

Abstract
Endogenous heat shock proteins (HSP) are decreased in disease states associated with insulin resistance and aging. Induction of HSPs has been shown to decrease oxidative stress, inhibit inflammatory pathways, and enhance metabolic characteristics in skeletal muscle. As such, HSPs have the potential to function as an important defense system against the development of insulin resistance and type 2 diabetes.
Keywords: insulin signaling, glucose uptake, oxidative stress, stress kinases, mitochondria, aging, obesity
INTRODUCTION
A small study done by Hooper (11) more than 10 yrs ago caught our interest for its simplicity and seeming improbability. Patients with type 2 diabetes were subjected to hot tub “heat therapy” over the course of 3 wks and demonstrated modest improvements in fasting plasma glucose and glycosylated hemoglobin levels. We recently performed a similar study in rodents and found that heat treatment prevented the development of peripheral insulin resistance in response to a high-fat diet (HFD) (8). As we continue to learn more about the heat stress response and the functions of heat shock proteins (HSPs) in insulin-sensitive tissues, the initial findings from Hooper seem less and less incredulous. HSPs serve as one of the body’s major endogenous defense systems against oxidative stress. Currently, there is widespread clinical interest in HSP function in a number of human pathologies, including cancer, neurodegenerative conditions, aging, cardiovascular disease, and more recently, type 2 diabetes. Treatment strategies aimed at enhancing the body’s endogenous defense system of HSPs could have a profound impact on future approaches to disease prevention and treatment.
Given the multitude of cellular responses that occur as a result of heat stress, there are numerous potential mechanisms for the beneficial effects of heat treatment on insulin sensitivity observed in our rodent model. We hypothesize that increased HSP expression results in a reduction of inflammatory pathways known to inhibit insulin signaling. In addition, HSP could mimic the beneficial effects of exercise on skeletal muscle by increasing demand for adenosine triphosphate (ATP). Increased energy demand would serve to maintain mitochondrial function via enhanced flux through the tricarboxylic acid cycle (TCA) cycle and reduce oxidative stress in the presence of excess nutrition. These hypotheses regarding the effects of HSPs on skeletal muscle insulin action and glucose metabolism are examined in the following review. Given the increasing prevalence of type 2 diabetes and obesity today, a combination of lifestyle modification and pharmaceutical intervention will be necessary to prevent and treat type 2 diabetes and its complications. Although insulin sensitizers like metformin and the thiazolidinediones (TZD) have been very effective, the recent cardiovascular complications identified with TZD create increased demand for glucose-lowering drugs that work through a new target pathway. O-(3-piperidino-2-hydroxy-1-propyl) nicotinic amidoxime dihydrochloride (BGP-15), one of a class of hydroximic acid derivatives shown to activate HSP72, has demonstrated an insulin-sensitizing effect in humans in early clinical trials (20).
TYPE 2 DIABETES IS CHARACTERIZED BY DECREASED HSP EXPRESSION
A progressive decline in the body’s ability to detect and respond to stress could result in the development and/or acceleration of a number of disease states associated with oxidative stress. Aging, hyperlipidemia, and type 2 diabetes are associated with a diminished heat stress response, as measured by decreased expression of HSP72. Best known for their chaperone function, HSPs aid in protein refolding, target damaged proteins for degradation, protect proteins against aggregation, and maintain the overall integrity of cellular components (23). HSPs also have less well-characterized roles as antioxidants and in the inhibition of apoptosis and inflammation. In the first study to examine HSPs in diabetes, Kurucz et al. (16) demonstrated that reduced skeletal muscle HSP72 gene expression correlates with the degree of insulin resistance in individuals with type 2 diabetes. These findings were further supported by Bruce et al. (3) and, more recently, Chung and colleagues (4) demonstrated that HSP72 protein expression is decreased in skeletal muscle from obese insulin-resistant humans. Evidence from animal models support these findings in humans and demonstrate decreased HSP72 and HSP25 in insulin-resistant muscle from aged rats (6) and impaired HSP72 synthesis in rats with streptozotocin (STZ)-induced diabetes (1). Atalay and colleagues (1) further showed that although endurance exercise training up-regulated HSP72 expression in STZ rats, the response was significantly blunted compared with nondiabetic rats.
A blunted HSP response and decreased HSP expression in insulin-resistant tissue could be the result of inflammatory inhibition of the primary HSP transcription factor, heat shock factor 1 (HSF-1). HSF-1 has several layers of regulation including negatively regulated feedback control through interaction with HSPs (23) and phosphorylation by protein kinases (Fig. 1). Overactivity of stress kinases capable of phosphorylating HSF-1 on serine residues may repress HSF-1 activation in insulin-resistant tissue. Glycogen synthase kinase 3 (GSK-3), extracellular regulated kinase (ERK), and c-Jun NH2-terminal kinase (JNK), protein kinases closely associated with the development of insulin resistance, are known to negatively regulate HSF-1 by phosphorylation on serine residues 303, 307, and 363, respectively (13). Constitutive phosphorylation of HSF-1 on serine residues holds HSF-1 in an inactive state under normal physiological growth conditions. The release of HSF-1 serine phosphorylation causes HSF-1 to translocate to the nucleus, trimerize, and bind to heat shock elements (HSE) necessary for new HSP synthesis. Regulation of HSF-1 and inhibition of the heat stress response in a rodent model of insulin resistance has yet to be examined.
Figure 1.
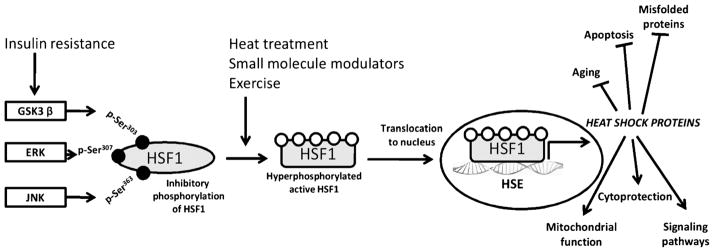
Regulation of the HSP defense mechanism. Stress (physical/chemical), change in physiological or pathophysiological states, or small-molecule activators such as bimoclomol/BGP-15 cause activation of the master regulator of the heat stress response, HSF-1. HSF-1 is a transcription factor maintained in the cytoplasm under tight negative regulation by phosphorylation. Phosphorylation by stress kinases GSK-3β, ERK, and JNK occurs on serine residues (Ser) 303, 307, and 363, respectively. The heat stress response is diminished in insulin-resistant tissue, where the activity of these kinases is increased, causing HSF-1 to remain inactive in the cytoplasm. Activation of HSF-1 results in nuclear translocation, binding to HSE, and transcription of new HSPs. HSPs function as mediators of the HSF-HSP defense system by reducing aging processes and apoptosis, enhancing signaling pathways and cytoprotection, and preventing misfolding of proteins. Stress kinases present a therapeutic target to improve the HSP defense response. HSF-1, heat shock factor 1; GSK-3β, glycogen synthase kinase 3; ERK, extracellular regulated kinase; JNK, c-Jun NH2-terminal kinase; HSE, heat shock elements; HSP, heat shock proteins.
THE INSULIN-SENSITIZING EFFECTS OF HEAT TREATMENT
Although the ability of heat therapy to modestly lower blood glucose levels in humans was demonstrated more than 10 yr ago, the first evidence to show that heat treatment could prevent insulin resistance in a rodent model in vivo was demonstrated only recently by Chung and colleagues (4). In response to heat treatment (core body temperature increased to 41.5°C for 15 min), a transient increase in HSP72 expression was observed in skeletal muscle, liver, and adipose tissue that decreased by 24 h. This heat stress response was blunted in rodents placed on an high fat diet (HFD). Despite a blunted acute heat stress response in mice fed an HFD, weekly heat treatment attenuated the detrimental effects of an HFD in mice (4) and rats (8). In a study from our laboratory, male Wistar rats were fed an obesity-inducing HFD for 12 wk to elicit peripheral insulin resistance and whole body glucose intolerance (8). At the same time, a subgroup of rats was subjected to lower body immersion in hot water (41°C) for 20 min once a week for the course of the diet. Heat treatment completely prevented the development of glucose intolerance, fat deposition, and peripheral insulin resistance in the rats fed an HFD (Fig. 2). Our results demonstrate the ability of heat treatment to effectively lower fasting insulin levels and improve glucose clearance in response to a glucose tolerance test. Epididymal fat pad weight increased in rats fed an HFD, and this effect was blunted in rats subjected to weekly heat treatment. Body weight and food intake did not differ between HFD sham and heat-treated rats, suggesting increased metabolism in rats receiving heat treatment. Data from other laboratories in vivo (14) and in L6 muscle cells in our laboratory (8) show that heat treatment improves fatty acid utilization. In this manner, heat treatment could simulate the effects of exercise on whole-body metabolism by increasing energy demand and decreasing the accumulation of free fatty acids and adipose tissue in the presence of an HFD.
Figure 2.

Weekly heat treatment improves characteristics of high fat (HF) diet-induced insulin resistance. Male Wistar rats were given a chow or HF diet for 12 weeks. The HF rats received either a lower body heat treatment ((HT) HF + HT, 41°C for 20 min) or sham treatment ((ST) HF + ST, 36°C for 20 min) once per week during the diet regimen. Intraperitoneal glucose tolerance test (IPGTT): Overnight fasted rats were injected with a glucose load of 2 g kg−1 body weight, intraperitoneally. Blood glucose was measured at time 0, 15, 30, 60, 90, and 120 min after injection using a glucometer. IPGTT is expressed as glucose area under the curve (AUC) in panel A. Epididymal fat pad weights (B) were measured (***P < 0.001, **P < 0.01 HF + ST vs chow and ##P < 0.01, #P < 0.05 HF + HT vs HF + ST). Values are means ± standard error (SE) for 5–7 rats per group. Insulin-stimulated glucose transport: Soleus (C) and extensor digitorum longus (EDL) (D) from chow, HF + ST and HF + HT rats were incubated in the presence or absence of 1 mU·mL−1 insulin with 2[1,2-3H]-deoxyglucose and [14C]-mannitol, and 2-deoxyglucose uptake into the muscles was determined. Values are means ± SE for 3–5 muscles for the HF and HF + HT groups, 11–12 muscles per group for chow. Open bars represent basal (noninsulin-stimulated) measurements, and filled bars represent insulin-stimulated measurements (*P < 0.05 basal vs insulin treated, #P < 0.05). (Reprinted from Gupte AA, Bomhoff GL, Swerdlow RH, Geiger PC. Heat treatment improves glucose tolerance and prevents skeletal muscle insulin resistance in rats fed an HFD. Diabetes. 2009; 58:567–78. Copyright © 2009 American Diabetes Association. Used with permission.)
Skeletal muscle is responsible for the majority (75%) of insulin-mediated glucose disposal in the body, making this tissue vital in maintaining whole-body insulin action and glucose homeostasis. Our recent findings demonstrate that heat treatment can effectively increase insulin-stimulated glucose uptake and insulin signaling intermediates in skeletal muscles from HFD-fed rats (8). Furthermore, a single acute bout of heat treatment can increase insulin-stimulated glucose uptake in aged skeletal muscle (Geiger P, unpublished observations, 2010), much like a single bout of exercise training. Despite these findings, very little is known about the actions of HSP in skeletal muscle. HSP also were increased in the liver and adipose tissue as a result of lower body heat treatment, and metabolic changes in these tissues likely contributed to some of the improvements in whole-body glucose metabolism observed with heat treatment. The strong correlation between HSPs expression in skeletal muscle and whole-body glucose tolerance (4,14) has directed our initial focus on the role of HSPs in skeletal muscle insulin action. Given the importance of skeletal muscle in mediating whole-body glucose uptake, this may be the ideal target tissue for the protective effects of HSPs.
ANTIINFLAMMATORY ACTION OF HSP IN SKELETAL MUSCLE
Chronic hyperglycemia is a well-established hallmark of diabetes and is strongly linked to enhanced reactive oxygen species (ROS) formation, advanced glycation end products and lipid peroxidation. Considerable support also exists for a role of ROS in modulating the aging process (2,30). Both aging and diabetes are associated with a decrease in plasma antioxidant levels (30). Collectively, the imbalance of increased intracellular ROS and decreased antioxidant defense mechanisms has been strongly implicated in the development of insulin resistance. New findings indicate that oxidative stress activates multiple inflammatory signaling pathways capable of increasing serine phosphorylation of insulin receptor substrate 1 (IRS-1) and inducing insulin resistance. These include JNK, p38 mitogen-activated protein (MAP) kinase, inhibitor of nuclear factor κB (IKK/NF-κB), and GSK-3 (17). Of these, JNK and IKKβ are strongly implicated in the development of obesity and genetic models of insulin resistance. JNK and IKKβ also have been associated with human pathology, and patients with diabetes have been shown to have elevated DNA binding of NF-κB, a reflection of increased IKKβ activity. The strong evidence linking these inflammatory pathways with the development of insulin resistance has led to ongoing clinical trials using JNK and IKKβ inhibitors. Prevention of insulin resistance with heat treatment was associated with activation of HSP72 and HSP25 and a decrease in the stress kinases JNK and IKKβ in skeletal muscle. Evidence in the literature suggests a direct role for inhibition of JNK and IKKβ by HSP72 and HSP25, respectively.
HSPs are known to interact with and regulate many transcription factors, signaling molecules, and kinases, including IKKβ, Raf1, Akt, and JNK. Many of the HSPs exhibit anti-apoptotic functions, and previous studies indicate that the antiapoptotic actions of HSP72 specifically target the JNK signaling pathway (22). Evidence for HSP72 inhibition of JNK is based on studies in cell culture. Pretreatment of NIH 3T3 cells (fibroblasts) with mild heat shock suppressed UV-stimulated JNK activity, whereas the use of HSP72 antisense oligonucleotides in these cells blocked HSP72 induction and abolished the suppressive effect of heat shock on JNK activation (24). In response to an HFD, JNK phosphorylation was increased in both slow-twitch soleus and fast-twitch extensor digitorum longus (EDL) skeletal muscle, with the greatest increase seen in the EDL ((8) Fig. 3). Consistent with results from Chung et al. (4), prevention of insulin resistance via increased HSP72 expression with heat treatment was closely associated with a decrease in JNK activation. HSP72 induction independent of heat treatment also results in decreased JNK activation. Previous studies in our laboratory examined the ability of the antioxidant lipoic acid to increase HSPs and prevent diet-induced insulin resistance in skeletal muscle (7). Lipoic acid has demonstrated beneficial effects in several disease states characterized by increased oxidative stress, including insulin resistance, but it had not been previously identified as an inducer of the HSP response. Our findings revealed that daily lipoic acid treatment increased skeletal muscle HSP72 expression and decreased JNK activation to prevent diet-induced insulin resistance. Further evidence of HSP72-mediated JNK inhibition was observed by Chung et al. (4) using HSP72 transgenic mice. In response to an HFD, mice overexpressing HSP72 did not show an increase in JNK activation in skeletal muscle.
Figure 3.

Heat treatment induces heat shock protein (HSP)72 expression and a concomitant c-Jun NH2-terminal kinase (JNK) inactivation. Soleus and extensor digitorum longus (EDL) muscles were dissected from chow, high fat (HF) + sham treatment (ST), and HF + heat treatment (HT) rats and snap frozen in liquid nitrogen. Lysates were subjected to Western blot analysis for HSP72/tubulin in soleus (A) and EDL (B) and for phosphorylation of JNK, (p)-JNK/JNK in soleus (C) and EDL (D) (*P < 0.05 HF + ST versus chow, ###P < 0.001, #P < 0.05 HF + HT vs HF + ST). Values are means ± standard error (SE) for 5–7 muscles per group. (Reprinted from Gupte AA, Bomhoff GL, Swerdlow RH, Geiger PC. Heat treatment improves glucose tolerance and prevents skeletal muscle insulin resistance in rats fed an HFD. Diabetes. 2009; 58:567–78. Copyright © 2009 American Diabetes Association. Used with permission.)
There are several possible mechanisms that could account for the HSP72 inhibitory effect on JNK activation. Direct binding of HSP72 to JNK could prevent activation of JNK by upstream kinases SAPK/Erk kinase 1 (SEK1) and MAP kinase kinase 7 (MKK7) (24). Direct protein interaction also could inhibit the enzymatic activity of JNK or inhibit the interaction between JNK and its substrate, c-Jun. We have observed HSP72-JNK interaction in immunoprecipitation studies (Geiger, P., unpublished observations, 2010) in agreement with results shown in cell culture (24). Although these data suggest an interaction between HSP72 and JNK, a mechanism for JNK inhibition cannot be inferred from these results. Additional inhibitory mechanisms include the ability of HSP72 to act on upstream phosphatases of JNK like MAP kinase phosphatase-1 (MKP-1) and MKP-3 or on another kinase upstream of JNK, dual leucine zipper-bearing kinase (DLK) (5,22). A recent study demonstrated HSP72 interaction with the carboxyl terminus of HSP70-interacting protein (CHIP), a ubiquitin ligase known to negatively regulate DLK expression and activity (5).
Heat treatment and lipoic acid effectively increased phosphorylation of HSP25 and decreased IKKβ in skeletal muscle from HFD-fed rats (7,8). Although less well characterized than the HSP72-JNK interaction, several studies have demonstrated an ability of HSP25 to inhibit IKKβ, either by direct binding or through an, as yet, uncharacterized signaling intermediate (25). Our results indicate that IKKβ was increased with an HFD in both soleus and EDL muscles in rats (8), whereas Chung et al. (4) observed an increase in liver p-IKKβ in mice fed an HFD with no increase in skeletal muscle. Different measures of IKKβ in the two studies, as well as possible species and muscle fiber-type differences could readily account for this difference. However, even in our own muscle fiber-type comparison, the ability of heat treatment to decrease IKKβ varied. Phosphorylation of HSP25, a measure of its activation, was increased with heat treatment in the EDL muscle, and a corresponding decrease in IKKβ was observed. In contrast, although IKKβ activation was increased in response to an HFD, neither HSP25 phosphorylation nor IKKβ activation was altered with heat treatment in soleus muscle. These findings suggest that IKKβ may not play a significant role in soleus muscle insulin resistance because insulin signaling and glucose uptake were restored with heat treatment despite the lack of effect on IKKβ. Constitutive levels of HSPs differ in fast-twitch and slow-twitch muscle, and HSP induction and function may depend on the oxidative capacity of the muscle fiber type.
HSP EXPRESSION AND SKELETAL MUSCLE OXIDATIVE CAPACITY
Results from our laboratory (6,8) and others (21) demonstrate that HSP expression correlates with muscle oxidative capacity. HSP expression levels are considerably higher in the more oxidative soleus muscle compared with the fast-twitch EDL or epitrochlearis (EPI) muscles, for example (Fig. 4). Our results also demonstrate a consistent finding of increased stress kinase activation in fast-twitch compared with slow-twitch muscles (6,8). We hypothesize that the combination of higher stress kinase activation and lower HSP expression, as occurs with age or obesity, could result in increased susceptibility to conditions of insulin resistance in skeletal muscle. This agrees with previous studies demonstrating that skeletal muscle HSP72 expression correlates with the degree of insulin resistance (3,16).
Figure 4.
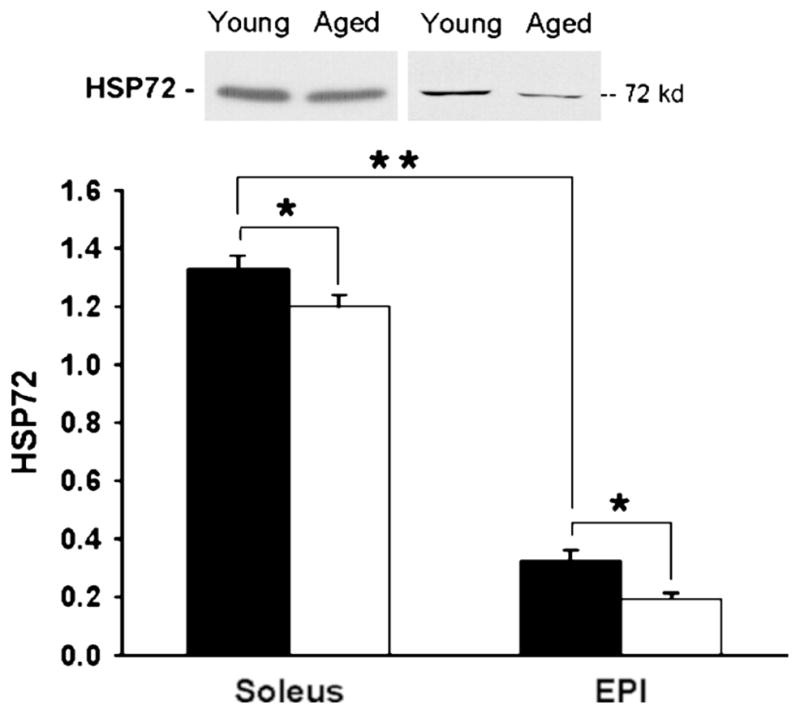
Heat shock protein (HSP) expression in young and aged muscles. Soleus and Epi muscles from young and aged rats were dissected, clamp frozen, and homogenized. Lysates were analyzed by Western blot for HSP72. Filled bars represent young muscle values, and open bars represent aged muscles values. Values are means ± standard error (SE) for 8–15 muscles per group. *P < 0.05, **P < 0.001. (Reprinted from Gupte AA, Bomhoff GL, Geiger PC. Age-related differences in skeletal muscle insulin signaling: the role of stress kinases and HSP. J. Appl. Physiol. 2008; 105:839–48. Copyright © 2008 The American Physiological Society. Used with permission.)
HSP72 mRNA expression also shows a positive correlation with mitochondrial enzyme activity in human skeletal muscle (3). HSPs facilitate import of nuclear encoded proteins into the mitochondria, and are thus crucial in maintaining the structural and functional integrity of mitochondria (35). This becomes especially relevant when mitochondria are stressed with nutrient overload and oxidative stress as occurs with obesity and aging. In cardiac cells, heat stress has been shown to enhance activities of the mitochondrial electron transport complexes, affording prolonged preservation of cardiac cells for transplantation (27). In skeletal muscle, we demonstrated that heat treatment increased mitochondrial enzyme activity in the presence of an HFD (8). As indicated, fast-twitch EDL muscles express low levels of HSPs compared with slow-twitch soleus muscles. However, heat treatment effectively increased HSPs in the EDL to levels similar to that of the more oxidative soleus muscles. Correlative to the robust HSP protein induction, mitochondrial enzyme activities and insulin sensitivity also were enhanced in the EDL muscles as a result of heat treatment compared with muscles from rats given an HFD and no heat treatment. Thus, improved mitochondrial integrity, enhanced mitochondrial energetic flux, and increased oxidative capacity resulting in enhanced mitochondrial enzyme activity may be an additional means by which increased HSP expression protects from skeletal muscle insulin resistance.
It is interesting to speculate that exercise- and heat treatment-induced HSPs protect mitochondrial function and insulin action in the skeletal muscle through similar mechanisms. Although the role of mitochondrial dysfunction in the development of insulin resistance is disputed (10), it is well known that increased oxidative stress caused by dys-regulation of mitochondrial pathways is characteristic of insulin-resistant and diabetic muscle. Increased beta oxidation in response to excess fatty acid supply with an HFD, without a concomitant increase in downstream mitochondrial pathways and ATP demand, can result in a buildup of beta oxidation intermediates that increase proton leak and oxidative stress in the mitochondria (15). This oxidative stress can in turn elevate stress kinase activation, further increasing the likelihood of insulin resistance. One way in which exercise training is thought to alleviate insulin resistance is through coordinated activation of mitochondrial pathways, such that the downstream electron transport chain (ETC) and TCA cycle intermediates are increased to a similar extent with beta oxidation. In this manner, beta oxidation and downstream TCA cycle metabolism are matched. Heat treatment, like exercise, could increase the demand of muscle cells for energy such that fatty acid uptake and utilization are matched, reducing the accumulation of free fatty acids and adipose tissue in the presence of an HFD (Fig. 5). This could account for the decreased epididymal fat pad weight in HFD-fed rats given heat treatment, despite no difference in body weight or food intake in these rats compared with sham-treated HFD-fed rats with much higher fat mass. Increased citrate synthase (CS) and cytochrome oxidase (COX) activity with heat treatment in EDL muscle from HFD-fed rats suggests increased activity of downstream mitochondrial pathways. In addition, our data from L6 muscle cells suggest that heat treatment can increase fatty acid oxidation and mitochondrial oxygen consumption (8). Future studies are needed to assess the ability of chronic heat treatment to alter skeletal muscle fatty acid metabolism in conjunction with an HFD.
Figure 5.

Exercise-like effects of heat treatment improve mitochondrial function in skeletal muscle. Fatty acid oxidation (FAO) is a rich source of mitochondria-derived adenosine triphosphate (ATP) in healthy skeletal muscle. However, conditions of nutrient excess in obesity increase beta oxidation out of proportion with the demand for ATP in the muscle. As a result, oxidation products of lipids do not enter the tricarboxylic acid (TCA) cycle and electron transport chain (ETC) because of limited cycling of intermediates. This in turn causes mitochondrial accumulation of incomplete oxidation products, increased reactive oxygen species (ROS), and mitochondrial stress. Our data suggest that heat treatment increases FAO and also activities of downstream mitochondrial pathways as indicated by citrate synthase (CS) and cytochrome oxidase (COX) activities and enhanced oxygen (O2) consumption. Uncoupling protein 3 (UCP3), a protein involved with ROS dissipation, also is increased as a result of heat treatment. Heat treatment mimics exercise by creating a demand for ATP that increases the flux through mitochondrial processes and balances increased FAO in the presence of excess lipid supply.
THERAPEUTIC POTENTIAL OF MODULATING THE HSP RESPONSE
Our findings suggest that greater constitutive HSP expression, and correspondingly lower stress kinase activation, at least partially protects slow-twitch muscle from age-related and obesity-induced insulin resistance. As a result, we hypothesize that maintenance or enhanced expression of HSPs could be a powerful tool in the prevention of insulin resistance and diabetes. The HSP response is a robust phenomenon induced by many stimuli, making it in some ways an ideal therapeutic target pathway. Hydroxylamine derivatives including bimoclomol and arimoclomol have demonstrated effectiveness against diabetic complications and in delaying neuro degeneration in rats (32). BPG-15, a hydroxylamine derivative, has been shown to have insulin-sensitizing effects in ob/ob mice and Goto-Kakizaki rats (4). Treatment of ob/ob mice with BGP-15 for 15 days resulted in increased HSP72 expression and decreased JNK phosphorylation in skeletal muscle, as well as increased insulin sensitivity as measured by hyperinsulinemic euglycemic clamp (4). These findings demonstrate that pharmacological induction of HSP72 can improve insulin action in a genetic model of obesity in vivo. BGP-15 is not a member of the glitazone family, and its glucose lowering action is thought to be as a chaperone inducer or coinducer of the HSP response, as well as by induction of nitric oxide synthases. As a coinducer, the drug augments the naturally occurring heat stress response, rather than directly inducing HSPs (32). Coinducers essentially act like “smart drugs” by selectively interacting only with cells subjected to stress, and these multitarget low-affinity drugs may be more efficient than single-target high-affinity drugs created through targeted drug design (31). The efficacy and safety of BGP-15 were recently tested in insulin-resistant patients in a 28-day dose-ranging study (20). Treatment with BGP-15 had no adverse affects and significantly improved insulin sensitivity in insulin-resistant nondiabetic patients compared with placebo. Further clinical studies with BGP-15 and other co-inducers of HSPs are warranted based on these findings.
Other pharmacological means of increasing HSP expression exist, including the previously mentioned antioxidant lipoic acid. Lipoic acid does not modulate insulin action in lean or chow-fed rats (7), which is indicative of an HSP response designed to mitigate adverse effects of an HFD rather than directly stimulate insulin action. Lipoic acid may directly activate HSF-1, although an exact mechanism has not been established. Resveratrol and curcumin are some examples of phytochemicals shown to improve glucose metabolism and increase lifespan (18,33). Interestingly, both of these chemicals also induce HSP72 expression (12,26). It has been proposed that phytochemicals mediate their action by stimulating production of antioxidant enzymes and protein chaperones as a protective mechanism against additional stress. These compounds demonstrate hormetic mechanisms of action, with beneficial effects at low doses that would have detrimental effects at higher doses. Some evidence exists to suggest that resveretrol inhibits the stress kinases JNK and GSK-3 that repress HSF-1 activation (34). Interestingly, resveretrol acts to lower the temperature threshold of the heat stress response. This function of resveretrol would seem to pair well with heat therapy or exercise training as a combined therapeutic approach in humans.
Perhaps of greater interest, one of the best inducers of the HSP response is not pharmacological. Exercise elicits a number of metabolic adaptations and is a powerful tool in the prevention and treatment of type 2 diabetes. Exercise training also is a stimulus for increased HSP expression, although the underlying mechanism(s) for exercise-induced HSP expression are currently unknown. Exercise-associated hyperthermia is commonly suggested as the stimulus responsible for inducing an increase in HSP after exercise (19). However, several studies suggest that elevations in muscle and core temperature may not be the sole factors responsible for exercise-induced HSP expression. For example, HSP72 content was increased in rat soleus and gastrocnemius muscles after treadmill running, independent of an increase in core temperature (29), and soleus muscle HSP72 production after exercise was enhanced when exercise was performed under elevated ambient temperature. If the HSP response to exercise occurs through a pathway independent of heat stress, then the combination of heat treatment and exercise, or pharmacological induction of HSPs in combination with exercise, may have an additive effect on HSP induction and dramatically improve insulin action in skeletal muscle. One possible mechanism for the exercise-mediated HSP response is via inhibition of GSK-3 and subsequent activation of HSF-1. Previous studies suggest that activation of HSP72 in the heart occurs through phosphatidylinositol 3-kinase–mediated activation of Akt and subsequent inhibition of GSK-3 (28). Given the variable expression and response of HSPs in insulin-sensitive tissue, distinct regulatory pathways for the HSP response are likely.
CONCLUSIONS
There is still much we do not know about HSP function in skeletal muscle and other insulin-sensitive tissues. Future studies are needed to determine the complex interactions between HSPs, stress kinases, and insulin-signaling intermediates (Fig. 6). Considerable evidence exists to suggest that modulating the HSP response in skeletal muscle could be a powerful tool in the prevention and treatment of insulin resistance. Lifelong overexpression of HSP72 in mice resulted in decreased age-related oxidative stress and protection from muscle damage (2). Similar longevity studies on the effects of HSP on insulin action are needed. Animal studies have so far demonstrated that up-regulation of HSP72, regardless of the stimulus, results in protection from diet-induced and genetic models of insulin resistance. Promising new clinical trials demonstrate the safety and efficacy of HSP-inducing compounds to improve insulin sensitivity in humans. Future strategies aimed at enhancing the body’s endogenous defense system of HSPs could have a profound impact on disease prevention and treatment.
Figure 6.

A. Inhibitory stress kinase regulation of insulin signaling and heat shock protein (HSP) expression. Oxidative stress in muscle activates stress kinases c-Jun NH2-terminal kinase (JNK) and inhibitor of nuclear factor (IKKβ). These stress kinases serine phosphorylate the insulin receptor substrate-1 (IRS-1) and decrease insulin signaling by blocking IRS-1 tyrosine phosphorylation. Decreased signaling through Akt leads to increased activation of glycogen synthase kinase 3 (GSK-3), which can feedback and further increase IRS-1 serine phosphorylation. GSK-3 and JNK also can serine phosphorylate the HSP transcription factor heat shock factor 1 (HSF-1), on serine residues (Ser) 303 and 363, respectively. Chronic serine phosphorylation of HSF-1 by GSK-3 and JNK can inhibit HSF-1 and the HSP response. A decreased HSP response could result in the development and/or acceleration of a number of oxidative stress-related disease states, including insulin resistance. Open shapes represent inactive proteins, dark filled shapes represent active proteins. B. Modulating HSPs to inhibit stress kinases and improve insulin signaling. Heat stress, exercise, and pharmacological interventions increase HSPs expression in skeletal muscle. Exercise could stimulate HSP expression through a pathway independent of heat stress, whereas GSK-3–dependent activation of HSF-1 may be a common pathway of HSP activation. These pathways indicate the complex interregulatory relationship of stress kinases, insulin signaling, and HSP in skeletal muscle. Open shapes represent inactive proteins, dark filled shapes represent active proteins. ROS, reactive oxygen species.
Acknowledgments
This work was supported by grants P20 RR016475 from the National Center for Research Resources (NCRR) and AG031575 from the National Institutes of Health.
References
- 1.Atalay M, Oksala NK, Laaksonen DE, et al. Exercise training modulates heat shock protein response in diabetic rats. J Appl Physiol. 2004;97:605–11. doi: 10.1152/japplphysiol.01183.2003. [DOI] [PubMed] [Google Scholar]
- 2.Broome CS, Kayani AC, Palomero J, et al. Effect of lifelong over-expression of HSP70 in skeletal muscle on age-related oxidative stress and adaptation after nondamaging contractile activity. FASEB J. 2006;20:1549–51. doi: 10.1096/fj.05-4935fje. [DOI] [PubMed] [Google Scholar]
- 3.Bruce CR, Carey AL, Hawley JA, Febbraio MA. Intramuscular heat shock protein 72 and heme oxygenase-1 mRNA are reduced in patients with type 2 diabetes: evidence that insulin resistance is associated with a disturbed antioxidant defense mechanism. Diabetes. 2003;52:2338–45. doi: 10.2337/diabetes.52.9.2338. [DOI] [PubMed] [Google Scholar]
- 4.Chung J, Nguyen AK, Henstridge DC, et al. HSP72 protects against obesity-induced insulin resistance. Proc Natl Acad Sci U S A. 2008;105:1739–44. doi: 10.1073/pnas.0705799105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Daviau A, Proulx R, Robitaille K, et al. Down-regulation of the mixed-lineage dual leucine zipper-bearing kinase by heat shock protein 70 and its co-chaperone CHIP. J Biol Chem. 2006;281:31467–77. doi: 10.1074/jbc.M607612200. [DOI] [PubMed] [Google Scholar]
- 6.Gupte AA, Bomhoff GL, Geiger PC. Age-related differences in skeletal muscle insulin signaling: the role of stress kinases and heat shock proteins. J Appl Physiol. 2008;105:839–48. doi: 10.1152/japplphysiol.00148.2008. [DOI] [PubMed] [Google Scholar]
- 7.Gupte AA, Bomhoff GL, Morris JK, Gorres BK, Geiger PC. Lipoic acid increases heat shock protein expression and inhibits stress kinase activation to improve insulin signaling in skeletal muscle from high-fat-fed rats. J Appl Physiol. 2009;106:1425–34. doi: 10.1152/japplphysiol.91210.2008. [DOI] [PubMed] [Google Scholar]
- 8.Gupte AA, Bomhoff GL, Swerdlow RH, Geiger PC. Heat treatment improves glucose tolerance and prevents skeletal muscle insulin resistance in rats fed a high-fat diet. Diabetes. 2009;58:567–78. doi: 10.2337/db08-1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Reference deleted in proofs.
- 10.Hancock CR, Han DH, Chen M, et al. High-fat diets cause insulin resistance despite an increase in muscle mitochondria. Proc Natl Acad Sci U S A. 2008;105:7815–20. doi: 10.1073/pnas.0802057105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hooper PL. Hot-tub therapy for type 2 diabetes mellitus. N Engl J Med. 1999;341:924–5. doi: 10.1056/NEJM199909163411216. [DOI] [PubMed] [Google Scholar]
- 12.Kanitkar M, Bhonde RR. Curcumin treatment enhances islet recovery by induction of heat shock response proteins, Hsp70 and heme oxygenase-1, during cryopreservation. Life Sci. 2008;82:182–9. doi: 10.1016/j.lfs.2007.10.026. [DOI] [PubMed] [Google Scholar]
- 13.Kline MP, Morimoto RI. Repression of the heat shock factor 1 transcriptional activation domain is modulated by constitutive phosphorylation. Mol Cell Biol. 1997;17:2107–15. doi: 10.1128/mcb.17.4.2107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kokura S, Adachi S, Manabe E, et al. Whole body hyperthermia improves obesity-induced insulin resistance in diabetic mice. Int J Hyperthermia. 2007;23:259–65. doi: 10.1080/02656730601176824. [DOI] [PubMed] [Google Scholar]
- 15.Koves TR, Ussher JR, Noland RC, et al. Mitochondrial overload and incomplete fatty acid oxidation contribute to skeletal muscle insulin resistance. Cell Metab. 2008;7:45–56. doi: 10.1016/j.cmet.2007.10.013. [DOI] [PubMed] [Google Scholar]
- 16.Kurucz I, Morva A, Vaag A, et al. Decreased expression of heat shock protein 72 in skeletal muscle of patients with type 2 diabetes correlates with insulin resistance. Diabetes. 2002;51:1102–9. doi: 10.2337/diabetes.51.4.1102. [DOI] [PubMed] [Google Scholar]
- 17.Kyriakis JM, Avruch J. Sounding the alarm: protein kinase cascades activated by stress and inflammation. J Biol Chem. 1996;271:24313–6. doi: 10.1074/jbc.271.40.24313. [DOI] [PubMed] [Google Scholar]
- 18.Lagouge M, Argmann C, Gerhart-Hines Z, et al. Resveratrol improves mitochondrial function and protects against metabolic disease by activating SIRT1 and PGC-1alpha. Cell. 2006;127:1109–22. doi: 10.1016/j.cell.2006.11.013. [DOI] [PubMed] [Google Scholar]
- 19.Lancaster GI, Febbraio MA. Mechanisms of stress-induced cellular HSP72 release: implications for exercise-induced increases in extracellular HSP72. Exerc Immunol Rev. 2005;11:46–52. [PubMed] [Google Scholar]
- 20.Literati-Nagy B, Kulcsar E, Literati-Nagy Z, et al. Improvement of insulin sensitivity by a novel drug, BGP-15, in insulin-resistant patients: a proof of concept randomized double-blind clinical trial. Horm Metab Res. 2009;41:374–80. doi: 10.1055/s-0028-1128142. [DOI] [PubMed] [Google Scholar]
- 21.Locke M, Noble EG, Atkinson BG. Inducible isoform of HSP70 is constitutively expressed in a muscle fiber type specific pattern. Am J Physiol. 1991;261:C774–9. doi: 10.1152/ajpcell.1991.261.5.C774. [DOI] [PubMed] [Google Scholar]
- 22.Meriin AB, Yaglom JA, Gabai VL, et al. Protein-damaging stresses activate c-Jun N-terminal kinase via inhibition of its dephosphorylation: a novel pathway controlled by HSP72. Mol Cell Biol. 1999;19:2547–55. doi: 10.1128/mcb.19.4.2547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Morimoto RI. Regulation of the heat shock transcriptional response: cross talk between a family of heat shock factors, molecular chaperones, and negative regulators. Genes Dev. 1998;12:3788–96. doi: 10.1101/gad.12.24.3788. [DOI] [PubMed] [Google Scholar]
- 24.Park HS, Lee JS, Huh SH, Seo JS, Choi EJ. Hsp72 functions as a natural inhibitory protein of c-Jun N-terminal kinase. EMBO J. 2001;20:446–56. doi: 10.1093/emboj/20.3.446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Park KJ, Gaynor RB, Kwak YT. Heat shock protein 27 association with the I kappa B kinase complex regulates tumor necrosis factor alpha-induced NF-kappa B activation. J Biol Chem. 2003;278:35272–8. doi: 10.1074/jbc.M305095200. [DOI] [PubMed] [Google Scholar]
- 26.Putics A, Vegh EM, Csermely P, Soti C. Resveratrol induces the heat-shock response and protects human cells from severe heat stress. Antioxid Redox Signal. 2008;10:65–75. doi: 10.1089/ars.2007.1866. [DOI] [PubMed] [Google Scholar]
- 27.Sammut IA, Jayakumar J, Latif N, et al. Heat stress contributes to the enhancement of cardiac mitochondrial complex activity. Am J Pathol. 2001;158:1821–31. doi: 10.1016/S0002-9440(10)64138-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shinohara T, Takahashi N, Ooie T, et al. Phosphatidylinositol 3-kinase–dependent activation of akt, an essential signal for hyperthermia-induced heat-shock protein 72, is attenuated in streptozotocin-induced diabetic heart. Diabetes. 2006;55:1307–15. doi: 10.2337/db05-0266. [DOI] [PubMed] [Google Scholar]
- 29.Skidmore R, Gutierrez JA, Guerriero V, Jr, Kregel KC. HSP70 induction during exercise and heat stress in rats: role of internal temperature. Am J Physiol. 1995;268:R92–7. doi: 10.1152/ajpregu.1995.268.1.R92. [DOI] [PubMed] [Google Scholar]
- 30.Sohal RS, Orr WC. Relationship between antioxidants, pro-oxidants, and the aging process. Ann N Y Acad Sci. 1992;663:74–84. doi: 10.1111/j.1749-6632.1992.tb38651.x. [DOI] [PubMed] [Google Scholar]
- 31.Soti C, Csermely P. Pharmacological modulation of the heat shock response. Handb Exp Pharmacol. 2006:417–36. doi: 10.1007/3-540-29717-0_17. [DOI] [PubMed] [Google Scholar]
- 32.Vigh L, Literati PN, Horvath I, et al. Bimoclomol: a nontoxic, hydroxylamine derivative with stress protein-inducing activity and cytoprotective effects. Nat Med. 1997;3:1150–4. doi: 10.1038/nm1097-1150. [DOI] [PubMed] [Google Scholar]
- 33.Weisberg SP, Leibel R, Tortoriello DV. Dietary curcumin significantly improves obesity-associated inflammation and diabetes in mouse models of diabesity. Endocrinology. 2008;149:3549–58. doi: 10.1210/en.2008-0262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Woo JH, Lim JH, Kim YH, et al. Resveratrol inhibits phorbol myristate acetate–induced matrix metalloproteinase-9 expression by inhibiting JNK and PKC delta signal transduction. Oncogene. 2004;23:1845–53. doi: 10.1038/sj.onc.1207307. [DOI] [PubMed] [Google Scholar]
- 35.Young JC, Hoogenraad NJ, Hartl FU. Molecular chaperones Hsp90 and Hsp70 deliver preproteins to the mitochondrial import receptor Tom70. Cell. 2003;112:41–50. doi: 10.1016/s0092-8674(02)01250-3. [DOI] [PubMed] [Google Scholar]