérébrales (ARTC), the Institut National du Cancer (INCa; PL046) and the French Ministry of Higher Education and Research. In Germany, funding was provided to MS and JS by the Deutsche Forschungsgemeinschaft (Si552, Schr285), the Deutsche Krebshilfe (70-2385-Wi2,70-3163-Wi3,10-6262) and BONFOR. Funding for the WTCCC was provided by the Wellcome Trust (076113&085475). The KORA Augsburg studies are supported by grants from the German Federal Ministry of Education and Research (BMBF) and were mainly financed by the Helmholtz Zentrum München, German Research Center for Environmental Health, Neuherberg. This work was financed by the German National Genome Research Network (NGFN) and supported within the Munich Center of Health Sciences (MC Health) as part of LMUinnovativ.
Footnotes
This work is published under the standard license to publish agreement. After 12 months the work will become freely available and the license terms will switch to a Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License.
References
- Bondy ML, Scheurer ME, Malmer B, Barnholtz-Sloan JS, Davis FG, Il'yasova D, Kruchko C, McCarthy BJ, Rajaraman P, Schwartzbaum JA, Sadetzki S, Schlehofer B, Tihan T, Wiemels JL, Wrensch M, Buffler PA. Brain tumor epidemiology: consensus from the Brain Tumor Epidemiology Consortium. Cancer. 2008;113:1953–1968. doi: 10.1002/cncr.23741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardis E, Richardson L, Deltour I, Armstrong B, Feychting M, Johansen C, Kilkenny M, McKinney P, Modan B, Sadetzki S, Schuz J, Swerdlow A, Vrijheid M, Auvinen A, Berg G, Blettner M, Bowman J, Brown J, Chetrit A, Christensen HC, Cook A, Hepworth S, Giles G, Hours M, Iavarone I, Jarus-Hakak A, Klaeboe L, Krewski D, Lagorio S, Lonn S, Mann S, McBride M, Muir K, Nadon L, Parent ME, Pearce N, Salminen T, Schoemaker M, Schlehofer B, Siemiatycki J, Taki M, Takebayashi T, Tynes T, van Tongeren M, Vecchia P, Wiart J, Woodward A, Yamaguchi N. The INTERPHONE study: design, epidemiological methods, and description of the study population. Eur J Epidemiol. 2007;22:647–664. doi: 10.1007/s10654-007-9152-z. [DOI] [PubMed] [Google Scholar]
- Egan KM, Nabors LB, Olson JJ, Monteiro AN, Browning JE, Madden MH, Thompson RC. Rare TP53 genetic variant associated with glioma risk and outcome. J Med Genet. 2012;49:420–421. doi: 10.1136/jmedgenet-2012-100941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabriel SB, Schaffner SF, Nguyen H, Moore JM, Roy J, Blumenstiel B, Higgins J, DeFelice M, Lochner A, Faggart M, Liu-Cordero SN, Rotimi C, Adeyemo A, Cooper R, Ward R, Lander ES, Daly MJ, Altshuler D. The structure of haplotype blocks in the human genome. Science. 2002;296:2225–2229. doi: 10.1126/science.1069424. [DOI] [PubMed] [Google Scholar]
- Hemminki K, Tretli S, Sundquist J, Johannesen TB, Granstrom C. Familial risks in nervous-system tumours: a histology-specific analysis from Sweden and Norway. Lancet Oncol. 2009;10:481–488. doi: 10.1016/S1470-2045(09)70076-2. [DOI] [PubMed] [Google Scholar]
- Hepworth SJ, Schoemaker MJ, Muir KR, Swerdlow AJ, van Tongeren MJ, McKinney PA. Mobile phone use and risk of glioma in adults: case-control study. BMJ. 2006;332:883–887. doi: 10.1136/bmj.38720.687975.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hercberg S, Galan P, Preziosi P, Bertrais S, Mennen L, Malvy D, Roussel AM, Favier A, Briancon S. The SU.VI.MAX Study: a randomized, placebo-controlled trial of the health effects of antioxidant vitamins and minerals. Arch Intern Med. 2004;164:2335–2342. doi: 10.1001/archinte.164.21.2335. [DOI] [PubMed] [Google Scholar]
- Higgins JP, Thompson SG. Quantifying heterogeneity in a meta-analysis. Stat Med. 2002;21:1539–1558. doi: 10.1002/sim.1186. [DOI] [PubMed] [Google Scholar]
- Holle R, Happich M, Lowel H, Wichmann HE. KORA--a research platform for population based health research. Gesundheitswesen. 2005;67 (Suppl 1:S19–S25. doi: 10.1055/s-2005-858235. [DOI] [PubMed] [Google Scholar]
- Houlston RS, Ford D. Genetics of coeliac disease. QJM. 1996;89:737–743. doi: 10.1093/qjmed/89.10.737. [DOI] [PubMed] [Google Scholar]
- Hunter DJ, Kraft P, Jacobs KB, Cox DG, Yeager M, Hankinson SE, Wacholder S, Wang Z, Welch R, Hutchinson A, Wang J, Yu K, Chatterjee N, Orr N, Willett WC, Colditz GA, Ziegler RG, Berg CD, Buys SS, McCarty CA, Feigelson HS, Calle EE, Thun MJ, Hayes RB, Tucker M, Gerhard DS, Fraumeni JF, Jr, Hoover RN, Thomas G, Chanock SJ. A genome-wide association study identifies alleles in FGFR2 associated with risk of sporadic postmenopausal breast cancer. Nat Genet. 2007;39:870–874. doi: 10.1038/ng2075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Idbaih A, Boisselier B, Marie Y, Sanson M, El Hallani S, Criniere E, Fourtassi M, Paris S, Carpentier C, Rousseau A, Mokhtari K, Combadiere C, Laigle-Donadey F, Hoang-Xuan K, Delattre JY. Influence of MDM2 SNP309 alone or in combination with the TP53 R72P polymorphism in oligodendroglial tumors. Brain Res. 2008;1198:16–20. doi: 10.1016/j.brainres.2008.01.027. [DOI] [PubMed] [Google Scholar]
- Idbaih A, Boisselier B, Sanson M, Criniere E, Liva S, Marie Y, Carpentier C, Paris S, Laigle-Donadey F, Mokhtari K, Kujas M, Hoang-Xuan K, Delattre O, Delattre JY. Tumor genomic profiling and TP53 germline mutation analysis of first-degree relative familial gliomas. Cancer Genet Cytogenet. 2007;176:121–126. doi: 10.1016/j.cancergencyto.2007.04.012. [DOI] [PubMed] [Google Scholar]
- Jha P, Pathak P, Chosdol K, Suri V, Sharma MC, Kumar G, Singh M, Mahapatra AK, Sarkar C. TP53 polymorphisms in gliomas from Indian patients: study of codon 72 genotype, rs1642785, rs1800370 and 16 base pair insertion in intron-3. Exp Mol Pathol. 2011;90:167–172. doi: 10.1016/j.yexmp.2010.11.002. [DOI] [PubMed] [Google Scholar]
- Klein J,MM. Statistics for Biology and Health Berlin. Springer; 1997. Survival analysis: techniques for censored and truncated data. [Google Scholar]
- Krawczak M, Nikolaus S, von Eberstein H, Croucher PJ, El Mokhtari NE, Schreiber S. PopGen: population-based recruitment of patients and controls for the analysis of complex genotype-phenotype relationships. Community Genet. 2006;9:55–61. doi: 10.1159/000090694. [DOI] [PubMed] [Google Scholar]
- Myers S, Bottolo L, Freeman C, McVean G, Donnelly P. A fine-scale map of recombination rates and hotspots across the human genome. Science. 2005;310:321–324. doi: 10.1126/science.1117196. [DOI] [PubMed] [Google Scholar]
- Nobusawa S, Watanabe T, Kleihues P, Ohgaki H. IDH1 mutations as molecular signature and predictive factor of secondary glioblastomas. Clin Cancer Res. 2009;15:6002–6007. doi: 10.1158/1078-0432.CCR-09-0715. [DOI] [PubMed] [Google Scholar]
- Ohgaki H, Kleihues P. Genetic pathways to primary and secondary glioblastoma. Am J Pathol. 2007;170:1445–1453. doi: 10.2353/ajpath.2007.070011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohgaki H, Kleihues P. Genetic profile of astrocytic and oligodendroglial gliomas. Brain Tumor Pathol. 2011;28:177–183. doi: 10.1007/s10014-011-0029-1. [DOI] [PubMed] [Google Scholar]
- Rajaraman P, Wang SS, Rothman N, Brown MM, Black PM, Fine HA, Loeffler JS, Selker RG, Shapiro WR, Chanock SJ, Inskip PD. Polymorphisms in apoptosis and cell cycle control genes and risk of brain tumors in adults. Cancer Epidemiol Biomarkers Prev. 2007;16:1655–1661. doi: 10.1158/1055-9965.EPI-07-0314. [DOI] [PubMed] [Google Scholar]
- Sanson M, Hosking FJ, Shete S, Zelenika D, Dobbins SE, Ma Y, Enciso-Mora V, Idbaih A, Delattre JY, Hoang-Xuan K, Marie Y, Boisselier B, Carpentier C, Wang XW, Di Stefano AL, Labussiere M, Gousias K, Schramm J, Boland A, Lechner D, Gut I, Armstrong G, Liu Y, Yu R, Lau C, Di Bernardo MC, Robertson LB, Muir K, Hepworth S, Swerdlow A, Schoemaker MJ, Wichmann HE, Muller M, Schreiber S, Franke A, Moebus S, Eisele L, Forsti A, Hemminki K, Lathrop M, Bondy M, Houlston RS, Simon M. Chromosome 7p11.2 (EGFR) variation influences glioma risk. Hum Mol Genet. 2011;20:2897–2904. doi: 10.1093/hmg/ddr192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shete S, Hosking FJ, Robertson LB, Dobbins SE, Sanson M, Malmer B, Simon M, Marie Y, Boisselier B, Delattre JY, Hoang-Xuan K, El Hallani S, Idbaih A, Zelenika D, Andersson U, Henriksson R, Bergenheim AT, Feychting M, Lonn S, Ahlbom A, Schramm J, Linnebank M, Hemminki K, Kumar R, Hepworth SJ, Price A, Armstrong G, Liu Y, Gu X, Yu R, Lau C, Schoemaker M, Muir K, Swerdlow A, Lathrop M, Bondy M, Houlston RS. Genome-wide association study identifies five susceptibility loci for glioma. Nat Genet. 2009;41:899–904. doi: 10.1038/ng.407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi M, Huang R, Pei C, Jia X, Jiang C, Ren H. TP53 codon 72 polymorphism and glioma risk: a meta-analysis. Oncol Lett. 2012;3:599–606. doi: 10.3892/ol.2011.521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon M, Hosking FJ, Marie Y, Gousias K, Boisselier B, Carpentier C, Schramm J, Mokhtari K, Hoang-Xuan K, Idbaih A, Delattre JY, Lathrop M, Robertson LB, Houlston RS, Sanson M. Genetic risk profiles identify different molecular etiologies for glioma. Clin Cancer Res. 2010;16:5252–5259. doi: 10.1158/1078-0432.CCR-10-1502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stacey SN, Sulem P, Jonasdottir A, Masson G, Gudmundsson J, Gudbjartsson DF, Magnusson OT, Gudjonsson SA, Sigurgeirsson B, Thorisdottir K, Ragnarsson R, Benediktsdottir KR, Nexo BA, Tjonneland A, Overvad K, Rudnai P, Gurzau E, Koppova K, Hemminki K, Corredera C, Fuentelsaz V, Grasa P, Navarrete S, Fuertes F, Garcia-Prats MD, Sanambrosio E, Panadero A, De Juan A, Garcia A, Rivera F, Planelles D, Soriano V, Requena C, Aben KK, van Rossum MM, Cremers RG, van Oort IM, van Spronsen DJ, Schalken JA, Peters WH, Helfand BT, Donovan JL, Hamdy FC, Badescu D, Codreanu O, Jinga M, Csiki IE, Constantinescu V, Badea P, Mates IN, Dinu DE, Constantin A, Mates D, Kristjansdottir S, Agnarsson BA, Jonsson E, Barkardottir RB, Einarsson GV, Sigurdsson F, Moller PH, Stefansson T, Valdimarsson T, Johannsson OT, Sigurdsson H, Jonsson T, Jonasson JG, Tryggvadottir L, Rice T, Hansen HM, Xiao Y, Lachance DH, ON BP, Kosel ML, Decker PA, Thorleifsson G, Johannsdottir H, Helgadottir HT, Sigurdsson A, Steinthorsdottir V, Lindblom A, Sandler RS, Keku TO, Banasik K, Jorgensen T, Witte DR, Hansen T, Pedersen O, Jinga V, Neal DE, Catalona WJ, Wrensch M, Wiencke J, Jenkins RB, Nagore E, Vogel U, Kiemeney LA, Kumar R, Mayordomo JI, Olafsson JH, Kong A, Thorsteinsdottir U, Rafnar T, Stefansson K. A germline variant in the TP53 polyadenylation signal confers cancer susceptibility. Nat Genet. 2011;43:1098–1103. doi: 10.1038/ng.926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wichmann HE, Gieger C, Illig T. KORA-gen--resource for population genetics, controls and a broad spectrum of disease phenotypes. Gesundheitswesen. 2005;67 (Suppl 1:S26–S30. doi: 10.1055/s-2005-858226. [DOI] [PubMed] [Google Scholar]
- Wrensch M, Jenkins RB, Chang JS, Yeh RF, Xiao Y, Decker PA, Ballman KV, Berger M, Buckner JC, Chang S, Giannini C, Halder C, Kollmeyer TM, Kosel ML, LaChance DH, McCoy L, O'Neill BP, Patoka J, Pico AR, Prados M, Quesenberry C, Rice T, Rynearson AL, Smirnov I, Tihan T, Wiemels J, Yang P, Wiencke JK. Variants in the CDKN2B and RTEL1 regions are associated with high-grade glioma susceptibility. Nat Genet. 2009;41:905–908. doi: 10.1038/ng.408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeager M, Orr N, Hayes RB, Jacobs KB, Kraft P, Wacholder S, Minichiello MJ, Fearnhead P, Yu K, Chatterjee N, Wang Z, Welch R, Staats BJ, Calle EE, Feigelson HS, Thun MJ, Rodriguez C, Albanes D, Virtamo J, Weinstein S, Schumacher FR, Giovannucci E, Willett WC, Cancel-Tassin G, Cussenot O, Valeri A, Andriole GL, Gelmann EP, Tucker M, Gerhard DS, Fraumeni JF, Jr., Hoover R, Hunter DJ, Chanock SJ, Thomas G. Genome-wide association study of prostate cancer identifies a second risk locus at 8q24. Nat Genet. 2007;39:645–649. doi: 10.1038/ng2022. [DOI] [PubMed] [Google Scholar]