

Abstract
Background
Toll-like receptors (TLRs) detect endogenous ligands released after trauma and contribute to the proinflammatory response to injury. Post-traumatic mortality correlates with the extent of the immuno-inflammatory response to injury which is comprised of a complex regulation of innate and adaptive immune responses. Although TLRs are known to modulate innate immune responses, their role in the suppression of lymphocyte responses following traumatic tissue injury is unclear.
Methods
This study used a murine model of severe peripheral tissue injury, involving muscle crush injury and injection of fracture components, to evaluate the roles of TLR2, 4 and 9 in the early and delayed immuno-inflammatory phenotype. Post-traumatic immune dysfunction was measured in our trauma model using the following parameters: ex-vivo splenocyte proliferation, Th1 cytokine release and iNOS induction within splenic myeloid-derived suppressor cells (MDSC). Systemic inflammation and liver damage were determined by circulating interleukin-6 levels and hepatocellular injury.
Results
Suppression of splenocyte responses after injury was dependent on TLR4 and 9 signaling as was post-traumatic iNOS upregulation in splenic MDSC. TLR2 was found to have only a partial role through contribution to inhibition of splenocyte proliferation. This study also reveals the involvement of TLR2 and 4 in the initial systemic inflammatory response to traumatic tissue injury, however, this response was found to be TLR9-independent.
Conclusions
These findings demonstrate the previously unidentified role of TLR2, 4 and 9 in the T-cell associated immune dysfunction following traumatic tissue injury. Importantly, this study also illustrates that TLRs play differing and selective roles in both the initial proinflammatory response and adaptive immune response after trauma. Furthermore, the results in the TLR9-deficient mice establishes that the upregulation of early pro-inflammatory markers do not always correlate with the extent of sustained immune dysfunction. This suggests potential for targeted therapies that could limit the immune dysfunction through selective inhibition of receptor function following injury.
Keywords: Injury, Toll-like receptor, lymphocyte, MDSC, immune dysfunction
Background
Traumatic tissue injury releases endogenous ligands which serve as triggers for the immuno-inflammatory response following injury. Exaggerated and sustained levels of this post-traumatic response have been correlated with subsequent patient morbidity and mortality1. The early pro-inflammatory component of this response involves excessive activation of innate immune pathways whereas immune dysfunction is described as a depression in adaptive immunity and includes in part T-cell dysfunction2.
Toll-like receptors (TLRs) are pattern-recognition receptors that were discovered based on their central role in the recognition of microbial molecules as part of host defense3. A subset of TLRs also recognize endogenous molecules and can trigger activation of inflammatory pathways in the setting of sterile inflammation4,5,6. In the setting of trauma TLR27, TLR48, and TLR99 have been shown to selectively participate in the pro-inflammatory response to hemorrhagic shock or tissue trauma. It is unknown if these same TLRs are involved in the regulation of pathways that lead to the sustained suppression of the immune responses, specifically lymphocyte responses, seen after trauma.
Here, we carried out experiments in a murine model of severe peripheral tissue injury to establish if TLR2, TLR4 and/or TLR9 contribute to either the early pro-inflammatory response or sustained immune dysfunction after injury. Our findings show that TLR4 is strongly involved in both components of the response. In contrast TLR2 was found to mediate systemic inflammation with little effect on immune dysfunction while TLR9 was shown to only contribute to the suppression of the adaptive T-cell function. Notably, our findings highlight a disconnect between the TLR-driven mechanisms and suggest a receptor-specific separation of the components of the immuno-inflammatory response to sterile tissue injury.
Methods
Animal Care
This research protocol complied with the regulations regarding the care and use of experimental animals published by the National Institutes of Health, and was approved by the Institutional Animal Care and Use Committee of the University of Pittsburgh.
All mice used in this study were male, aged 8–12 weeks. TLR4 knockout (TLR4−/−), TLR2−/− and TLR9-mutant (TLR9CpG1/CpG1) mice23, all on a C57BL/6 background, were bred at the University of Pittsburgh and originally provided by Dr. R. Medzhitov (Howard Hughes Medical Institute, Yale University), Dr. J. Kolls (University of Pittsburgh) and Dr. B.Beutler (The Scripps Research Institute) respectively. Age-matched wild type (WT) C57BL/6 mice were obtained from Charles River Laboratories International (Wilmington, MA). TLR4-mutant mice (C3H/HeJ) and the corresponding WT strain (C3H/HeOuJ) were obtained from Jackson Laboratories (Bar Harbor,ME).
Experimental Groups
The mice were randomly allocated to trauma or control groups. Control untraumatized mice were used to obtain physiologic baseline results. Prior to being euthanized under inhalational anesthesia, the control untraumatized mice receive no experimental manipulation and no other anesthesia. The traumatized mice underwent pseudofracture (PF), a peripheral tissue injury model under general anesthesia with pentobarbital sodium (70mg/kg, IP; Ovation Pharmaceuticals, Deerfield, IL) and with supplemental inhalational isoflurane (Abbott Laboratories, Chicago, IL) as needed. Buprenorphine (0.1mg/kg SC; Bedford Laboratories, Bedford, OH) was administered after recovery from anesthesia as an analgesic.
The pseudofracture model was performed as previously described10,11. In brief, soft tissue injury in the form of a muscle crush to both posterior thighs was followed by an injection into the crushed tissue of a suspension of pulverized bone. The bone suspension was prepared from both pulverized femurs and tibias of age- and weight-matched syngeneic donor mice resuspended in phosphate buffered saline (PBS). The animals were allowed full freedom after the procedure. Mortality of the procedure was 0%.
Mice that received control or inhibitory CpG oligodeoxynucleotides (100 μg) were injected subcutaneously both at time of and at 24hr after trauma.
Reagents
Saponin was from Sigma-Aldrich Co. (St.Louis, MO); mouse Interleukin-6 (IL-6), IL-2, Interferon-γ (IFN-γ) enzyme-linked immunosorbent assay (ELISA) kits from R&D systems Inc. (Minneapolis, MN); RPMI 1640 and L-Glutamine from Lonza BioWhittaker (Walkersville, MD); heat-inactivated fetal bovine serum (FBS), non-essential amino acids (NEAA) and sodium pyruvate from Hyclone Lab., Fisher Scientific, (Logan, UT); penicillin-streptomycin (pen/strep) and 2-mercaptoethanol (2-ME) from Gibco, Life Technologies Corp., (Grand Island, NY); concanavalin A (Con A) from GE healthcare Corp. (Piscataway, NJ) and antiCD3ε mAb (clone 145-2C11), anti-CD16/CD32, BD cytofix/cytoperm from BD Biosciences (San Jose, CA, USA). Fluorescently-labeled antibodies for flow cytometry were purchased from either eBiosciences (San Diego, CA) or BD Biosciences (San Jose, CA). Control CpG (ODN2088 Control; 5′-TCCTGAGCTTGAAGT –3prime;) and inhibitory CpG (ODN2088 miCpG; 5prime;-TCCTGGCGGGGAAGT -3prime;) oligodeoxynucleotides were purchased from Fisher Scientific Eurofins MWG Operon (Huntsville, AL).
Sample collection and preparation
Traumatized mice were euthanized 6, 12 or 48hr after trauma and compared with unmanipulated controls. At the conclusion of each experiment, animals were sacrificed with an overdose of isoflurane, then blood was collected by cardiac puncture and spleens were aseptically harvested.
Plasma was measured for levels of alanine aminotransferase (ALT) and aspartate aminotransferase (AST) with HESKA Dri-chem 4000 (Heska Loveland CO; slides from FUJIFILM Corp., Japan) analysis and IL-6 levels by ELISA, performed according to the manufacturer’s instructions.
Single cell suspensions were prepared from spleens for both culture and flow cytometric analysis as previously described12. In brief, the separated splenocytes were resuspended in RPMI 1640 supplemented with 10%FBS, l-glutamine, pen/strep, NEAA, sodium pyruvate, 2-ME and cell viability of 90% was confirmed with trypan blue stain. Parallel cultures were carried out in 96-well round-bottom tissue-culture plates (BD Falcon, BD Biosciences (San Jose, CA) at 1 x 105 viable cells/well and were stimulated with T-cell specific mitogens concanavlin A (2.5 μg/ml) or antiCD3ε mAb (1μg/ml) for 72hr at 37°C. Splenocyte culture supernatant was collected at 48hr for cytokine analysis. Tritiated thymidine (1μCi/well; Perkin Elmer Inc., Waltham, MA) was added for the last 18hr of culture. Proliferation was assessed through thymidine incorporation using a Topcount scintillation counter (Perkin Elmer Inc., Waltham, MA) and measured as counts per minute (cpm).
For flow cytometric analysis, splenocytes were incubated with anti-CD16/CD32 (Fc block) for 10min at 4°C. Cells were then stained for surface markers for 20min at 4°C with the following fluorescently labeled mAbs: anti-CD19, anti-CD90.2, anti-CD11b, anti-Gr-1. Cells were then incubated with BD cytofix/cytoperm for 20min at 4°C and washed with 0.1% saponin, stained with anti-iNOS (clone 6/iNOS/NOS Type II) for intracellular staining at room temperature for 1hr, washed with 0.1% saponin buffer and immediately analyzed. A Becton Dickinson LSR II flow cytometer was used for analysis of samples; FlowJo v.7.6.5 software (Tree Star Inc., Ashland, OR) was used for data analysis.
Statistical analysis
Statistical significance (p<0.05) was assessed by Student’s t-test and ANOVA analysis using SigmaPlot 11.0 software (Systat Software Inc., San Jose, California, USA). The results presented in the study are expressed as the mean ± standard error or mean (SEM).
Results
Peripheral tissue trauma elicits an early TLR4- and TLR2-dependent systemic inflammatory response with hepatocellular injury
The pseudofracture model (PF) was established and validated to simulate the severe peripheral tissue trauma seen with bilateral femur fracture10. However, because no bones are broken, the model permits the study of both early and delayed time points. We have previously shown that the early (6hr) systemic inflammatory response and remote organ injury in this model is TLR4-dependent13. Here, we tested whether TLR2 and TLR9, receptors known to be involved in the pro-inflammatory response to either hemorrhagic shock9 or tissue trauma7, are involved in the early responses in the PF model. Circulating IL-6 levels were used as a representative biomarker of the systemic inflammatory response and hepatocellular injury as a marker of liver damage, as an example of one form of remote organ damage. We have previously shown that PF leads to a significant elevation of both IL-6 and ALT levels within the circulation at 6hr following injury in wild type C57Bl/6 mice12,13, hence, this study uses the 6hr time point after PF to evaluate these markers. As shown in Figure 1, PF led to significant elevations in both IL-6 and ALT levels at 6hr, in comparison with uninjured controls. Also, as expected, this was dependent on TLR4 signaling as TLR4−/− mice exhibited minimal increases in IL-6 and ALT levels following PF. Injured TLR2−/− mice also exhibited minimal increases in IL-6 and ALT levels, while injured TLR9-mutant mice showed significantly elevated levels similar to those observed in WT mice after injury. Thus, the initial pro-inflammatory response following severe peripheral tissue injury is dependent on both TLR2 and TLR4, but independent of TLR9 signaling.
Figure 1. Early systemic hyperinflammatory response to peripheral tissue trauma is TLR4-and TLR2-dependent, but TLR9-independent.

Plasma IL-6 (A) and plasma alanine aminotranferase (ALT) (B) in WT, TLR9-mutant, TLR4−/−, TLR2−/− mice at 6hr following pseudofracture (PF) or in unmanipulated controls. Data shown as mean±SEM; * = p < 0.05 vs. respective control; n=4–9 mice/group at each time point; representative of 2 independent experiments.
Post trauma suppression in splenocyte function is TLR4 dependent
The capacity of splenocytes isolated from injured animals to respond to known mitogens is frequently used to assess immune dysfunction after trauma2. We have previously shown that PF leads to a profound suppression of splenocyte, specifically T-lymphocyte, proliferation and Th1 cytokine production in response to Con A and anti-CD3 antibody12. The nadir in the responses is reached at 48hr post injury12, as such, this study uses this time point after PF to evaluate the splenocyte responses. To determine if TLR4 is also involved in the trauma-induced immune dysfunction, we examined splenocyte responses in global TLR4−/− mice, and mice with a mutant form of TLR4 that is defective in TLR4 signaling (TLR4-mutant (C3H/HeJ)). As shown in Figure 2, WT mouse strains, C57BL/6 (control for TLR4−/−) and C3H/HeOuJ (control for TLR4-mutant) mice exhibited the expected suppression in splenocyte proliferation as well as IFN-γ and IL-2 release (in response to Con A) at 48hr after PF. In contrast, the TLR4−/− and TLR4-mutant strains exhibited no decrease in proliferation or cytokine production after PF. In fact, proliferation (TLR4-mutant) and IL-2 release (TLR4−/−) was increased significantly after injury in the TLR4-deficient strains. These data provide evidence that TLR4 signaling is also involved in the altered splenocyte responses characteristic of trauma models.
Figure 2. TLR4-dependent post-traumatic lymphocyte dysfunction.

Splenocyte function at 48h after pseudofracture (PF) or in unmanipulated controls after concanavalin A (2.5μg/mL) stimulation: proliferation measured in counts per minute (cpm) of tritiated thymidine uptake (A), IFN- release (B) and IL-2 release (C). TLR4−/− mice compared to background C57BL/6 (WT B6) strain. TLR4-mutant mice (C3H/HeJ) compared to background strain C3H/HeOuJ (WT C3H). Data presented as percent change from baseline control. Data shown as mean±SEM; * = p < 0.05 vs. respective control; n=4–10 mice/group; representative of 2 independent experiments.
The role of TLR2 in depressed splenocyte proliferation after injury
To determine whether the role of TLR2 in the early post-traumatic response was associated with a role in late post-traumatic immune suppression we evaluated splenocyte responses from control and PF injured WT C57BL/6 and TLR2−/− mice. Unlike splenocytes from injured WT mice, splenocytes from injured TLR2−/− mice showed no decrease in proliferative capacity (Fig. 3A). However, cytokine release from stimulated splenocytes of injured TLR2−/−mice was significantly depressed when compared to that of uninjured TLR2−/− mice, a finding similar to the WT C57BL/6 response (Fig. 3B,C). These findings identify a role for TLR2 in dysfunctional lymphocyte proliferation after peripheral tissue injury, but no role in lymphocyte Th1-cytokine release. Despite the involvement of TLR2 in the systemic inflammatory response to injury, a disconnect is identified here where only a partial role is observed in the late splenocyte dysfunction.
Figure 3. A partial role for TLR2 in depressed lymphocyte responses after injury: splenocyte proliferation but not IFN-g, IL-2 release involves TLR2.

Splenocyte function in WT and TLR2−/− mice at 48h after pseudofracture (PF) or in unmanipulated controls after concanavalin A (2.5μg/ml) stimulation: proliferation measured in counts per minute (cpm) of tritiated thymidine uptake (A), IFN-g release (B) and IL-2 release (C). Data shown as mean±SEM; * = p < 0.05 vs. respective control; n=4–10 mice/group.
TLR9-dependent immune dysfunction following peripheral tissue injury
Although TLR9 was not found to be involved in the early pro-inflammatory response in PF, we sought to determine if it had a role in the altered splenocyte responses following PF. Both injured and control WT (C57BL/6) and TLR9-mutant mice were sacrificed at 48hr after injury. Baseline values of splenocyte proliferation in TLR9-mutant mice were found to be similar to WT mice (Fig. 4). In contrast, the reduction in splenocyte proliferative activity seen in injured WT mice, was not found in TLR9-mutant mice at 48hr following PF. Similarly, the post-traumatic suppression of splenocyte cytokine production seen in WT mice was not observed in injured TLR9-mutant mice.
Figure 4. Depressed lymphocyte responses following trauma are TLR9-dependent.

Splenocyte function of WT, TLR9-mutant and WT mice +CpG oligonucleotides at 48h after pseudofracture (PF) or in unmanipulated controls. Splenocyte responses after antiCD3e (1mg/mL) stimulation: proliferation measured in counts per minute (cpm) of tritiated thymidine uptake (A), IFN-g release (B), IL-2 release (C). WT mice that received control or inhibitory CpG oligodeoxynucleotides (100 μg) were injected subcutaneously both at time of and at 24h after trauma. Data shown as mean±SEM; * = p < 0.05 vs. respective control; n=6–8 mice/group; representative of 3 independent experiments.
To further confirm the role of TLR9 in depressed lymphocyte responses after trauma, we injected WT mice with either control or inhibitory CpG oligonucleotides to selectively block the activation of TLR9. Injured WT mice that received the control CpG showed the expected depression in splenocyte responses at 48hr following trauma. However, the WT mice that received inhibitory CpG-injections did not show the injury-induced depression of splenocyte responses. Collectively, these results show that TLR9 blockade attenuates depressed T lymphocyte responses induced by peripheral tissue trauma.
A role for TLR4 and TLR9 signaling in the upregulation of iNOS within splenic Gr-1hi-CD11bhi MDSC after trauma
An upregulation of iNOS following injury is well-described and we have previously shown the contribution of iNOS to the depressed lymphocyte responses after peripheral tissue injury12. To determine if TLRs contribute to iNOS expression after PF we evaluated the intracellular iNOS expression of the splenic Gr-1hi-CD11bhi MDSC subset in TLR4−/−, TLR2−/−and TLR9-mutant mice. These MDSC are a cell type known to show iNOS upregulation at 12hr following trauma12.
As expected the injured WT mice showed an upregulation of intracellular iNOS by flow cytometry at 12hr following trauma (Fig. 5). In parallel to these findings, splenic Gr-1hi-CD11bhi MDSC from injured TLR2−/− mice showed a similar increase in iNOS expression. In contrast, TLR9-mutant mice did not show any change in iNOS expression in spleen cells at 12hr after injury in comparison with respective uninjured controls. However, splenic Gr-1hi-CD11bhi MDSC subset from TLR4−/− mice actually showed reduced iNOS expression at the 12hr time point following PF in comparison with respective baseline controls. Overall, these data show an association between TLR4 and TLR9 signaling and the upregulation of iNOS in MDSC in the spleen after peripheral tissue trauma.
Figure 5. iNOS induction in splenic Gr-1hi-CD11bhi MDSC at 12h after trauma is TLR4-and TLR9-dependent.
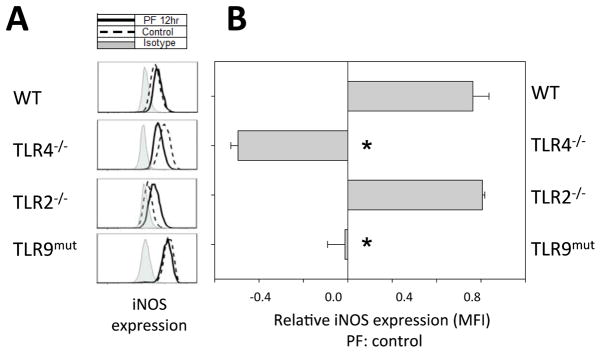
(A) FACS analysis of intracellular iNOS expression in splenic Gr-1hi-CD11bhi MDSC from WT, TLR4−/−, TLR2−/− and TLR9-mutant mice at 12h following pseudofracture (PF) or in unmanipulated controls. Cells were gated through a CD19+CD90.2+ exclusion gate. (B) Relative change in mean fluorescence intensity (MFI) of iNOS expression at 12h after trauma in comparison to respective unmanipulated control. Data shown as mean±SEM; * = p < 0.05 vs. WT; n=2–3 mice/group; representative of 2 independent experiments.
Discussion
Traumatic injury induces an immuno-inflammatory response, the degree of which has been shown to correlate with subsequent morbidity and mortality1. Exaggerated responses are thought to contribute to multiple organ dysfunction and/or a state of immunosuppression5. The early pro-inflammatory component of this response involves excessive activation of innate immune pathways whereas immune dysfunction is described as a depression in adaptive immunity including T-cell dysfunction2,14. Here, we show that TLR2, 4 and 9 play selective roles in both the early pro-inflammatory and sustained immune dysfunction after extremity injury in mice.
Toll-like receptors (TLRs) are important pattern-recognition receptors that serve the function of detecting pathogens and initiating the immune response3. TLRs are expressed on many cell types and have been shown to both activate innate immunity and also modulate adaptive immunity3. In the setting of sterile tissue injury TLRs recognize endogenous ligands termed DAMPS (damage-associated molecular patterns) and initiate a proinflammatory response2,4,5,6 as has been demonstrated after hemorrhagic shock, trauma and ischemia15,16,17. Nevertheless, the involvement of each of the TLRs in injury-induced immunosuppression remains unclear. Our study demonstrates that TLRs play differing and selective roles in both the systemic inflammatory response and in the adaptive immune response to sterile tissue trauma. For example, we have shown that TLR4 serves to mediate systemic inflammation, the suppression of T-cell proliferation, Th1-cytokine release and iNOS upregulation. In contrast, TLR2 has little effect on immune dysfunction despite its role in mediating systemic inflammation. Quite the opposite, TLR9 has almost no effect on early systemic inflammatory responses to peripheral tissue trauma but can act to inhibit lymphocyte proliferation and Th1-cytokine release while having a minimal role in iNOS upregulation.
The role of TLRs in the early proinflammatory response to acute tissue injury has been well-recognized. Experimental models of hemorrhagic shock8,15,18, femur fracture16, tissue injury13 and ischemia17,19,20 have all shown evidence of TLR4 mediated danger signaling. Although the triggers of TLR4 signaling following trauma are continually under investigation, many different ligands including high mobility group box 1 (HMGB1)4 have been identified. Studies using TLR2−/− mice indicate that TLR2 also contributes to trauma-induced responses as seen in pulmonary contusion7 and burns21. Our findings also suggest a role for both TLR4 and TLR2 in the early proinflammatory response in our model of sterile tissue injury.
TLR9 is a receptor that has been shown to be solely responsible for the recognition of CpG DNA motifs, as TLR9-deficient and -mutant mice are unresponsive to CpG DNA22,23. Recent studies from our laboratory and others added TLR9 to the growing list of pattern recognition receptors involved in the hemorrhagic shock-induced pro-inflammatory response9,24. We therefore hypothesized that TLR9 blockade following PF would attenuate systemic hyperinflammation. Thus, unlike TLR4 which appears to contribute to the induction of inflammation both during ischemic insults and to tissue trauma, TLR9 appears to be only involved in the pro-inflammatory response to ischemia and shock. This may be the result of the release of DNA from mitochondria injured or rendered dysfunctional in the setting of ischemia24. It is interesting to find that TLR9 does contribute to the altered splenocyte responses after peripheral tissue injury. Our results do not identify the source of the TLR9 ligands, but the effect of the inhibitory oligonucleotides suggest that an endogenous source of DNA may be involved.
In this study we have identified a unique role for TLR4 and 9 and also a partial role for TLR2 signaling in suppressed splenocyte, specifically T-lymphocyte, responses following trauma. Multiple mechanisms have been implicated in TLR modulation of adaptive immune responses. It is well-established that TLR signaling indirectly modulates T-cell mediated adaptive immunity through antigen presenting cells (APC)25,27,31 of the innate immune system. However TLRs have recently been shown to be functionally expressed on T-cells28,29 resulting in modulation of T-cell activation directly by TLR ligands26,30. After trauma, a depression of the function of cells from both the innate and adaptive immune systems has been shown2. TLR involvement in innate cell dysfunction has been described32,33, 34 and must also be considered due to significant contribution of these cells to the adaptive response. We expect that the mechanisms are specific to each receptor and may involve responses of several cell types.
Regulatory effects of TLRs on T-cell function have also been described through myeloid-derived suppressor cells (MDSC) in the context of murine tumor models35,36,37. We and others have shown MDSC to be implicated in depressed T-cell function after trauma12,38. We have previously established the contribution of iNOS-derived NO to T-cell dysfunction following peripheral tissue injury and identified iNOS-induction within a subpopulation of MDSCs. Interestingly, in this study, analysis of MDSC in this model has shown that TLR4 and TLR9 modulate the production of NO by upregulation of iNOS in MDSC suggesting that one mechanism for TLR4 and 9 mediated depression of splenocyte responses could be through iNOS upregulation in MDSC.
In summary, this study demonstrates the previously unidentified roles of TLR2, 4 and 9 in the T-cell mediated adaptive immune dysfunction following traumatic tissue injury. Taken in the context of previous studies demonstrating a role for TLRs in similar trauma-induced immunosuppression models, a paradigm emerges where several TLRs driven by diverse DAMPs are involved in the host immune imbalance to injury. Table 1 summarizes the relationships between the early pro-inflammatory response induced by peripheral tissue to the sustained depression in splenocyte responses. Whereas the two responses appear to be at least partially linked for TLR2 and 4, there is a clear disconnect for TLR9. Thus, it can be concluded that the early IL-6 elevations are not always proportional to the extent of sustained immune suppression. These results also suggest that targeted therapies could be developed to limit the immune dysfunction by selectively inhibiting receptor function at some interval after injury.
Table 1.
The comparative involvement of TLRs 4, 2 and 9 in the immuno-inflammatory response to peripheral tissue trauma.
| TLR 4 | TLR 2 | TLR 9 | |
|---|---|---|---|
| Early systemic inflammation | +++ | +++ | − |
| Late splenocyte dysfunction | +++ | + | +++ |
Acknowledgments
The authors would like to thank Debra Williams, Richard Shapiro and Lauryn Kohut for their generous technical assistance.
Source of Funding:
This study was performed under the funding provided through National Institute of Health Grant 5P50 GM053789 (TRB).
Footnotes
Conflicts of Interest:
For all authors, no conflicts of interest are declared.
List of meetings at which the paper was presented:
98th Annual American College of Surgeons Clinical Congress Oct 2012, Chicago, IL, USA.
Author Contribution:
Sophie S. Darwiche = literature search, study conception, design and implementation, execution of experiments and data collection, data analysis, data interpretation, writing, critical revision
Xiangcai Ruan = literature search, study design and implementation, execution of experiments and data collection, data analysis, data interpretation, writing, critical revision
Marcus K. Hoffman = literature search, study implementation, data interpretation, critical revision
Kent R. Zettel = literature search, data interpretation, writing, critical revision
Aaron P.Tracy = literature search, execution of experiments and data collection, data analysis
Linda Schroeder = literature search, execution of experiments and data collection
Changchun Cai = literature search, critical revision
Rosemary A. Hoffman = study design, data analysis, data interpretation, critical revision
Melanie J. Scott = data analysis, data interpretation, critical revision
Hans-Christoph Pape = study design, data analysis, data interpretation, critical revision
Timothy R. Billiar = study conception and design, data analysis, data interpretation, writing, critical revision
Contributor Information
Sophie S. Darwiche, Email: darwiches@upmc.edu.
Xiangcai Ruan, Email: xc_ruan@hotmail.com.
Marcus K. Hoffman, Email: hoffmanmk@upmc.edu.
Kent R. Zettel, Email: zettelkr@upmc.edu.
Aaron P. Tracy, Email: aptracy17@gmail.com.
Linda M.I. Schroeder, Email: Linda.Schroeder@rwth-aachen.de.
Changchun Cai, Email: caic@upmc.edu.
Rosemary A. Hoffman, Email: hoffmanr3@upmc.edu.
Melanie J. Scott, Email: scotmx@upmc.edu.
Hans-Christoph Pape, Email: papehc@aol.com.
Timothy R. Billiar, Email: billiartr@upmc.edu.
References
- 1.Xiao W, Mindrinos MN, Seok J, Cuschieri J, Cuenca AG, Gao H, Hayden DL, Hennessy L, Moore EE, Minei JP, et al. Inflammation and Host Response to Injury Large-Scale Collaborative Research Program. A genomic storm in critically injured humans. J Exp Med. 2011;208(13):2581–90. doi: 10.1084/jem.20111354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Flohé SB, Flohé S, Schade FU. Invited review: deterioration of the immune system after trauma: signals and cellular mechanisms. Innate Immun. 2008;14(6):333–44. doi: 10.1177/1753425908100016. [DOI] [PubMed] [Google Scholar]
- 3.Kawai T, Akira S. The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat Immunol. 2010;11(5):373–84. doi: 10.1038/ni.1863. [DOI] [PubMed] [Google Scholar]
- 4.Kaczorowski DJ, Mollen KP, Edmonds R, Billiar TR. Early events in the recognition of danger signals after tissue injury. J Leukoc Biol. 2008;83(3):546–52. doi: 10.1189/jlb.0607374. [DOI] [PubMed] [Google Scholar]
- 5.Stoecklein VM, Osuka A, Lederer JA. Trauma equals danger – damage control by the immune system. J Leukoc Biol. 2012;92(3):539–51. doi: 10.1189/jlb.0212072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Manson J, Thiemermann C, Brohi K. Trauma alarmins as activators of damage-induced inflammation. Br J Surg. 2012;99(S1):12–20. doi: 10.1002/bjs.7717. [DOI] [PubMed] [Google Scholar]
- 7.Hoth JJ, Hudson WP, Brownlee NA, Yoza BK, Hiltbold EM, Meredith JW, McCall CE. Toll-like receptor 2 participates in the response to lung injury in a murine model of pulmonary contusion. Shock. 2007;28(4):447–52. doi: 10.1097/shk.0b013e318048801a. [DOI] [PubMed] [Google Scholar]
- 8.DeMaria EJ, Pellicane JV, Lee RB. Hemorrhagic shock in endotoxin-resistant mice: improved survival unrelated to deficient production of tumor necrosis factor. J Trauma. 1993;35(5):720–4. discussion 724–5. [PubMed] [Google Scholar]
- 9.Gill R, Ruan X, Menzel CL, Namkoong S, Loughran P, Hackam DJ, Billiar TR. Systemic inflammation and liver injury following hemorrhagic shock and peripheral tissue trauma involve functional TLR9 signaling on bone marrow-derived cells and parenchymal cells. Shock. 2011;35(2):164–70. doi: 10.1097/SHK.0b013e3181eddcab. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Menzel CL, Pfeifer R, Darwiche SS, Kobbe P, Gill R, Shapiro RA, Loughran P, Vodovotz Y, Scott MJ, Zenati MS, et al. Models of lower extremity damage in mice: time course of organ damage and immune response. J Surg Res. 2011;166(2):e149–56. doi: 10.1016/j.jss.2010.11.914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Darwiche SS, Kobbe P, Pfeifer R, Kohut L, Pape H, Billiar T. Pseudofracture: An Acute Peripheral Tissue Trauma Model. JoVE. 2011;50 doi: 10.3791/2074. URL: http://www.jove.com/details.php?id=2074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Darwiche SS, Pfeifer R, Menzel C, Ruan X, Hoffman M, Cai C, Chanthaphavong RS, Loughran P, Pitt BR, Hoffman R, et al. Inducible Nitric Oxide Synthase Contributes to Immune Dysfunction following Trauma. Shock. 2012;38(5):499–507. doi: 10.1097/SHK.0b013e31826c5afe. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kobbe P, Kaczorowski DJ, Vodovotz Y, Tzioupis CH, Mollen KP, Billiar TR, Pape HC. Local exposure of bone components to injured soft tissue induces Toll-like receptor 4-dependent systemic inflammation with acute lung injury. Shock. 2008;30(6):686–91. doi: 10.1097/SHK.0b013e31816f257e. [DOI] [PubMed] [Google Scholar]
- 14.Brune IB, Wilke W, Hensler T, Holzmann B, Siewert JR. Downregulation of T helper type 1 immune response and altered pro-inflammatory and anti-inflammatory T cell cytokine balance following conventional but not laparoscopic surgery. Am J Surg. 1999;177(1):55–60. doi: 10.1016/s0002-9610(98)00299-2. [DOI] [PubMed] [Google Scholar]
- 15.Prince JM, Levy RM, Yang R, Mollen KP, Fink MP, Vodovotz Y, Billiar TR. Toll-like receptor-4 signaling mediates hepatic injury and systemic inflammation in hemorrhagic shock. J Am Coll Surg. 2006;202(3):407–17. doi: 10.1016/j.jamcollsurg.2005.11.021. [DOI] [PubMed] [Google Scholar]
- 16.Levy RM, Prince JM, Yang R, Mollen KP, Liao H, Watson GA, Fink MP, Vodovotz Y, Billiar TR. Systemic inflammation and remote organ damage following bilateral femur fracture requires Toll-like receptor 4. Am J Physiol Regul Integr Comp Physiol. 2006;291(4):R970–6. doi: 10.1152/ajpregu.00793.2005. [DOI] [PubMed] [Google Scholar]
- 17.Kaczorowski DJ, Nakao A, Vallabhaneni R, Mollen KP, Sugimoto R, Kohmoto J, Zuckerbraun BS, McCurry KR, Billiar TR. Mechanisms of Toll-like receptor 4 (TLR4)-mediated inflammation after cold ischemia/reperfusion in the heart. Transplantation. 2009;87(10):1455–63. doi: 10.1097/TP.0b013e3181a36e5e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Barsness KA, Arcaroli J, Harken AH, Abraham E, Banerjee A, Reznikov L, McIntyre RC. Hemorrhage-induced acute lung injury is TLR-4 dependent. Am J Physiol Regul Integr Comp Physiol. 2004;287(3):R592–9. doi: 10.1152/ajpregu.00412.2003. [DOI] [PubMed] [Google Scholar]
- 19.Tsung A, Hoffman RA, Izuishi K, Critchlow ND, Nakao A, Chan MH, Lotze MT, Geller DA, Billiar TR. Hepatic ischemia/reperfusion injury involves functional TLR4 signaling in nonparenchymal cells. J Immunol. 2005;175(11):7661–8. doi: 10.4049/jimmunol.175.11.7661. [DOI] [PubMed] [Google Scholar]
- 20.Oyama J, Blais C, Jr, Liu X, Pu M, Kobzik L, Kelly RA, Bourcier T. Reduced myocardial ischemia-reperfusion injury in toll-like receptor 4-deficient mice. Circulation. 2004;109(6):784–9. doi: 10.1161/01.CIR.0000112575.66565.84. [DOI] [PubMed] [Google Scholar]
- 21.Cairns BA, Barnes CM, Mlot S, Meyer AA, Maile R. Toll-like receptor 2 and 4 ligation results in complex altered cytokine profiles early and late after burn injury. J Trauma. 2008;64(4):1069–77. doi: 10.1097/TA.0b013e318166b7d9. [DOI] [PubMed] [Google Scholar]
- 22.Hemmi H, Takeuchi O, Kawai T, Kaisho T, Sato S, Sanjo H, Matsumoto M, Hoshino K, Wagner H, Takeda K, et al. A Toll-like receptor recognizes bacterial DNA. Nature. 2000;408(6813):740–5. doi: 10.1038/35047123. [DOI] [PubMed] [Google Scholar]
- 23.Tabeta K, Georgel P, Janssen E, Du X, Hoebe K, Crozat K, Mudd S, Shamel L, Sovath S, Goode J, et al. Toll-like receptors 9 and 3 as essential components of innate immune defense against mouse cytomegalovirus infection. Proc Natl Acad Sci U S A. 2004;101(10):3516–21. doi: 10.1073/pnas.0400525101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang Q, Raoof M, Chen Y, Sumi Y, Sursal T, Junger W, Brohi K, Itagaki K, Hauser CJ. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature. 2010;464(7285):104–7. doi: 10.1038/nature08780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Abdelsadik A, Trad A. Toll-like receptors on the fork roads between innate and adaptive immunity. Hum Immunol. 2011;72(12):1188–93. doi: 10.1016/j.humimm.2011.08.015. [DOI] [PubMed] [Google Scholar]
- 26.Imanishi T, Hara H, Suzuki S, Suzuki N, Akira S, Saito T. Cutting edge: TLR2 directly triggers Th1 effector functions. J Immunol. 2007;178(11):6715–9. doi: 10.4049/jimmunol.178.11.6715. [DOI] [PubMed] [Google Scholar]
- 27.Medzhitov R. Toll-like receptors and innate immunity. Nat Rev Immunol. 2001;1(2):135–45. doi: 10.1038/35100529. [DOI] [PubMed] [Google Scholar]
- 28.Caramalho I, Lopes-Carvalho T, Ostler D, Zelenay S, Haury M, Demengeot J. Regulatory T cells selectively express toll-like receptors and are activated by lipopolysaccharide. J Exp Med. 2003;197(4):403–11. doi: 10.1084/jem.20021633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gelman AE, Zhang J, Choi Y, Turka LA. Toll-like receptor ligands directly promote activated CD4+ T cell survival. J Immunol. 2004;172(10):6065–73. doi: 10.4049/jimmunol.172.10.6065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Landrigan A, Wong MT, Utz PJ. CpG and non-CpG oligodeoxynucleotides directly costimulate mouse and human CD4+ T cells through a TLR9- and MyD88-independent mechanism. J Immunol. 2011;187(6):3033–43. doi: 10.4049/jimmunol.1003414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kabelitz D. Expression and function of Toll-like receptors in T lymphocytes. Curr Opin Immunol. 2007;19(1):39–45. doi: 10.1016/j.coi.2006.11.007. [DOI] [PubMed] [Google Scholar]
- 32.Kawasaki T, Choudhry MA, Schwacha MG, Bland KI, Chaudry IH. Effect of interleukin-15 on depressed splenic dendritic cell functions following trauma-hemorrhage. Am J Physiol Cell Physiol. 2009;296(1):C124–30. doi: 10.1152/ajpcell.00447.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Versteeg D, Dol E, Hoefer IE, Flier S, Buhre WF, de Kleijn D, van Dongen EP, Pasterkamp G, de Vries JP. Toll-like receptor 2 and 4 response and expression on monocytes decrease rapidly in patients undergoing arterial surgery and are related to preoperative smoking. Shock. 2009;31(1):21–7. doi: 10.1097/SHK.0b013e31817d43bf. [DOI] [PubMed] [Google Scholar]
- 34.Patenaude J, D’Elia M, Hamelin C, Bernier J. Selective effect of burn injury on splenic CD11c(+) dendritic cells and CD8alpha(+)CD4(−)CD11c(+) dendritic cell subsets. Cell Mol Life Sci. 2010;67(8):1315–29. doi: 10.1007/s00018-009-0251-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bunt SK, Clements VK, Hanson EM, Sinha P, Ostrand-Rosenberg S. Inflammation enhances myeloid-derived suppressor cell cross-talk by signaling through Toll-like receptor 4. J Leukoc Biol. 2009;85(6):996–1004. doi: 10.1189/jlb.0708446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zoglmeier C, Bauer H, Nörenberg D, Wedekind G, Bittner P, Sandholzer N, Rapp M, Anz D, Endres S, Bourquin C. CpG blocks immunosuppression by myeloid-derived suppressor cells in tumor-bearing mice. Clin Cancer Res. 2011;17(7):1765–75. doi: 10.1158/1078-0432.CCR-10-2672. [DOI] [PubMed] [Google Scholar]
- 37.Shirota H, Klinman DM. Effect of CpG ODN on monocytic myeloid derived suppressor cells. Oncoimmunology. 2012;1(5):780–782. doi: 10.4161/onci.19731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Makarenkova VP, Bansal V, Matta BM, Perez LA, Ochoa JB. CD11b+/Gr-1+ myeloid suppressor cells cause T cell dysfunction after traumatic stress. J Immunol. 2006;176(4):2085–94. doi: 10.4049/jimmunol.176.4.2085. [DOI] [PubMed] [Google Scholar]