

Abstract
Cardiac mitochondrial matrix (m) free Ca2+ ([Ca2+]m) increases primarily by Ca2+ uptake through the Ca2+ uniporter (CU). Ca2+ uptake via the CU is attenuated by extra-matrix (e) Mg2+ ([Mg2+]e). How [Ca2+]m is dynamically modulated by interacting physiological levels of [Ca2+]e and [Mg2+]e and how this interaction alters bioenergetics is not well understood. We postulated that as [Mg2+]e modulates Ca2+ uptake via the CU, it also alters bioenergetics in a matrix Ca2+–induced and matrix Ca2+–independent manner. To test this, we measured changes in [Ca2+]e, [Ca2+]m, [Mg2+]e and [Mg2+]m spectrofluorometrically in guinea pig cardiac mitochondria in response to added CaCl2 (0–0.6 mM; 1 mM EGTA buffer) with/without added MgCl2 (0–2 mM). In parallel, we assessed effects of added CaCl2 and MgCl2 on NADH, membrane potential (ΔΨm), and respiration. We found that ≥0.125 mM MgCl2 significantly attenuated CU-mediated Ca2+ uptake and [Ca2+]m. Incremental [Mg2+]e did not reduce initial Ca2+uptake but attenuated the subsequent slower Ca2+ uptake, so that [Ca2+]m remained unaltered over time. Adding CaCl2 without MgCl2 to attain a [Ca2+]m from 46 to 221 nM enhanced state 3 NADH oxidation and increased respiration by 15%; up to 868 nM [Ca2+]m did not additionally enhance NADH oxidation or respiration. Adding MgCl2 did not increase [Mg2+]m but it altered bioenergetics by its direct effect to decrease Ca2+ uptake. However, at a given [Ca2+]m, state 3 respiration was incrementally attenuated, and state 4 respiration enhanced, by higher [Mg2+]e. Thus, [Mg2+]e without a change in [Mg2+]m can modulate bioenergetics independently of CU-mediated Ca2+ transport.
Keywords: Cardiac mitochondria, Inner mitochondrial membrane, Bioenergetics, Ca2+ uniporter, Ca2+ uptake, Mg2+ inhibition
Introduction
Mitochondria are not only the major source of ATP production but are also vital to cellular homeostasis and function. Mitochondria must be able to adjust ATP production to meet the cell’s ever changing energy demand. A widely held hypothesis is that mitochondrial matrix free Ca2+ ([Ca2+]m) modulates ATP production; this is because an increase in [Ca2+]m was reported to differentially stimulate the activities of three mitochondrial dehydrogenases in the tricarboxylic acid (TCA) cycle (Denton 2009, McCormack et al 1990). In cardiomyocytes, where an increase in extra-matrix free Ca2+ ([Ca2+]e) is linked to muscle contraction during the process of Ca2+–induced Ca2+ release by the sarcoplasmic reticulum (SR), an increase in [Ca2+]m would provide an elegant feed-forward signal for mitochondrial bioenergetics to adapt to increased workload. More recent studies also suggest that F1Fo-ATPase activity is post-translationally regulated by [Ca2+]m (Hubbard et al 1996, Territo et al 2001), although there are no reported binding sites for Ca2+ on F1Fo-ATPase (Abrahams et al 1994), and Ca2+ does not directly stimulate F1Fo-ATPase (Balaban 2009).
There are other proposed mechanisms that may control mitochondrial energy production, such as changes in ADP/ATP and Pi, based on computational evaluation (Beard 2006) of original data (Katz et al 1989), as well as a change in [Ca2+]e via Ca2+ binding domains on regulatory proteins extending into the intermembrane space (IMS) (Gellerich et al 2010, Satrustegui et al 2007). These mechanisms are not likely mutually exclusive. Because oxidative phosphorylation changes rapidly depending on energy demand, it must be highly regulated; thus it is possible that these several mechanisms are synergistic, with distinct mechanisms dominating under different metabolic conditions.
The main route for matrix Ca2+ influx is the ruthenium red (RR) sensitive Ca2+ uniporter (CU) (Bernardi 1999). Another accessory pathway for Ca2+ uptake has been proposed, i.e. the rapid mode (RaM) of Ca2+ uptake (Buntinas et al 2001, Sparagna et al 1995), which may occur at a different site, or be due to a conformational change in the CU. The structural identity of the CU was recently reported (Baughman et al 2011, De Stefani et al 2011). The CU is a 40 kDa protein that forms oligomers in the inner mitochondrial membrane (IMM) and resides within a large molecular weight complex; the CU has two predicted trans-membrane helices. The protein MICU1 is an EF-hand-containing protein (Baughman et al 2011, Perocchi et al 2010) that physically interacts with the CU and serves as a putative sensor of external Ca2+ for its uptake by the CU.
The transport of Ca2+ via the CU is differently affected by other divalent cations, e.g. Mg2+, Mn2+, as well as by the IMM pH gradient, either directly or indirectly. Mg2+ is an essential and abundant intracellular cation involved in many cellular processes. Mg2+ is well known to antagonize Ca2+ uptake (Denton et al 1980, McCormack et al 1980, Panov et al 1996), specifically by impeding Ca2+ uptake via the CU (Favaron et al 1985). A change in extra-matrix Mg2+ ([Mg2+]e) might also alter matrix Mg2+ ([Mg2+]m) to modulate mitochondrial bioenergetics. Like [Ca2+]m, [Mg2+]m may modulate TCA cycle activity directly, e.g. by effects on mitochondrial dehydrogenases (Rodriguez-Zavala et al 1998); alternatively, this effect of Mg2+ could be due to an indirect effect on Ca2+ uptake. Because the uptake of Mg2+ into mitochondria is normally very slow (Brierley et al 1987), it is unclear how it could have any rapid effects on TCA cycle activity. But Mg2+–induced regulation of Ca2+ uptake may help to attenuate Ca2+ overload and to reduce its pathological consequences (Romani et al 1990). Although Mg2+ can modulate mitochondrial activity indirectly by altering [Ca2+]m due to its direct effect to restrict Ca2+ uptake via the CU, it is possible that a change in [Mg2+]e itself may alter mitochondrial activity in a way that is unrelated to restricting Ca2+ up-take.
The kinetic mechanisms associated with CU–mediated Ca2+ influx and Mg2+ inhibition of CU function have been characterized using both initial velocity studies (Bragadin et al 1979, Crompton et al 1976, Scarpa et al 1973, Vinogradov et al 1973) and mathematical models (Pradhan et al 2011). Experimentally, the presence of MgCl2 was shown to alter the kinetics of Ca2+ uptake with added CaCl2 from a hyperbolic relationship in the absence of Mg2+ to a sigmoidal relationship in the presence of Mg2+, with an increase in sigmoidicity at higher [Mg2+]e (Bragadin et al 1979, Crompton et al 1976, Scarpa et al 1973, Vinogradov et al 1973). However, these studies in isolated mitochondria used rather high [Ca2+]e (up to 400 μM) and [Mg2+]e (up to 5 mM), which are above the physiological ranges of 0.1–1 μM for [Ca2+]e and 0.25–1 mM for [Mg2+]e (Rotevatn et al 1989). Thus the effect of [Mg2+]e on matrix Ca2+ uptake is not well characterized at physiological levels of [Ca2+]e or [Mg2+]e. Although [Mg2+]m and [Ca2+]m have been proposed to alter mitochondrial function independently (Panov et al 1996, Rodriguez-Zavala et al 1998), the effect of [Mg2+]e on dynamic responses of both [Ca2+]e and [Ca2+]m was not investigated. Also it is not known if [Mg2+]e alters mitochondrial bioenergetics in a manner that is not due to its direct effect to reduce matrix Ca2+ uptake via the CU at physiological levels of these ions.
We hypothesized a) that Ca2+ uptake via the CU at physiological levels of [Ca2+]e is significantly impeded at even low [Mg2+]e, and b) that an increase in [Mg2+]e can alter the bioenergetic state independent of its effect to decrease [Ca2+]m via the CU. Thus the aim of this study was two-fold: first, to dynamically characterize the extent that MgCl2 impedes Ca2+ uptake via the CU when adding physiological levels of CaCl2, and second, to assess the extent that changes in [Mg2+]e and [Ca2+]e, directly ([Ca2+]m–dependent), or indirectly ([Ca2+]m–independent), modulate bioenergetics. Specifically, we determined if an increase in [Mg2+]e can also modulate the bioenergetic state of mitochondria at a given [Ca2+]m. To carry out these aims, we used fluorescence spectrophotometry to monitor dynamic changes in [Ca2+]e, [Ca2+]m, [Mg2+]e, [Mg2+]m, NADH and membrane potential (ΔΨm) in isolated mitochondria from guinea pig hearts during extra-matrix addition of a range of physiological [CaCl2] and [MgCl2]; in parallel experiments, we measured O2 consumption with and without added ADP at the same ranges of [CaCl2] and [MgCl2]. To prevent Ca2+ extrusion via Na+–Ca2+ exchange (NCXm) we used only Na+–free solutions and compounds.
Materials and methods
Mitochondrial isolation
All experiments conformed to the Guide for the Care and Use of Laboratory Animals and were approved by the Institutional Animal Care and Utilization Committee. Guinea pig heart mitochondria were isolated as described before (Blomeyer et al 2012, Haumann et al 2010, Riess et al 2005). Guinea pigs (250–400 g) were anesthetized by intra-peritoneal injection of 30 mg ketamine along with 700 units of the anticoagulant heparin to prevent clotting. Each heart was excised and minced to approximately 1 mm3 pieces in ice-cold isolation buffer containing in mM: mannitol 200, sucrose 50, KH2PO4 5, 3-(N-morpholino) propanesulfonic acid (MOPS) 5, EGTA 1, BSA 0.1%, at pH 7.15 (adjusted with KOH). The buffer was decanted and the minced heart was suspended in 2.65 ml buffer with 5U/ml protease (Bacillus licheniformis), and was homogenized at low speed using a CAT X homogenizer for 30 s; next 17 ml isolation buffer was added and this suspension was again homogenized for 30 s and then was centrifuged at 8000 g for 10 min. The supernatant was discarded and the pellet was resuspended in 25 ml isolation buffer and centrifuged at 900 g for 10 min. The supernatant was centrifuged once more at 8000 g to yield the final mitochondrial pellet, which was suspended in 0.5 ml isolation buffer and kept on ice until used. Mitochondrial protein concentration was measured using the Bradford method (Bradford 1976). The suspension volume was adjusted to obtain a concentration of 12.5 mg protein/ml isolation buffer. All experiments were conducted at room temperature (25°C) in a Na+–free respiration buffer (0.5 mg protein/ml) containing in mM: KCl 130, K2HPO4 5, MOPS 20, EGTA 1, BSA 0.1% and at pH 7.15 (adjusted with KOH). Trial experiments were conducted in the presence of 25 μM CGP 37157 (Tocris Bioscience), a NCX inhibitor, to verify that Na+ was not present in the respiration buffer.
Measurements of [Ca2+]m and [Ca2+]e
Fluorescence spectrophotometry (Qm-8, Photon Technology International) was used to measure [Ca2+]m and [Ca2+]e. To measure [Ca2+]m, isolated mitochondria (5 mg/ml) were incubated with indo-1 acetoxymethyl (indo-1-AM) (Invitrogen) (5 μM in DMSO) for 20 min at room temperature (25 °C), followed by addition of 25 ml ice-cold isolation buffer and repeated centrifugation at 8000 g. The AM form of indo-1 is taken up into the matrix where it is de-esterified and retained. The dye-loaded pellet was re-suspended in 0.5 ml isolation buffer, and protein concentration was measured again and diluted to 12.5 mg mitochondrial protein/ml. For [Ca2+]m measurements, the dye loaded mitochondria were suspended in the Na+–free respiration buffer (0.5 mg protein/ml).
The emission wavelengths (λem) of indo-1 at an excitation wavelength of 350 nm (λex) were 456 nm and 390 nm. The ratio (R) between the two λem’s relative to the ratios obtained when all indo-1 is bound to Ca2+ (Rmax) and when no Ca2+ is bound to indo-1 (Rmin) corresponds to [Ca2+]m and also serves to correct for variations that affect both signals equally, e.g. differences in the amount of dye taken up into mitochondria and small fluctuations in excitation intensity. For each preparation Rmax was measured in the presence of 500 nM cyclosporine A plus 10 mM CaCl2, and Rmin was measured after adding A23187 (Ca2+-ionophore) plus 1 mM EGTA. Because there may be considerable background autofluorescence (AF) at these wavelengths, which is largely attributable to NADH, mitochondria AF was measured in mitochondria not loaded with indo-1-AM but with its vehicle DMSO, using the same procedure as described above; these AF signals were then subtracted from the indo-1 signals before R was calculated. [Ca2+]m was then calculated using the equation (Grynkiewicz et al 1985):
The Kd value for indo-1-AM binding to Ca2+ under our conditions was determined as 326 nM (see Figs. S1 and S2 of Supplemental Materials). Sf2 is the signal intensity of free indo-1 measured at 456 nm; Sb2 is the signal intensity of Ca2+-saturated indo-1 measured at 456 nm. Each fluorescence signals was measured every second.
[Ca2+]e was measured using the same procedure, but with indo-1 penta-potassium salt (indo-1-PP) instead of indo-1-AM; indo-1-PP is relatively impermeable to IMM. Mitochondria were isolated as above for the indo-1-AM experiments, but were incubated for 20 min at 25°C with an equivalent amount of the vehicle, DMSO, to mimic conditions of the indo-1-AM experiments. Indo-1-PP was present in the respiration buffer at a concentration of 1 μM. The signal was corrected for AF and [Ca2+]e was calculated using the same formula as for [Ca2+]m. With 1 mM MgCl2 in the respiration buffer [Ca2+]e was not altered after adding CaCl2, which verifies that Mg2+ does not interfere with the indo-1 fluorescence signal. The Kd value for indo-1-PP binding to Ca2+ was determined as 311 nM.
Measurements of [Mg2+]m and [Mg2+]e
[Mg2+]m was measured with Mag-fura-2-AM and [Mg2+]e was measured with Mag-fura-2 tetra-potassium salt (mag-fura-2-K) using fluorescence spectrophotometry. The λex’s for Mag-fura-2 at λem 490 nm were 385 and 340 nm. [Mg2+]m was calculated from the ratio between λex 385 and 340 nm using the same formula as for indo-1. Rmin was generated by permeabilizing mitochondria with A23187 and 0.005% (v/v) Triton X-100 in respiratory buffer containing 1 mM EGTA (Rodriguez-Zavala et al 1998). Rmax was obtained after further addition of 100 mM MgCl2. For Mag-fura-2 binding to Mg2+ the Kd 2.9 mM (Howarth et al 1995) was used.
Measurements of mitochondrial NADH and membrane potential
NADH is a measure of reduction/oxidation (redox) potential. Unlike NAD+, NADH molecules have natural fluorescence properties. NADH AF was measured ratiometrically at λem 390 nm and λem 456 at λex 350 nm after incubating with DMSO for 20 min. The ratio of the signals, λem 456 nm/λem 390 nm reflects an increase in the ratio of NADH to NAD+, i.e. a shift to a more reduced state (Brandes et al 1996, Haumann et al 2010). ΔΨm was measured using the lipophilic dye TMRM in a ratiometric excitation approach (Blomeyer et al 2012, Scaduto et al 1999). TMRM (1 μM), dissolved in DMSO, was separately added to the experimental buffer. Fluorescence changes were detected by two λex (546 and 573 nm) and one λem (590 nm). The calculated ratio of both excitation wavelengths (573/546) is proportional to ΔΨm and has the advantage of a broader dynamic range when compared to a single wavelength technique (Scaduto et al 1999). Measured ratios were scaled for each group to their average photon counts at t = 100 s (for protocol and timeline see Experimental protocol).
Measurement of mitochondrial respiration
O2 consumption rate was measured using a Clark-type polarographic O2 sensor (Oxygraph-2K from OROBOROS Instruments, Austria) at 25 °C (Haumann et al 2010). Mitochondria (0.125 mg protein/ml) from 5 hearts were suspended in Na+-free respiration buffer. State 2 respiration was defined as O2 consumption in the presence of high substrate and low ADP levels (i.e. after addition of 0.5 mM pyruvic acid (PA)), state 3 respiration was defined as O2 consumption in the presence of high substrate and ADP levels (i.e. after adding ADP), and state 4 respiration was defined as O2 consumption after all added ADP was phosphorylated to ATP; O2 consumption was expressed as pmol/s/mg protein. Experiments were conducted in mitochondria that exhibited a respiratory control index (RCI; state 3 respiration/state 4 respiration in the absence of added Ca2+ and Mg2+) consistently greater than 16 with 0.5 mM PA and added ADP. At this RCI, mitochondria are tightly coupled with a fully polarized ΔΨm. At the end of each experiment, lasting up to 6 h, RCI’s remained greater than 12.
Experimental protocol
All experiments followed the same timeline protocol as shown in Fig. 1. At t = −120 s, the experiment was initiated with the Na+–free buffer containing 0 (de-ionized H2O), 0.125, 0.25, 0.5, 1 or 2 mM MgCl2 and 1 mM EGTA. At t = −90 s mitochondria were added to the Na+–free respiration buffer and at t = 0 s mitochondria were energized using 0.5 mM PA (Na+–free). At t = 120 s, either 0 (control, de-ionized H2O), 0.25, 0.40, 0.50 or 0.60 mM CaCl2 was added and responses were measured for another 120 s. Concentrations above 0.60 mM (i.e. 0.75 mM) CaCl2 did not further increase [Ca2+]m but decreased NADH levels, indicating mCa2+ overload (preliminary data). Finally, at t = 240 s ADP (250 μM) was added. At t = 480 s the experiment was concluded, or the uncoupler CCCP (carbonyl cyanide m-chlorophenyl-hydrazone, 4 μM) was added to fully depolarize the mitochondria. Figures are presented starting at t = 0 s. For some figures only the data with 0 mM (no added) MgCl2, or 0.25 and 1 mM added MgCl2 are shown. The order of experiments was randomized during the course of each day. Data for comparison of groups were collected and averaged at the following time points: 225–235 s (state 2), 245–255 s (state 3), and 360–370 s (state 4).
Fig. 1.
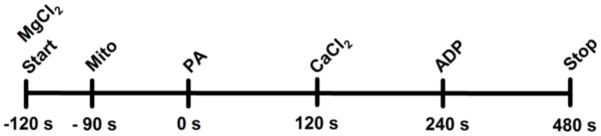
Timeline for adding substances to experimental buffer. Figures are presented from addition of PA (t=0 s); 500 μM PA, Na+-free pyruvic acid; 250 μM ADP, (adenosine diphosphate).
Statistical analyses
All data are presented as mean (±SEM). ANOVA followed by a post hoc analysis using Student-Newman-Keuls’ test was performed to determine statistically significant differences between and within groups using Sigmaplot 11 software (Systat Software, Inc., USA). A P value < 0.05 (two-tailed) was considered significant. Statistical comparisons are not shown for all time-collected data but are shown for key interrelationship summary data.
Results
Effect of extra-matrix MgCl2 on [Mg2+]e and [Mg2+]m
[Mg2+]e, measured using mag-fura-2-K, was undetected prior to adding MgCl2 and proportional to the added MgCl2 (Fig. 2A). [Mg2+]e rose rapidly but remained constant over 10 min. [Mg2+]m (Fig. 2B), measured using mag-fura-2-AM, was 0.35±0.09, 0.34±0.08 and 0.34±0.09 mM (state 2) in the 0.5, 1, and 2 mM MgCl2 groups, respectively. There was no significant change in [Mg2+]m from these baseline values over 10 min indicating no Mg2+ uptake. Adding ADP at 240 s had no effect on either [Mg2+]e or [Mg2+]m. These data indicated that extra-matrix Mg2+ was not taken up into the matrix during this time so that any effects of Mg2+ on Ca2+ uptake or mitochondrial bioenergetics originated from the extra-matrix side.
Fig. 2.

Changes in external free [Mg2+]e on addition of MgCl2 to the buffer containing isolated mitochondria (A); [Mg2+]e was slightly less than the amount of added MgCl2. Measure of matrix [Mg2+]m on addition of MgCl2 (B). Note that over 10 min [Mg2+]m did not increase with the increase in [Mg2+]e. Adding ADP had no effect to alter either [Mg2+]e or [Mg2+]m.
Effect of extra-matrix CaCl2 and MgCl2 on Ca2+ uptake into mitochondria
We first determined to what extent MgCl2 alters Ca2+ uptake via the CU. Mitochondria were energized with the Na+–free substrate PA to prevent Ca2+ extrusion via NCXm. At 0.25, 0.40, 0.50 or 0.60 mM CaCl2, with no added MgCl2 and in the presence of 1 mM EGTA, [Ca2+]e increased rapidly to 60±3, 120±9, 173±10 and 254±36 nM, respectively, compared to the 0 mM (control) CaCl2 value of 6±1 nM (Fig. 3). There were no further changes in [Ca2+]e over time among the corresponding CaCl2 groups when adding any concentration of MgCl2 to the buffer (data not shown). Although Ca2+ enters the mitochondria via the CU, [Ca2+]e changed little because of the high buffering effect of 1 mM EGTA and the much larger extra-matrix volume than matrix volume. However, at 0.50 and 0.60 mM CaCl2, there was a small initial transient decline in [Ca2+]e (Fig. 3), signifying a rapid increase in Ca2+ uptake. Test experiments conducted in the presence of CGP 37157 gave similar results thus verifying that the buffer was free of Na+ and that Ca2+ extrusion via NCXm did not occur. Adding ADP at 240 s did not change [Ca2+]e (not shown).
Fig. 3.

Changes in dynamics of [Ca2+]e in response to adding PA, CaCl2 and ADP as shown in Fig. 1 timeline. Adding CaCl2 in the presence of 1 mM EGTA caused a rapid and sustained increase in [Ca2+]e that remained unchanged due to buffering by EGTA. Steady state [Ca2+]e ranged from 5±1 to 268±30 nM for 0 to 0.60 mM CaCl2, respectively. Adding ADP at 240 s did not alter [Ca2+]e.
The increases in [Ca2+]e resulted in a two-phase concentration-dependent increase in [Ca2+]m (Fig. 4); an initial faster uptake of Ca2+ (t= 120–125 min) followed by a slower, more gradual uptake (t = 125–230 s) (Fig. 4A–C). Most Ca2+ uptake occurred during the initial faster (5–10 s) phase, particularly in the presence of added MgCl2, which attenuated only the slower phase of Ca2+ uptake. In the absence of added MgCl2 (Fig. 4A), the increase in [Ca2+]e (above) in response to added CaCl2 increased [Ca2+]m from 46±9 nM (no added CaCl2) to 221±24, 509±51, 775±27 and 868±25 nM (0.25, 0.40, 0.50, 0.60 mM CaCl2), respectively, by the end of state 2 respiration (t = 230 s). In the presence of 0.125, 0.25, 0.5, 1 and 2 mM MgCl2, Ca2+ uptake was significantly and equivalently reduced, which resulted in lower [Ca2+]m at the end of state 2 respiration (Fig. 4B–F). Interestingly, the decrease in Ca2+ uptake was apparent with as little as 0.125 mM MgCl2 and higher [MgCl2] did not appreciably enhance this decrease in Ca2+ uptake.
Fig. 4.

Changes in dynamics of [Ca2+]m after adding PA, CaCl2 and ADP as shown in Fig. 1 timeline. Each left panel (A–F) shows five traces corresponding to added CaCl2 in the presence of 0 mM (no added) MgCl2 (A) and added MgCl2 (B, 0.125; C, 0.25; D, 0.5; E, 1; F, 2 mM). There appeared to be two phases of Ca2+ uptake via the CU at these levels of MgCl2: a rapid uptake phase (5–10 s) and a slow uptake phase (1–2 min). See Fig 5 for statistics.
Effect of extra-matrix MgCl2 on [Ca2+]m as a function of [Ca2+]e
We next plotted [Ca2+]m as a function of [Ca2+]e with/without added MgCl2 at the end of state 2 respiration (t = 230 s) (Fig. 5). [Ca2+]m rose up to 3.5 fold higher than [Ca2+]e in the absence of MgCl2. With no added MgCl2, [Ca2+]m reached a plateau level of about 868 nM at 0.50 mM CaCl2 so that at 0.60 mM CaCl2 (254 nM [Ca2+]e) there was no further increase in [Ca2+]m. All MgCl2 groups higher than 0 mM (0.125–2 mM) reduced [Ca2+]m similarly at a given [Ca2+]e, such that each [MgCl2] from 0.125 mM and higher reduced [Ca2+]m between 45% and 55%, respectively, at 254 nM [Ca2+]e. Thus the strength of attenuation of Ca2+ uptake by Mg2+ was very steep, indicating that even adding very little MgCl2 (0.125 mM) could reduce the maximal amount of Ca2+ uptake through the CU.
Fig. 5.

[Ca2+]m as a function of [Ca2+]e at increasing [Mg2+]e during state 2 respiration (at t=230 s). In the absence of added MgCl2, [Ca2+]m was 3.5 fold greater than [Ca2+]e; in the presence of MgCl2, [Ca2+]m remained greater than [Ca2+]e. Note the near maximal effect of added MgCl2 on reducing [Ca2+]m.*P<0.05 each [Ca2+]m with added CaCl2 vs. at 0 mM CaCl2; #P<0.05 each [Ca2+]m with added MgCl2 vs. 0 mM (no added) MgCl2 at a given [Ca2+]e.
Effects of [Ca2+]e and [Mg2+]e on mitochondrial NADH and membrane potential
We next assessed changes in two important markers of bioenergetics, NADH and ΔΨm, that could be affected by increases in buffer [CaCl2] and [MgCl2]. NADH levels were measured (Fig. 6) following the same timeline as for the Ca2+ and Mg2+ experiments (Fig. 1) to correlate changes in [Ca2+]m to changes in mitochondrial bioenergetics in real time. Adding PA increased state 2 NADH markedly and adding CaCl2 further increased NADH significantly (P<0.05 during state 2 at t = 230 s) in the absence of MgCl2 (Fig. 6A). The presence of 0.25 and 1 mM MgCl2 (Fig. 6B,C) attenuated the increases in state 2 NADH after adding CaCl2 to values not significantly different (P>0.05) between 0 mM (control) and added CaCl2 groups. Initiation of state 3 respiration by adding ADP led to a large transient oxidation of NADH (decreased NADH/NAD+ ratio) as NADH was consumed by the electron transport chain to maintain the electrochemical gradient. There was no difference in NADH oxidation levels during state 3 respiration among the CaCl2 groups (P>0.05) but state 3 duration decreased with increasing CaCl2 added in the absence of MgCl2 (Fig. 6A); this indicated faster electron transfer and ADP phosphorylation. After consumption of all ADP (state 4), and in the absence of MgCl2, NADH returned to pre-ADP levels (state 2 respiration) for all levels of CaCl2; NADH was higher after adding CaCl2 compared to no CaCl2 (P>0.05). At 0.25 or 1 mM MgCl2, NADH levels during state 4 respiration remained relatively more oxidized (decreased NADH) in all CaCl2 groups (P<0.05) compared to pre-ADP levels (state 2 respiration). Unlike their effects to alter redox state (NADH), adding MgCl2 or CaCl2 had no appreciable effect to alter ΔΨm (Fig. 6D–F). Adding ADP caused a transient, submaximal depression of ΔΨm.
Fig. 6.

Changes in dynamics of mitochondrial NADH redox state (autofluorescence) and ΔΨm (ratio of TMRM signals) after adding PA, CaCl2 and ADP as shown in the timeline scheme (Fig. 1). Each left panel (A,B,C) for NADH and each right panel (D,E,F) for TMRM shows traces corresponding to added CaCl2 in the presence of 0 mM (no added) MgCl2 (A,D) and 0.25 mM (B,E) and 1 MgCl2 (C,F). Tracings show that NADH increased markedly after adding PA and less so after adding CaCl2 (t=120 s). Between 0.25 and 0.6 mM CaCl2 there was no significant increase in state 2 NADH for all added [MgCl2]. Adding 1 mM MgCl2 (C) decreased the Ca2+–induced rise in NADH compared to corresponding CaCl2 groups without added MgCl2 (A) at each respiratory state. NADH was similar during states 4 and 2 respiration in the 0 mM (no added) MgCl2 group, but in the added MgCl2 groups NADH was lower during state 4 than in state 2 respiration (P<0.05). Adding ADP (state 3 respiration) transiently and similarly decreased NADH as it was consumed to maintain the proton gradient in each CaCl2 and MgCl2 group. Note that state 3 duration in the group without added CaCl2 and MgCl2 (A) was longer than in the groups with added CaCl2 and MgCl2, reflecting slower state 3 respiration. Adding CaCl2 or MgCl2 had no effect on ΔΨm (D–F); only adding ADP caused a transient fall in ΔΨm. At 480 s CCCP was given to completely depolarize the membrane potential. Other statistics are given in text.
Effects of [Ca2+]e and [Mg2+]e on mitochondrial O2 consumption rates
O2 consumption (Fig. 7) was measured following the same timeline (Fig. 1) to correlate this data with changes in [Ca2+]m, NADH, and ΔΨm over time. Representative traces of O2 concentration (Fig. 7A,B) and summarized O2 consumption rates (Fig. 7C,D) showed faster state 3 respiration at 0.60 mM CaCl2 vs. no added CaCl2 (control) with no added MgCl2. State 2 respiration did not differ among CaCl2 groups in the presence or absence of MgCl2. Adding ADP induced state 3 respiration as shown by the marked increase in O2 consumption in all groups with or without added MgCl2; moreover, adding CaCl2 further enhanced state 3 respiration in a concentration-independent manner. Adding MgCl2 reduced state 3 respiration (Fig. 7D) at all concentrations of added CaCl2 (P<0.05) compared to corresponding groups with no added MgCl2 (Fig. 7D vs. C). In contrast, adding MgCl2 resulted in faster state 4 respiration independent of added CaCl2 (Fig. 7D vs. C). Re-arranging part of these data to plot effects of added MgCl2 at a constant CaCl2 (0 or 0.6 mM CaCl2) shows more readily how MgCl2 reduced state 3 respiration while enhancing state 4 respiration (Fig. 8A–D).
Fig. 7.

Representative traces of O2 concentration for 0 (solid black line) and 0.60 mM (solid blue line) CaCl2 without added MgCl2 (A) or with 1 mM MgCl2 (B) in response to adding PA, CaCl2, and ADP as shown in the timeline scheme (Fig. 1). Dotted lines are the first derivatives showing the maximal rates of change during state 3. Calculated mean O2 consumption rates (C,D) increased in the presence 0.25 mM CaCl2 during state 3 respiration; higher [CaCl2] did not increase state 3 respiration further. Adding MgCl2 attenuated state 3 respiration. Adding MgCl2 enhanced state 4 respiration at all [CaCl2], which was compatible with the decreased NADH during state 4 respiration (Fig. 6). See Fig. 9 for statistics.
Fig. 8.

Representative traces of O2 concentration for 0 (solid blue line) and 1 mM (solid black line) MgCl2 without added CaCl2 (A) or with 0.6 mM CaCl2 (B) in response to adding PA, CaCl2, and ADP. Dotted lines are the first derivatives showing the maximal rates of change during state 3. Calculated mean O2 consumption rates (C,D) decreased in the presence 1 mM MgCl2 during state 3 respiration (B); higher [CaCl2] did not increase state 3 respiration further. Adding CaCl2 enhanced state 3 respiration. Adding MgCl2 enhanced state 4 respiration at all [CaCl2], which was compatible with decreased NADH during state 4 respiration (Fig. 5). See Fig. 9 for statistics.
Effects of MgCl2 on O2 consumption as a function of extra-matrix and matrix [Ca2+]
Lastly, we plotted changes in states 3 (Fig. 9A,C) and 4 (Fig. 9B,D) O2 consumption rates as a function of end state 2 [Ca2+]e and [Ca2+]m in the presence and absence of added MgCl2 to illustrate their inter-relationships. In the absence of added MgCl2, state 3 respiration was faster with an increase in [Ca2+]e or [Ca2+]m compared to after adding MgCl2 (A,C). Adding as little as 0.25 mM CaCl2 (≈ 221 nM [Ca2+]m) maximally enhanced respiration in the absence of MgCl2, compared to the absence of CaCl2. Importantly, at a given [Ca2+]e or [Ca2+]m, each added [MgCl2] had a concentration-dependent effect to slow the rate of state 3 respiration. At the highest [Ca2+]e or [Ca2+]m, 1 mM MgCl2 significantly depressed the rate of state 3 respiration. These reductions in states 3 respiratory rate by added MgCl2 at a given [Ca2+]e or [Ca2+]m indicated that an increase in [Mg2+]e, but not an increase in [Mg2+]m (as it did not change), had an effect to reduce state 3 respiration. Unlike during state 3, state 4 respiration was not affected by an increase in [Ca2+]e or [Ca2+]m in the absence or presence of MgCl2 (Fig. 9B,D). But adding as little as 0.25 mM MgCl2 maximally increased state 4 respiration by over two fold at any given [Ca2+]e or [Ca2+]m. Note that the presence of MgCl2 increased state 4 respiration with no change in ΔΨm (Fig. 6D–F), indicating there was enhanced proton conductance. (see Supplemental Material (Figs. S5–7) for changes in respiration in the presence of ruthenium red to block CU.)
Fig. 9.

State 3 (A,C) and state 4 (B,D) O2 consumption rates as a function of [Ca2+]e (A,B) and [Ca2+]m (C,D) in the absence and presence of 0.25 and 1 mM MgCl2. In the absence of MgCl2 state 3 respiration was enhanced between 6 to 60 nM [Ca2+]e, but not by higher [Ca2+]e up to 254 nM (A). At a given [Ca2+]e or [Ca2+]m at the end of state 2, increasing MgCl2 (from 0 to 1 mM) reduced state 3 respiration (A,C) in a stepwise manner. State 4 respiration (B,D) was not affected by adding CaCl2 in the absence or presence of MgCl2 but was enhanced similarly by both 0.25 and 1 mM MgCl2. *P<0.05 added CaCl2 vs. no added CaCl2 at a given MgCl2; #P<0.05 added MgCl2 vs. no added MgCl2 at a given CaCl2.
Discussion
An increase in cytosolic [Ca2+]e leads to mitochondrial Ca2+ uptake via the CU, particularly if ΔΨm is high; much of this Ca2+ is buffered in the matrix; some is extruded via the NCXm. Cytosolic Ca2+ over-load, as occurs in ischemia-reperfusion injury, can disrupt mitochondrial function and induce apoptotic mechanisms (Bernardi 1999, Camara et al 2010, Duchen 2000). Because Mg2+ is a modulator of CU –mediated Ca2+ uptake, its impact on [Ca2+]m is therefore important. Moreover, Mg2+ may have effects on bioenergetics unrelated to its modulation of [Ca2+]m.
Our aims were: a) to directly compare effects of increasing [Mg2+]e and [Ca2+]e on [Ca2+]m dynamics over a physiologic range, and b) to assess the effects of these cations on three bioenergetics markers, ΔΨm, NADH redox state, and O2 consumption. We found that very small increases in [Mg2+]e near maximally attenuated the CU-mediated increase in [Ca2+]m for the range of [Ca2+]e examined (0–0.6 mM CaCl2 with 1 mM EGTA); further increases in [Mg2+]e (from 0.125 to 2 mM) did not significantly alter the dynamics of Ca2+ uptake. Also, a CaCl2–induced increase in [Ca2+]m from 46 to 221 nM was associated with a 5% increase in state 2 NADH (redox state) and a 15% increase in state 3 respiration in the absence of added MgCl2; greater than 221 nM [Ca2+]m had no additional effect on NADH or O2 consumption. The presence of MgCl2 resulted in a concentration–dependent fall in state 3 NADH and O2 consumption due in part to the concomitant decrease in [Ca2+]m. However, at a given [Ca2+]m (e.g. 221 nM), 1 mM MgCl2 nearly abolished the increases in NADH and O2 consumption due to the increase in [Ca2+]m from 46 to 221 nM. In contrast, the presence of Mg2+ markedly enhanced state 4 respiration at any given [Ca2+]m.
These results suggest that the modulatory effects of Mg2+ on states 3 and 4 bioenergetics appear to be dependent not only on [Mg2+]e to reduce Ca2+ uptake and decrease [Ca2+]m, but also on an increase in [Mg2+]e by matrix Ca2+–independent mechanisms. Indeed, bioenergetic function was reduced foremost because Mg2+ attenuated Ca2+ uptake, even at higher [Ca2+]e or [Ca2+]m. However, because neither adding MgCl2 nor ADP increased [Mg2+]m, Mg2+ had to have an indirect, i.e. extra-matrix, effect to attenuate state 3 respiration and NADH, while it enhanced state 4 respiration. Because adding MgCl2 or CaCl2 had no effect on ΔΨm, these cations likely had no direct effect to alter proton conductance (“H+ leak”). Therefore, the slower state 3 respiration and faster state 4 respiration with increasing [Mg2+]e was due not only to an effect of Mg2+ to reduce [Ca2+]m but, by exclusion, also due to effects mediated outside the matrix.
Mg2+reduces maximal mitochondrial Ca2+ uptake induced by increasing external Ca2+
The main route for Ca2+ uptake is likely the CU, which can be considered a low affinity influx pathway (Bragadin et al 1979, Crompton et al 1976, Scarpa et al 1973, Vinogradov et al 1973). Patch clamp experiments show that the CU is a highly selective ion channel (Kirichok et al 2004), and its protein structure is now known (Baughman et al 2011, De Stefani et al 2011); an associated protein, MICU1, is required for Ca2+ uptake (Perocchi et al 2010). Though the CU is the major route for Ca2+ uptake, the rate of uptake may be regulated by a different distinct channel or a different conformational state of the CU (Bazil et al 2011, Buntinas et al 2001, Sparagna et al 1995). Our experiments were designed to dynamically measure extra-matrix and matrix free [Ca2+] and [Mg2+] to better assess how [Mg2+] affects movement of Ca2+ across the IMM via CU. Earlier studies (Denton et al 1980, McCormack et al 1990, Wan et al 1989) examined only static levels of Ca2+ and correlated them with added extra-matrix CaCl2. We show here how the maximal uptake of Ca2+ was significantly affected by [Mg2+]e at physiological levels of [Ca2+]e.
The range of [Ca2+]m we examined lies within a physiological range of 100 to 900 nM (up to 0.60 mM added CaCl2); this range of added CaCl2 slightly enhanced respiration but did not decrease NADH. Most ionized Ca2+ entering the matrix is buffered by soluble phosphate complexes (Bataille et al 1994, Morgan et al 2006, Starkov 2010) that are influenced by proteins (e.g. annexins) and lipids (e.g. cardiolipin), so total loading of Ca2+ into the matrix from the added CaCl2 is much greater than that seen from the less than 4 fold higher [Ca2+]m than [Ca2+]e; a [Ca2+]m between 700 and 1100 nM, as measured in our intact heart model (Aldakkak et al 2011, Camara et al 2007, Rhodes et al 2012), may indicate excess Ca2+ loading and a nearly exhausted Ca2+ buffering capacity, so that mitochondrial permeability transition pore (mPTP) opening and apoptosis may occur. Furthermore, above a certain [Ca2+]m the increase in respiration might actually be due to the uncoupling effects of excess mCa2+.
Matrix Ca2+–induced changes in mitochondrial bioenergetics are sensitive but limited
Several possible mechanisms by which matrix Ca2+ is believed to regulate bioenergetics have been reviewed (Balaban 2009, Griffiths et al 2009). The first proteins observed to be activated by Ca2+ were three dehydrogenases in the TCA cycle. Since then Ca2+ has been proposed to regulate a number of substrate transporters, as well as F1F0-ATPase and cytochrome c oxidase (Balaban 2009). We showed that an increase in [Ca2+]m is accompanied by an immediate, but small, increase in NADH levels during state 2 respiration without an increase in O2 consumption. This effect was quite small and active only at very low [Ca2+]m. The small increase in redox potential, combined with the possible effects of [Ca2+]m on some mitochondrial enzymes could underlie, in part, the higher state 3 respiration. However, the importance of [Ca2+]m on mitochondrial dehydrogenase activities is strongly dependent on the substrate used. For example, it was reported that pyruvate dehydrogenase is activated at almost 100%, regardless of the [Ca2+]m, and that α-ketoglutarate dehydrogenase is a more physiologically relevant target of Ca2+ regulation (Vinnakota et al 2011, Wan et al 1989). This means that the greatest effect of Ca2+ on bioenergetics would be with α-ketoglutarate as the substrate.
For this study, we chose PA as our substrate because it is effectively (through acetyl CoA) the two-carbon donor to oxaloacetate; when the six-carbon product isocitrate and other TCA cycle intermediates become oxidized this reduces NAD+ to NADH used for proton pumping to maintain ΔΨm. This might explain in part why we observed only a small effect of Ca2+ to increase bioenergetic activity compared to previous studies using α-ketoglutarate and other TCA cycle substrates. The substrate glutamate+malate may provide a greater NADH response to increasing [Ca2+]e (Vinnakota et al 2011); moreover, higher concentrations of substrates to energize mitochondria may saturate ADP-independent TCA cycle flux. We used 0.5 mM PA, a lesser amount than used in many studies, which may have led to our observation of only a small, stimulated respiratory response to Ca2+ uptake. In contrast, an increase in cardiac work demand can increase cardiac O2 consumption by up to 8 fold, so it is doubtful that this is solely due to the reported modest increases in [Ca2+]m.
Mg2+–induced attenuation of mitochondrial Ca2+ uptake occurs at the Ca2+ entry site
That Ca2+ uptake is attenuated by [Mg2+]e, as shown by the fall in [Ca2+]m at a constant [Ca2+]e (Fig. 5A), agrees qualitatively with earlier findings (Favaron et al 1985, Litsky et al 1997). McCormack et al. (McCormack et al 1990) that adding 1–2 mM MgCl2 reduced [Ca2+]m by about 8 fold at 250 nM [Ca2+]e. We show, however, that even at a very low physiologic range of [Mg2+]e, this effect is already close to a maximum but can be partially overcome by adding more CaCl2 (Fig. 4B). This trend of [Mg2+]e to blunt, but not to completely inhibit, the increases in [Ca2+]m was also observed recently (Wei et al 2012). As noted above, a temporal characteristic of Ca2+ uptake is that at any [Ca2+]e, adding MgCl2 reduced the net Ca2+ uptake, but not the initial uptake phase (Fig. 4). Interestingly, adding MgCl2 did not at all increase [Mg2+]m over many minutes (Fig. 2). Thus our study indicates that Mg2+ inhibits CU-mediated Ca2+ uptake by interfering with Ca2+ only at the extra-matrix side of the CU. Although it was reported that Mg2+ does not appreciably enter the matrix in the absence of ADP/ATP exchange (Chinopoulos et al 2009), we observed that adding ADP indeed did not cause a net movement of Mg2+ into the matrix.
Mg2+ is proposed to modulate mitochondrial cation and substrate transport systems; but the dynamic interaction of physiological concentrations of Mg2+ with Ca2+ inside or outside the mitochondrion has not been rigorously examined. Earlier Ca2+ uptake experiments in isolated mitochondria were conducted at very high [Ca2+]e of up to 400 μM, and high [Mg2+]e of up to 5 mM (Bragadin et al 1979, Crompton et al 1976, Scarpa et al 1973, Vinogradov et al 1973), which are well above their physiologic ranges. Although these studies provided useful information on how Ca2+ and Mg2+ interacted at the putative CU, and on the type of Mg2+ inhibition (e.g. competitive, non-competitive or mixed-type), these effects were not examined within the lower physiological ranges of [Mg2+] or [Ca2+] and the dynamics of Mg2+ on the time course of Ca2+ uptake were not examined. Importantly, in these earlier studies [Mg2+]m was not directly measured during the duration of Ca2+ uptake; interestingly, in our study [Mg2+]m did not change over 10 min at any added MgCl2. Therefore, it has remained largely unknown how [Mg2+]m could dynamically alter CU-mediated Ca2+ transport. Our study demonstrates clearly how normal levels of [Ca2+]e and [Mg2+]e interact to modulate [Ca2+]m via the CU. A recent study in permeabilized HEK cells demonstrated that physiological changes in cytosolic Mg2+ attenuated Ca2+ uptake (Szanda et al 2009), which is consistent with our findings and those of others. But our study additionally provides detailed information on the characteristics of this Mg2+–induced attenuation of Ca2+ uptake that can now be utilized to construct more accurate computational models of Ca2+ transport systems and their regulation.
External Mg2+ alters bioenergetics by both mCa2+–dependent and independent mechanisms
The physiological range of cytoplasmic [Ca2+]e in cardiac cells fluctuates between about 100 and 400 nM with each heart beat and increases only moderately with inotropic stimulation (Rhodes et al 2006). In a normal cardiac cell, average [Ca2+]m is about 150–200 nM (Riess et al 2002). An increase in [Ca2+]e to 250 nM led only to an increase in [Ca2+]m of 870 nM at the end of state 2 respiration (Figs. 4A and 5A) at 0.60 mM CaCl2 in the absence of MgCl2. We attempted to measure the dynamics of [Ca2+]m in response to higher CaCl2 (e.g. at 0.75 mM CaCl2 equivalent to 1236±218 nM [Ca2+]m) to increase [Ca2+]m further. However, we observed a detrimental effect of high [Ca2+]m on state 3 respiration; i.e. state 3 respiration was slower, state 3 duration was longer, and in some experiments a faster state 3 respiration did not occur at all after adding ADP (data not shown). The finding that higher [Ca2+]e led to dysfunction is likely related to the absence of Na+ in our buffer to extrude excess Ca2+ via NCXm. We showed previously that a lack of buffer Na+ abolishes NCXm similar to inhibiting the NCXm with CGP 37157 (Agarwal et al 2012, Blomeyer et al 2012). Because NCXm is the major route for Ca2+ extrusion in cmitochondria, the presence of extra-matrix Na+ clearly exerts a diminishing effect on the net rise in [Ca2+]m (Maack et al 2006) when [Ca2+]e increases. Moreover, because excess Mg2+ was reported to inhibit mPTP opening (Bernardi et al 1993), a lack of added MgCl2 might contribute to earlier mPTP opening when Ca2+uptake is large.
It would seem unclear how a change in [Mg2+]e (not [Mg2+]m) could exert any direct, i.e. not via changes in [Ca2+]m, effects on bioenergetics. However, a change in [Mg2+]m was reported to independently modulate oxidative phosphorylation (Klingenberg 2008, Panov et al 1996, Rodriguez-Zavala et al 1998) among other effects, because of actions on succinate and glutamate dehydrogenase (Romani 2007). Changes in [Mg2+]m were shown to occur in response to hormonal stimulation (Romani 2007) and altered metabolic state (Jung et al 1990). The process of very slow Mg2+ uptake into mitochondria is much less clearly defined than for Ca2+ uptake and is more difficult to examine because trans-membrane Mg2+ levels do not vary greatly. Mg2+ uptake was reported to be dependent on the activity of the ATP-Mg2+/Pi carrier, but this is many times lower than the activity of the adenine nucleotide transporter (ANT) (Chinopoulos et al 2009). A proposed channel, mrs2p, may be present in mitochondria to transport Mg2+ (Schindl et al 2007), but it is not believed to contribute significantly to Mg2+ transport (Chinopoulos et al 2009). Regardless, other reports (Brierley et al 1987, Jung et al 1990), like ours, show that Mg2+ flux through the IMM is very slow or negligible, at least at the physiological concentrations we used. In fact, in another study (Rodriguez-Zavala et al 1998) where the effects of changes in [Mg2+]m were observed, the ionophore A23187 was used to equilibrate trans-membrane Mg2+.
From historical studies we expected that adding MgCl2 would increase [Mg2+]m during the course of the experiments (Fig. 2). One report (Jung et al 1990) indicated that adding MgCl2 increased [Mg2+]m and that adding ADP resulted in an even higher [Mg2+]m; it was suggested that enhanced metabolism (state 3 respiration) was associated with Mg2+ uptake. However, it was reported later (Chinopoulos et al 2009) that added Mg2+ did not appreciably enter the matrix, except in the presence of ADP/ATP exchange; this group showed that when increasing amounts of ADP3− were exchanged with ATP4− via ANT, [Mg2+]m decreased; conversely, an increase in [Mg2+]m with added MgCl2 caused a decrease in ADP/ATP exchange. However, our results support the notion that an increase in [Mg2+]e, rather than an increase in [Mg2+]m, indirectly attenuates electrogenic transport by the ADP/ATP carrier ANT (Gropp et al 1999, Klingenberg 2008) to retard mitochondrial respiration. Our preliminary work (see Fig. S3 of Supplementary Materials) supports an earlier finding (Kramer 1980) that extra-matrix Mg2+ attenuated state 3 ADP/ATP exchange rate mediated by the ANT per se; it is likely that this effect results from Mg2+ binding to nucleotides, which reduces the amount of free ADP3− for exchange with free ATP4− by ANT.
Thus, our results in part contradict the two prior reports (Chinopoulos et al 2009, Jung et al 1990) that adding either MgCl2 or ADP causes an acute increase in [Mg2+]m. In our study, adding MgCl2 did not increase [Mg2+]m from its baseline value over 10 min, irrespective of Ca2+ uptake; moreover, adding ADP neither decreased [Mg2+]e nor increased [Mg2+]m. First, the Mg2+–induced reduction of CU-mediated Ca2+ uptake occurred only at the extra-matrix side of the CU. Second, it follows that the apparent attenuating (state 3) and enhancing (state 4) effects of Mg2+ on mitochondrial bioenergetics in our study was not exerted directly on matrix enzymes. In studies by Rodriguez-Zavala (Rodriguez-Zavala et al 1998) and Panov (Panov et al 1996), a higher [Mg2+]m was associated with faster oxidative phosphorylation. In contrast, [Mg2+]m did not change in our study, but rather an increase in [Mg2+]e attenuated state 3 and accentuated state 4 at any given, constant [Ca2+]m (Fig. 9).
External Mg2+ may alter non-CU Ca2+ binding sites to modulate bioenergetics
Our data show that extra-matrix Mg2+ has a concentration-dependent effect to decrease state 3 respiration independently of induced changes in [Ca2+]m. We suggest that one mechanism is an indirect effect of extra-matrix Mg2+, rather than matrix Mg2+ (Chinopoulos et al 2009), to decrease ATP generation by attenuating ADP/ATP transport. Several groups (Gellerich et al 2010, Satrustegui et al 2007) have reviewed possible mechanisms by which [Ca2+]e (vs. [Ca2+]m) might modulate bioenergetics. For example, extra-matrix Ca2+ may regulate glutamate-dependent state 3 respiration by modifying the supply of glutamate via aralar, a glutamate-aspartate carrier (Pardo et al 2006), thereby affecting oxidative phosphorylation indirectly via a change in extra-matrix Ca2+. Ca2+ activation of aralar occurs by increasing Vmax rather than by decreasing Km (Contreras et al 2007). Moreover, some enzymes, like the ATP-Mg2+/Pi carrier, have EF-hand Ca2+-binding motifs that are localized in the IMS where they sense extra-matrix [Ca2+]e (Satrustegui et al 2007). The ATP-Mg2+/Pi carrier can slowly increase or decrease the matrix content of adenine nucleotides in an extra-matrix Ca2+–dependent manner (Traba et al 2008); but this is likely too slow to occur in our studies. These reports and others indicate that extra-matrix Ca2+ may alter oxidative phosphorylation via Ca2+–sensing sites on several enzymes located in the IMS in a manner that is independent of the bioenergetics effects resulting from a change in [Ca2+]m (Gellerich et al 2010).
We also observed that extra-matrix Mg2+ decreased state 4 respiration independently of induced changes in [Ca2+]m. In preliminary experiments (see Fig. S4 of Supplementary Materials) we observed that the MgCl2-induced increase in state 4 respiration was reversed by the F1Fo ATPsynthase(ase) inhibitor oligomycin. State 4 is generally faster than state 2 in part because of the presence of ATPase in the buffer that converts the generated ATP to ADP as substrate for the F1Fo ATPsynthase, thus inducing an inward proton leak at a constant ΔΨm. Because Mg2+ is required for ATPase activity (Pedersen et al 1987), the increase in extra-matrix Mg2+ by adding MgCl2 may enhance the amount of Mg2+-ATP available for hydrolysis so that more ADP becomes available. In light of our study, how extra-matrix [Mg2+] retards state 3 respiration and enhances state 4 respiration by an effect on mitochondrial Ca2+–binding proteins, Mg2+-inhibited nucleotide transport, or Mg2+-requiring ATPases, requires more investigation.
Summary and conclusions
A small increase in [Ca2+]m, but limited only to the low nM range, stimulates mitochondrial bioenergetics, most likely as part of a larger regulatory system to adapt ATP production to workload and ATP consumption. However, under physiological conditions, this role of matrix Ca2+ might be more sensitive, but weaker, and the role of cytosolic Mg2+ stronger, but fixed, than previously believed. More work is needed to exactly define under which conditions this extra-matrix Mg2+regulatory system plays a role. However, Mg2+ appears to be limited in its capacity to regulate matrix Ca2+ uptake via the CU, which itself should reduce bioenergetic activity, because its maximal effect already occurs at a low [Mg2+]e. Modulation of respiratory activity by [Mg2+]e, at a given [Ca2+]m, may be mediated alternatively by Mg2+ to compete with Ca2+ to attenuate ADP/ATP transport (state 3) to enhance ATPase activity (state 4), or to compete at one or more extra-matrix Ca2+-sensitive sites. Finally, attenuation of Ca2+ uptake by physiological elevations of [Mg2+]e renders the Ca2+-Mg2+ interrelationship more complex and hence, incorporation of our collected data into computational models should yield clearer insights into how extra-matrix Mg2+ modulates [Ca2+]m as well as bioenergetic activity that is both independent and dependent on [Ca2+]m.
Supplementary Material
Acknowledgments
This work was supported by grants from the National Institutes of Health (5R01-HL095122-03, 5R01-HL089514-03) and Veterans Administration (Merit Review 8204-05P).
Footnotes
Disclosures
No conflicts of interest, financial or otherwise, are declared by the authors.
References
- Abrahams JP, Leslie AG, Lutter R, Walker JE. Structure at 2.8 A resolution of F1-ATPase from bovine heart mitochondria. Nature. 1994;370(6491):621–628. doi: 10.1038/370621a0. [DOI] [PubMed] [Google Scholar]
- Agarwal B, Camara AK, Stowe DF, Bosnjak ZJ, Dash RK. Enhanced charge-independent mitochondrial free Ca2+ and attenuated ADP-induced NADH oxidation by isoflurane: Implications for cardioprotection. Biochim Biophys Acta. 2012;1817(3):453–465. doi: 10.1016/j.bbabio.2011.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aldakkak M, Camara AK, Heisner JS, Yang M, Stowe DF. Ranolazine reduces Ca2+ overload and oxidative stress and improves mitochondrial integrity to protect against ischemia reperfusion injury in isolated hearts. Pharmacol Res. 2011;64(4):381–392. doi: 10.1016/j.phrs.2011.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balaban RS. The role of Ca2+ signaling in the coordination of mitochondrial ATP production with cardiac work. Biochim Biophys Acta. 2009;1787(11):1334–1341. doi: 10.1016/j.bbabio.2009.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bataille N, Schmitt N, Aumercier-Maes P, Ollivier B, Lucas-Heron B, Lestienne P. Molecular cloning of human calmitine, a mitochondrial calcium binding protein, reveals identity with calsequestrine. Biochem Biophys Res Commun. 1994;203(3):1477–1482. doi: 10.1006/bbrc.1994.2351. [DOI] [PubMed] [Google Scholar]
- Baughman JM, Perocchi F, Girgis HS, Plovanich M, Belcher-Timme CA, Sancak Y, Bao XR, Strittmatter L, Goldberger O, Bogorad RL, Koteliansky V, Mootha VK. Integrative genomics identifies MCU as an essential component of the mitochondrial calcium uniporter. Nature. 2011;476(7360):341–345. doi: 10.1038/nature10234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bazil JN, Dash RK. A minimal model for the mitochondrial rapid mode of Ca2+ uptake mechanism. PLoS One. 2011;6(6):e21324. doi: 10.1371/journal.pone.0021324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beard DA. Modeling of oxygen transport and cellular energetics explains observations on in vivo cardiac energy metabolism. PLoS Comput Biol. 2006;2(9):e107. doi: 10.1371/journal.pcbi.0020107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernardi P. Mitochondrial transport of cations: channels, exchangers, and permeability transition. Physiol Rev. 1999;79(4):1127–1155. doi: 10.1152/physrev.1999.79.4.1127. [DOI] [PubMed] [Google Scholar]
- Bernardi P, Veronese P, Petronilli V. Modulation of the mitochondrial cyclosporin A-sensitive permeability transition pore. I. Evidence for two separate Me2+ binding sites with opposing effects on the pore open probability. J Biol Chem. 1993;268(2):1005–1010. [PubMed] [Google Scholar]
- Blomeyer CA, Bazil JN, Stowe DF, Pradhan RK, Dash RK, Camara AK. Dynamic buffering of mitochondrial Ca2+ during Ca2+ uptake and Na+ -induced Ca2+ release. J Bioenerg Biomembr. 2012 doi: 10.1007/s10863-012-9483-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- Bragadin M, Pozzan T, Azzone GF. Activation energies and enthalpies during Ca2+ transport in rat liver mitochondria. FEBS Lett. 1979;104(2):347–351. doi: 10.1016/0014-5793(79)80849-2. [DOI] [PubMed] [Google Scholar]
- Brandes R, Bers DM. Increased work in cardiac trabeculae causes decreased mitochondrial NADH fluorescence followed by slow recovery. Biophys J. 1996;71(2):1024–1035. doi: 10.1016/S0006-3495(96)79303-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brierley GP, Davis M, Jung DW. Respiration-dependent uptake and extrusion of Mg2+ by isolated heart mitochondria. Arch Biochem Biophys. 1987;253(2):322–332. doi: 10.1016/0003-9861(87)90185-8. [DOI] [PubMed] [Google Scholar]
- Buntinas L, Gunter KK, Sparagna GC, Gunter TE. The rapid mode of calcium uptake into heart mitochondria (RaM): comparison to RaM in liver mitochondria. Biochim Biophys Acta. 2001;1504(2–3):248–261. doi: 10.1016/s0005-2728(00)00254-1. [DOI] [PubMed] [Google Scholar]
- Camara AK, Aldakkak M, Heisner JS, Rhodes SS, Riess ML, An J, Heinen A, Stowe DF. ROS scavenging before 27°C ischemia protects hearts and reduces mitochondrial ROS, Ca2+ overload, and changes in redox state. Am J Physiol Cell Physiol. 2007;292(6):C2021–2031. doi: 10.1152/ajpcell.00231.2006. [DOI] [PubMed] [Google Scholar]
- Camara AK, Lesnefsky EJ, Stowe DF. Potential therapeutic benefits of strategies directed to mitochondria. Antioxid Redox Signal. 2010;13(3):279–347. doi: 10.1089/ars.2009.2788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chinopoulos C, Vajda S, Csanady L, Mandi M, Mathe K, Adam-Vizi V. A novel kinetic assay of mitochondrial ATP-ADP exchange rate mediated by the ANT. Biophys J. 2009;96(6):2490–2504. doi: 10.1016/j.bpj.2008.12.3915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Contreras L, Gomez-Puertas P, Iijima M, Kobayashi K, Saheki T, Satrustegui J. Ca2+ activation kinetics of the two aspartate-glutamate mitochondrial carriers, aralar and citrin: role in the heart malate-aspartate NADH shuttle. J Biol Chem. 2007;282(10):7098–7106. doi: 10.1074/jbc.M610491200. [DOI] [PubMed] [Google Scholar]
- Crompton M, Capano M, Carafoli E. Respiration-dependent efflux of magnesium ions from heart mitochondria. Biochem J. 1976;154(3):735–742. doi: 10.1042/bj1540735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crompton M, Sigel E, Salzmann M, Carafoli E. A Kinetic study of the energy-linked Influx of Ca2+ into heart mitochondria. Eur J Biochem. 1976;69(2):429–434. [Google Scholar]
- De Stefani D, Raffaello A, Teardo E, Szabo I, Rizzuto R. A forty-kilodalton protein of the inner membrane is the mitochondrial calcium uniporter. Nature. 2011;476(7360):336–340. doi: 10.1038/nature10230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denton RM. Regulation of mitochondrial dehydrogenases by calcium ions. Biochim Biophys Acta. 2009;1787(11):1309–1316. doi: 10.1016/j.bbabio.2009.01.005. [DOI] [PubMed] [Google Scholar]
- Denton RM, McCormack JG, Edgell NJ. Role of calcium ions in the regulation of intramitochondrial metabolism. Effects of Na+, Mg2+ and ruthenium red on the Ca2+-stimulated oxidation of oxoglutarate and on pyruvate dehydrogenase activity in intact rat heart mitochondria. Biochem J. 1980;190(1):107–117. doi: 10.1042/bj1900107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duchen MR. Mitochondria and calcium: from cell signalling to cell death. J Physiol. 2000;529(Pt 1):57–68. doi: 10.1111/j.1469-7793.2000.00057.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Favaron M, Bernardi P. Tissue-specific modulation of the mitochondrial calcium uniporter by magnesium ions. FEBS Lett. 1985;183(2):260–264. doi: 10.1016/0014-5793(85)80789-4. [DOI] [PubMed] [Google Scholar]
- Gellerich FN, Gizatullina Z, Trumbeckaite S, Nguyen HP, Pallas T, Arandarcikaite O, Vielhaber S, Seppet E, Striggow F. The regulation of OXPHOS by extramitochondrial calcium. Biochim Biophys Acta. 2010;1797(6–7):1018–1027. doi: 10.1016/j.bbabio.2010.02.005. [DOI] [PubMed] [Google Scholar]
- Griffiths EJ, Rutter GA. Mitochondrial calcium as a key regulator of mitochondrial ATP production in mammalian cells. Biochim Biophys Acta. 2009;1787(11):1324–1333. doi: 10.1016/j.bbabio.2009.01.019. [DOI] [PubMed] [Google Scholar]
- Gropp T, Brustovetsky N, Klingenberg M, Muller V, Fendler K, Bamberg E. Kinetics of electrogenic transport by the ADP/ATP carrier. Biophys J. 1999;77(2):714–726. doi: 10.1016/S0006-3495(99)76926-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grynkiewicz G, Poenie M, Tsien RY. A new generation of Ca2+ indicators with greatly improved fluorescence properties. J Biol Chem. 1985;260(6):3440–3450. [PubMed] [Google Scholar]
- Haumann J, Dash RK, Stowe DF, Boelens AD, Beard DA, Camara AK. Mitochondrial free [Ca2+] increases during ATP/ADP antiport and ADP phosphorylation: exploration of mechanisms. Biophys J. 2010;99(4):997–1006. doi: 10.1016/j.bpj.2010.04.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howarth FC, Singh J, Waring JJ, Hustler BI, Bailey M. Effects of monovalent cations, pH and temperature on the dissociation constant (KD) for the fluorescent indicator mag-fura-2 at different excitation wavelengths. Magnes Res. 1995;8(4):299–306. [PubMed] [Google Scholar]
- Hubbard MJ, McHugh NJ. Mitochondrial ATP synthase F1-beta-subunit is a calcium-binding protein. FEBS Lett. 1996;391(3):323–329. doi: 10.1016/0014-5793(96)00767-3. [DOI] [PubMed] [Google Scholar]
- Jung DW, Apel L, Brierley GP. Matrix free Mg2+ changes with metabolic state in isolated heart mitochondria. Biochemistry. 1990;29(17):4121–4128. doi: 10.1021/bi00469a015. [DOI] [PubMed] [Google Scholar]
- Katz LA, Swain JA, Portman MA, Balaban RS. Relation between phosphate metabolites and oxygen consumption of heart in vivo. Am J Physiol. 1989;256(1 Pt 2):H265–274. doi: 10.1152/ajpheart.1989.256.1.H265. [DOI] [PubMed] [Google Scholar]
- Kirichok Y, Krapivinsky G, Clapham DE. The mitochondrial calcium uniporter is a highly selective ion channel. Nature. 2004;427(6972):360–364. doi: 10.1038/nature02246. [DOI] [PubMed] [Google Scholar]
- Klingenberg M. The ADP and ATP transport in mitochondria and its carrier. Biochim Biophys Acta. 2008;1778(10):1978–2021. doi: 10.1016/j.bbamem.2008.04.011. [DOI] [PubMed] [Google Scholar]
- Kramer R. Influence of divalent cations on the reconstituted ADP, ATP exchange. Biochim Biophys Acta. 1980;592(3):615–620. doi: 10.1016/0005-2728(80)90104-8. [DOI] [PubMed] [Google Scholar]
- Litsky ML, Pfeiffer DR. Regulation of the mitochondrial Ca2+ uniporter by external adenine nucleotides: the uniporter behaves like a gated channel which is regulated by nucleotides and divalent cations. Biochemistry. 1997;36(23):7071–7080. doi: 10.1021/bi970180y. [DOI] [PubMed] [Google Scholar]
- Maack C, Cortassa S, Aon MA, Ganesan AN, Liu T, O’Rourke B. Elevated cytosolic Na+ decreases mitochondrial Ca2+ uptake during excitation-contraction coupling and impairs energetic adaptation in cardiac myocytes. Circ Res. 2006;99(2):172–182. doi: 10.1161/01.RES.0000232546.92777.05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCormack JG, Denton RM. Role of calcium ions in the regulation of intramitochondrial metabolism. Properties of the Ca2+-sensitive dehydrogenases within intact uncoupled mitochondria from the white and brown adipose tissue of the rat. Biochem J. 1980;190(1):95–105. doi: 10.1042/bj1900095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCormack JG, Halestrap AP, Denton RM. Role of calcium ions in regulation of mammalian intramitochondrial metabolism. Physiol Rev. 1990;70(2):391–425. doi: 10.1152/physrev.1990.70.2.391. [DOI] [PubMed] [Google Scholar]
- Morgan RO, Martin-Almedina S, Garcia M, Jhoncon-Kooyip J, Fernandez MP. Deciphering function and mechanism of calcium-binding proteins from their evolutionary imprints. Biochim Biophys Acta. 2006;1763(11):1238–1249. doi: 10.1016/j.bbamcr.2006.09.028. [DOI] [PubMed] [Google Scholar]
- Panov A, Scarpa A. Independent modulation of the activity of alpha-ketoglutarate dehydrogenase complex by Ca2+ and Mg2+ Biochemistry. 1996;35(2):427–432. doi: 10.1021/bi952101t. [DOI] [PubMed] [Google Scholar]
- Panov A, Scarpa A. Mg2+ control of respiration in isolated rat liver mitochondria. Biochemistry. 1996;35(39):12849–12856. doi: 10.1021/bi960139f. [DOI] [PubMed] [Google Scholar]
- Pardo B, Contreras L, Serrano A, Ramos M, Kobayashi K, Iijima M, Saheki T, Satrustegui J. Essential role of aralar in the transduction of small Ca2+ signals to neuronal mitochondria. J Biol Chem. 2006;281(2):1039–1047. doi: 10.1074/jbc.M507270200. [DOI] [PubMed] [Google Scholar]
- Pedersen PL, Williams N, Hullihen J. Mitochondrial ATP synthase: dramatic Mg2+-induced alterations in the structure and function of the F1-ATPase moiety. Biochemistry. 1987;26(26):8631–8637. doi: 10.1021/bi00400a021. [DOI] [PubMed] [Google Scholar]
- Perocchi F, Gohil VM, Girgis HS, Bao XR, McCombs JE, Palmer AE, Mootha VK. MICU1 encodes a mitochondrial EF hand protein required for Ca2+ uptake. Nature. 2010;467(7313):291–296. doi: 10.1038/nature09358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pradhan RK, Qi F, Beard DA, Dash RK. Characterization of Mg2+ inhibition of mitochondrial Ca2+ uptake by a mechanistic model of mitochondrial Ca2+ uniporter. Biophys J. 2011;101(9):2071–2081. doi: 10.1016/j.bpj.2011.09.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhodes SS, Camara AK, Heisner JS, Riess ML, Aldakkak M, Stowe DF. Reduced mitochondrial Ca2+ loading and improved functional recovery after ischemia-reperfusion injury in old vs. young guinea pig hearts. Am J Physiol Heart Circ Physiol. 2012;302(3):H855–863. doi: 10.1152/ajpheart.00533.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhodes SS, Ropella KM, Camara AK, Chen Q, Riess ML, Pagel PS, Stowe DF. Ischemia-reperfusion injury changes the dynamics of Ca2+-contraction coupling due to inotropic drugs in isolated hearts. J Appl Physiol. 2006;100(3):940–950. doi: 10.1152/japplphysiol.00285.2005. [DOI] [PubMed] [Google Scholar]
- Riess ML, Camara AK, Novalija E, Chen Q, Rhodes SS, Stowe DF. Anesthetic preconditioning attenuates mitochondrial Ca2+ overload during ischemia in Guinea pig intact hearts: reversal by 5-hydroxydecanoic acid. Anesth Analg. 2002;95(6):1540–1546. doi: 10.1097/00000539-200212000-00013. table of contents. [DOI] [PubMed] [Google Scholar]
- Riess ML, Kevin LG, McCormick J, Jiang MT, Rhodes SS, Stowe DF. Anesthetic preconditioning: the role of free radicals in sevoflurane-induced attenuation of mitochondrial electron transport in Guinea pig isolated hearts. Anesth Analg. 2005;100(1):46–53. doi: 10.1213/01.ANE.0000139346.76784.72. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Zavala JS, Moreno-Sanchez R. Modulation of oxidative phosphorylation by Mg2+ in rat heart mitochondria. J Biol Chem. 1998;273(14):7850–7855. doi: 10.1074/jbc.273.14.7850. [DOI] [PubMed] [Google Scholar]
- Romani A. Regulation of magnesium homeostasis and transport in mammalian cells. Arch Biochem Biophys. 2007;458(1):90–102. doi: 10.1016/j.abb.2006.07.012. [DOI] [PubMed] [Google Scholar]
- Romani A, Scarpa A. Hormonal control of Mg2+ transport in the heart. Nature. 1990;346(6287):841–844. doi: 10.1038/346841a0. [DOI] [PubMed] [Google Scholar]
- Rotevatn S, Murphy E, Levy LA, Raju B, Lieberman M, London RE. Cytosolic free magnesium concentration in cultured chick heart cells. Am J Physiol. 1989;257(1 Pt 1):C141–146. doi: 10.1152/ajpcell.1989.257.1.C141. [DOI] [PubMed] [Google Scholar]
- Satrustegui J, Pardo B, Del Arco A. Mitochondrial transporters as novel targets for intracellular calcium signaling. Physiol Rev. 2007;87(1):29–67. doi: 10.1152/physrev.00005.2006. [DOI] [PubMed] [Google Scholar]
- Scaduto RC, Jr, Grotyohann LW. Measurement of mitochondrial membrane potential using fluorescent rhodamine derivatives. Biophys J. 1999;76(1 Pt 1):469–477. doi: 10.1016/S0006-3495(99)77214-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scarpa A, Graziotti P. Mechanisms for intracellular calcium regulation in heart. I. Stopped-flow measurements of Ca2+ uptake by cardiac mitochondria. J Gen Physiol. 1973;62(6):756–772. doi: 10.1085/jgp.62.6.756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindl R, Weghuber J, Romanin C, Schweyen RJ. Mrs2p forms a high conductance Mg2+ selective channel in mitochondria. Biophys J. 2007;93(11):3872–3883. doi: 10.1529/biophysj.107.112318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sparagna GC, Gunter KK, Sheu SS, Gunter TE. Mitochondrial calcium uptake from physiological-type pulses of calcium. A description of the rapid uptake mode. J Biol Chem. 1995;270(46):27510–27515. doi: 10.1074/jbc.270.46.27510. [DOI] [PubMed] [Google Scholar]
- Starkov AA. The molecular identity of the mitochondrial Ca2+ sequestration system. FEBS J. 2010;277(18):3652–3663. doi: 10.1111/j.1742-4658.2010.07756.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szanda G, Rajki A, Gallego-Sandin S, Garcia-Sancho J, Spat A. Effect of cytosolic Mg2+ on mitochondrial Ca2+ signaling. Pflugers Arch. 2009;457(4):941–954. doi: 10.1007/s00424-008-0551-0. [DOI] [PubMed] [Google Scholar]
- Territo PR, French SA, Dunleavy MC, Evans FJ, Balaban RS. Calcium activation of heart mitochondrial oxidative phosphorylation: rapid kinetics of mVO2, NADH, and light scattering. J Biol Chem. 2001;276(4):2586–2599. doi: 10.1074/jbc.M002923200. [DOI] [PubMed] [Google Scholar]
- Traba J, Froschauer EM, Wiesenberger G, Satrustegui J, Del Arco A. Yeast mitochondria import ATP through the calcium-dependent ATP-Mg/Pi carrier Sal1p, and are ATP consumers during aerobic growth in glucose. Mol Microbiol. 2008;69(3):570–585. doi: 10.1111/j.1365-2958.2008.06300.x. [DOI] [PubMed] [Google Scholar]
- Vinnakota KC, Dash RK, Beard DA. Stimulatory effects of calcium on respiration and NAD(P)H synthesis in intact rat heart mitochondria utilizing physiological substrates cannot explain respiratory control in vivo. J Biol Chem. 2011;286(35):30816–30822. doi: 10.1074/jbc.M111.242529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vinogradov A, Scarpa A. The initial velocities of calcium uptake by rat liver mitochondria. J Biol Chem. 1973;248(15):5527–5531. [PubMed] [Google Scholar]
- Wan B, LaNoue KF, Cheung JY, Scaduto RC., Jr Regulation of citric acid cycle by calcium. J Biol Chem. 1989;264(23):13430–13439. [PubMed] [Google Scholar]
- Wei AC, Liu T, Winslow RL, O’Rourke B. Dynamics of matrix-free Ca2+ in cardiac mitochondria: two components of Ca2+ uptake and role of phosphate buffering. J Gen Physiol. 2012;139(6):465–478. doi: 10.1085/jgp.201210784. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.