

Abstract
Background
Thyroid hormone influences gene expression in virtually all vertebrates. Its action is initiated by the activation of T4 to T3, an outer ring deiodination reaction that is catalyzed by the type 1 or the type 2 iodothyronine selenodeiodinases (D1 or D2). Inactivation of T4 and T3 occurs via inner ring deiodination catalyzed by the type 3 iodothyronine selenodeiodinases (D3). The T4 concentration is generally quite stable in human plasma, with T3 levels also remaining constant. Deiodinase actions are tightly regulated in both pre- and post-natal life when they are required to make local adjustments of intracellular T3 concentrations in a precise spatio- and temporal manner. Although all the signals governing the dynamic expression of deiodinases in specific cell types are not known, many important regulatory factors have been deciphered.
Scope of review
This review provides striking examples from the recent literature illustrating how the expression of D2 and D3 is finely tuned during maturation of different organs, and how their action play a critical role in different settings to control intracellular T3 availability.
Major conclusions
Emerging evidence indicates that in various cell contexts, D2 and D3 are expressed in a dynamic balance, in which the expression of one enzyme is coordinately regulated with that of the other to tightly control intracellular T3 levels commensurate with cell requirements at that time.
General significance
Deiodinases control TH action in a precise spatio-temporal fashion thereby providing a novel mechanism for the local paracrine and autocrine regulation of TH action. This remarkable tissue-specific regulation of intracellular thyroid status remains hidden due to the maintenance of constant circulating TH concentrations by the hypothalamic–pituitary–thyroid axis. This article is part of a Special Issue entitled Thyroid hormone signalling.
Abbreviations: ABR, auditory-evoked brainstem potentials; D1, Type 1 seleno deiodinase; D2, Type 2 seleno deiodinase; D3, Type 3 seleno deiodinase; Mpc, muscle precursor cell; PTU, propyl-thio-uracil; TH, thyroid hormone; TR, thyroid hormone receptor; Shh, Sonic hedgehog
Keywords: Thyroid hormone, Deiodinase, Cellular differentiation
Highlights
► Deiodinases contribute to the control of TH availability in different cell types. ► Deiodinase actions are part of the TH-mediated control of differentiation. ► Deiodinases are critical in cochlear and retinal maturation in pre/post-natal life. ► In muscle cells, deiodinases are required for myogenesis and muscle regeneration.
1. Introduction
Thyroid hormone (TH) is important for the normal development of many tissues, and for the regulation of their function throughout life. Thyroid hormone can enter the cells via membrane transporters, and, while diffusing from the plasma membrane to the nucleus, its concentration can be modified by the action of the deiodinases. Many genes have been identified whose expression is under the positive or negative control of TH. Although TH-dependent processes are coordinated, they are not necessarily synchronized between tissues, and the proper function of the organism requires tissue-specific changes in TH action. These varying local requirements cannot be governed by global mechanisms such as an alteration of thyroid gland function or by modulation of hormone concentrations at plasma level, but instead require tissue-specific regulation. The tissue-specific-expression of deiodinases is gaining increased recognition in the field of TH action as a potent mechanism by which T3 levels can be controlled at the local level with D2 converting the prohormone T4 to the active hormone T3, while D3 converts T4 and T3 to inactive metabolites [1].
The iodothyronine deiodinase family of selenoproteins consists of three enzymes (D1, 2, and 3; or DIO1, 2, and 3) differentially expressed in several adult and fetal tissues.
All deiodinases are membrane-anchored proteins of 29–33 kDa that share substantial sequence homology, catalytic properties and contain selenocysteine (Sec) as the key residue within their catalytic centers. The unique feature of selenoproteins in general and deiodinases lies in the recoding of the UGA codon from a stop codon to Sec-insertion codon by the presence of the SECIS element in the 3′ UTR of the respective mRNAs. Iodinated contrast agents such as iopanoic acid inhibit all three deiodinases, while propylthiouracil is a relatively specific inhibitor for D1.
D1 is primarily expressed in the liver, the kidney and the thyroid. It is able to deiodinate both the inner and the outer ring of T4, and is essential to provide T3 for the circulation as well as serving as a scavenger enzyme for iodide in peripheral tissues [2].
D2 is localized in the endoplasmic reticulum and its action is directed almost exclusively at the efficient conversion of the prohormone T4 to the active T3 by 5′-deiodination. It is negatively regulated by thyroid hormone both pre- and posttranscriptionally [3]. Thus, in hypothyroidism, D2 activity is markedly up-regulated [4,5], and this increases the proportion of T3 formed locally in D2-expressing tissues.
D3 is localized in the plasma membrane and is considered as the major thyroid hormone-inactivating enzyme. It inactivates T4 and T3 by converting them to the inactive iodothyronines (rT3 and 3,3′-diiodothyronine, T2). D3 expression is decreased in hypothyroidism and increased in the thyrotoxic state [2].
Here we provide examples illustrating the importance of the deiodinases in the tissue-specific modulation of TH action that highlights the importance of thyroid hormone receptor ligand availability in controlling maturation and cellular proliferation/differentiation within different organs.
2. Deiodinases and the proliferation/differentiation balance
2.1. Deiodinases in skin
Skin, a major target for TH, is a large tissue where the intracellular TH concentration is modulated by a balance between the activating (D2) and inactivating (D3) selenodeiodinases. In skin, TH exerts profound effects on fetal epidermal differentiation, barrier formation, hair growth, sebum production, wound healing, epidermal oxygen consumption, keratinocyte proliferation, and keratin gene expression [6–12]. As a result, thyroid dysfunction is correlated with alterations in skin architecture and homeostasis [10,13] and changes in circulating TH levels, particularly hypothyroidism, are often associated with an edema-like-associated skin condition caused by increased glycosaminoglycan deposition. Thyroid hormone receptor expression has been found in different skin cell types, including keratinocytes, dermal fibroblasts, and sebaceous gland cells, and both D2 and D3 activities have also been detected in human skin [14–17].
The effects of TH on keratinocyte proliferation and epidermal differentiation are controversial, with divergent results [8]. Safer's group has found that systemic and topical TH administration cause diverging effects on keratinocyte proliferation. While topical T3 stimulates epidermal proliferation, hair growth and dermal thickening [18–20], systemic thyrotoxicosis leads to thinning of skin, hair and collagen loss [19,21]. The authors postulate that the different effects exerted by topical versus systemic TH treatment on epidermal cell can be explained by the T3-induced production of inhibitory factor(s) from fibroblasts with systemic T3-treatment.
In keeping with the ability to modulate growth and differentiation, it is not surprising that TH might play a critical role in skin tumorigenesis. TH treatment decreases the proliferation and cyclin D1 expression in transformed keratinocytes in a dose and time-dependent fashion [17]. In human and mouse basal cell carcinoma (BCC), a keratinocyte-derived tumor that is the most commonly diagnosed human cancer, D3 is overexpressed and lies downstream of the Sonic hedgehog (Shh) pathway [17]. Inappropriate activation of the Shh–Gli signaling pathway occurs in up to 25% of human tumors, and constitutively activated Shh in skin is the causal lesion and the molecular hallmark of BCCs. In primary keratinocytes, as well as in BCCs, Shh increases the expression of D3, acting via a conserved Gli2 binding site on the Dio3 promoter [17]. Indeed, D3, the main TH inactivating enzyme, is often referred to as an oncofetal protein because its expression is very low in most adult tissues, but increased in several human cancers and in human solid tumors. Inhibition of D3 expression diminishes cell proliferation by increasing nuclear T3 action [17]. The Shh-mediated induction of D3 strongly suggests the existence of a novel cross-talk in which Shh induces local attenuation of the TH signal, thus contributing to enhanced cell proliferation. Hh control of TH signaling has also been found to play an important role in the tibial growth plate, in which Indian Hedgehog induces the expression of WSB-1 and thus targets D2 for proteasomal degradation — a process that results in a local decrease in T3 levels and increased chondrocyte proliferation [22]. Thus, the functional interplay between the Hh pathways and TH metabolism identified in the tibial growth plate and BCCs [17,22] may have a much broader significance and relevance to other tissues or pathological conditions.
2.2. Deiodinases are critical for the development of normal auditory function
Hearing impairment has long been recognized as a clinical consequence of congenital hypothyroidism or severe iodine deficiency, in humans and various rodent models [23–25]. More recent studies indicated that inactivation of the gene encoding the beta isoform of the thyroid hormone receptor TRβ (Thrb) resulted in hearing deficiency [26,27], as demonstrated by defective auditory-evoked brainstem potentials (ABR), thereby indicating that normal auditory development required this isoform of the thyroid hormone receptor. Impaired hearing has also been described in patients affected by the syndrome of thyroid hormone resistance and bearing mutations in the Thrb gene [28]. Studies in knockout mice models further indicate that while the TRα1 isoform is not an absolute requirement for normal auditory development, there is a synergism between the α and β thyroid receptor subtypes for both normal function and the anatomical development of structures within the cochlea. Differential effects were further observed with TRβ versus TRα1 deletions, which indicate that the two receptors also exert cell-specific effects [29].
Early studies in the rat showed that monodeiodination of T4 by a propylthiouracil-insensitive deiodinase, D2, was responsible for approximately 80% of the T3 present in the cerebral cortex [30–32]. In addition, the T3 and T4-inactivating D3 was also found to be highly expressed, predominantly in neurons, and positively regulated by T3 [33–35]. The D2 mRNA in the cortex was reported to be negatively regulated by T4 and T3 at both transcriptional (T3) and post-translational levels, which suggests that these two deiodinases act in concert to facilitate T3 homeostasis in the central nervous system structures [33–35]. A similar process has also been found to occur during iodine deficiency [36].
The above observations have led to a series of studies to define the role played by these two deiodinases in the development and function of the cochlea, and to determine how they correlate with the expression of the TH receptors and transporters. These studies have revealed that the iodothyronine deiodinases play an essential role in controlling cochlear development. The following discussion focuses on the qualitative and quantitative aspects of D2 and D3 expression during the development of this complex organ. The interested reader is referred to the original articles referenced herein for details of the structural basis underlying the disruption of auditory function when the actions of these enzymes are blocked.
Initial studies determined that D2 activity in mouse cochlear tissue increases abruptly starting at P4, peaks at P7 at levels about 10-times the initial values, and returns to basal levels by P10–12 (Fig. 1). This occurs before the initiation of hearing, as monitored by ABR. Of note, the peak of D2 activity precedes the peak of circulating T4 and T3 in the neonatal mouse at P16. The increase in D2 activity is accompanied by an approximately 4- to 5-fold increase in the T3 content of the cochlea, as well as a 3-fold increase in cochlear T4 concentrations. This results in a higher T3/T4 ratio in this tissue than in the serum, indicating that these changes reflect both the increase in circulating thyroid hormones and an increase in outer ring monodeiodination of T4 by D2 since D1 is not found in the cochlea. In addition, in situ hybridization studies indicated that the expression of D2 was not in the sensory structures of the organ but predominantly in the supporting connective tissue [37]. Furthermore, D2 expression did not parallel that of either TRβ or TRα. These data suggested that specific TH-independent signals in the connective tissue of the cochlea were responsible for the marked increase in the local concentration of T3 generated by D2.
Fig. 1.

Postnatal D2 enzymatic activity in the cochlea. D2 activity is high in the early phases of postnatal cochlea maturation; it peaks at P7 and returns to basal levels by P10. Interestingly, the high D2 expression precedes the rise in cochlear T3 and T4, which indicates that local 5′ deiodination plays a critical role in the regulation of the intracellular T3 concentration.
Figure modified from [37].
The importance of D2 in this system was confirmed by data obtained in mice in which the Dio2 gene was inactivated [38]. Despite the fact that the circulating T4 concentration in the D2KO mouse is slightly increased and serum T3 concentrations are normal, impaired ABR and anatomical abnormalities occurred in these animals. Studies of the ABR of D2KO mice indicated that the absence of D2 resulted in auditory dysfunction comparable to that found in hypothyroid mice. This finding confirmed that local cochlear T3 production plays a critical role in normal development. It was also found that a decrease in D3 did not occur to compensate for the deficiency of cochlear T3 in the absence of D2. The functional abnormality in D2KO hearing was accompanied by delayed differentiation of the sensory epithelium and of the tectorial membrane, a structure that is critical in cochlear function. Interestingly, the impaired hearing was rescued by supplementing maternal drinking water with high quantities of T3 by P10, but hearing did not completely normalize when T3 supplementation was delayed until P16 or later. The improvement in hearing with maternal T3 supplementation was matched by normalization of cochlear anatomy in the D2KO mice, thus indicating that the rescued function was due to a direct effect on the cochlea rather than to normalization in the extra-cochlear neurological components of the auditory system, which could also be dependent on D2. Serum concentrations were 2- to 5-fold higher in offspring of nursing mothers receiving a T3 supplement than in untreated D2KO mice, confirming that physiological concentrations of circulating T3 are insufficient to provide the amount of T3 required for normal cochlear development. This observation is similar to findings obtained in the pituitary and skeletal muscle [39–41]. The fact that auditory function could be partly rescued by T3 treatment as late as P16 is thought to be due to the relatively long time required for complete development of hearing in the mouse, which extends beyond the spike in D2 activity at P4–P8 (Fig. 1).
The increase in brain tissue T4 at P8 seen in the initial studies suggested that there might be increases in thyroid hormone transporters, which could provide additional T4 for T3 production. To verify this hypothesis, Sharlin et al. evaluated the expression of Lat1, Mct8, Mct10, and Oatp1C1. Lat1 is a high-affinity transporter of T3 and T4, Oatp1C1 transports T4, while Mct8 and 10 can transport either iodothyronine but have higher affinities for T3 [42]. There were minimal relative changes in the expression of all but the Oatp1C1 mRNA, which increased 15-fold between E18 and P3 and about 80-fold by P15 (Fig. 2). This compared with a change in Dio2 expression of 400- to 500-fold between E18 and P7 (Fig. 2). There was, in addition, an anatomical overlap of Oatp1C1 and Dio2 expression in the modiolus, osseous spiral lamina, and spiral ligament of the cochlea. While the Oatp1C1 protein has been primarily identified in microvessels, in the cochlea, this mRNA appeared to be localized to fibrocytes in close proximity to blood vessels. These results suggest that the Oatp1C1 transporter is critical because it provides the T4 substrate for D2. Mct8 was also detected in fibrocytes, although its expression was not amplified during the developmental process. Sharlin et al., also noted that the Oatp1C1 gene is localized within a large segment of chromosome 12, which contains the Dfnb62 deafness locus — a finding consistent with a potential causal relationship between a defect in T4 uptake in D2-expressing tissues and impaired hearing [42].
Fig. 2.

TH transporter levels in cochlea. (A) The expression of Lat1, Mct8, Mct10, and Oatp1C1 was evaluated in cochlea during embryonic development and early postnatal phases. All transporters are upregulated during postnatal growth and their expression parallels the expression of D2 mRNA (B) thereby providing the T4 substrate for D2-mediated deiodination. Oatp, organic anion-transporting polypeptide, Mct, monocarboxylate transporter, and Lat, L-type amino acid transporter.
Figure modified from [42].
Since it is well established that D2 and D3 expression respond in opposite ways to changes in thyroid status or to morphogenic signals such as Sonic hedgehog, the authors also evaluated the expression of D3 in cochlear structures [43]. Interestingly, levels of D3 mRNA were high in the cochlea beginning at E13, and remained constant until birth, but then fell to low levels by P8, opposite to what occurred with D2 activity. Strikingly, in the D3KO mouse, there was severe impairment of hearing as evidenced by the ABR, and immunohistochemical studies indicated that there was premature differentiation of cochlear structures [43]. Mice with a combined elimination of Thrb and Dio3 did not develop the premature maturation characteristic of the D3KO animals, which indicates that this event was due to the actions of T3. Further testing to determine whether T3 toxicity per se could replicate the effects of local D3 deficiency showed that when T3 was administered between P0 and P3, there was a dose-dependent impairment of hearing that led to severe functional deficits. Both during these studies and in the D3KO mice, serum T3 concentrations were markedly elevated above normal, especially within a few hours of injection. Interestingly, combined in situ hybridization studies showed that Dio3 and Dio2 are expressed in different structures of the cochlea, with different patterns, and that TRβ mRNA expression overlaps that of D3 but not of D2 [43].
The elegant studies summarized above provide ample evidence that the expression of the deiodinases, thyroid hormone receptors, and the T3 and T4 transporters is coordinated during the development of the cochlea and that the specific disruption of either D2 or D3 will result in an impairment of both the normal structural development and the function of this organ. The next step in these studies will be to define the endogenous signals that regulate the complex and complementary modulation of local T3 production and degradation required for normal hearing.
2.3. Deiodinase-mediated effects during retinal development
Although D2 and D3 are expressed in the retinas of the chick, Xenopus laevis, and Rana catesbeiana, D2 (and D1) are absent in the mouse retina [44–46]. However, normal retinal development requires thyroid hormone acting through TRβ2, which induces M opsin expression in cones [46,47]. Studies in Xenopus have suggested that the asymmetric growth of the retina during metamorphosis is due to D3 expression in the dorsal portions of this tissue [45]. D3 is also found in the mouse retina but there does not appear to be any difference between its expression in the dorsal and ventral regions [46]. Surprisingly, studies in the D3KO mouse showed that, in the absence of D3, 80% of cones were lost through apoptosis in the first week of neonatal life [46]. In contrast, the absence of D3 did not affect rod photoreceptors. In subsequent studies, normal neonatal mice were given graded doses of T3 to determine if exogenous T3 could overwhelm the capacity of cone D3 to protect these cells. High doses of T3 (leading to ~ 70- to 300-fold increases in circulating T3) caused cone apoptosis. As was the case in T3-induced dysregulation of the auditory system discussed above, ablation of TRβ2 expression eliminated the apoptosis of the cones induced by excessive T3 thereby indicating that this was a TR-dependent effect. Only extremely high levels of T3 resulted in cone destruction and the sensitivity was limited to the first week of neonatal life. Interestingly, in combined TRβ2/D3KO mice, the cones appeared normal but did not express M opsin.
These studies are in an early phase and it is not yet clear what the physiological ramifications might be for human retinal development. However, it is apparent that D3 has a role in modulating the T3 concentration in cones, and that thyroid hormone-dependent destruction of these cells may occur in its absence. Thus, both reduced and excessive intra cellular T3 impairs normal retinal function, which illustrates the critical role of deiodinases in the development of the visual system.
2.4. Deiodinases in skeletal myogenesis
2.4.1. Thyroid hormone and muscle physiology
Skeletal muscle is a major peripheral tissue that accounts for ~ 40% of the total body mass. It serves several purposes: movement, posture, stability, heat production, and cold tolerance. Furthermore, it has a paramount role in energy balance, and is the primary tissue of insulin-dependent glucose uptake and storage, and of regulation of circulating and stored lipid flux.
The overall contractile and metabolic feature of any single muscle depends upon the properties of its fiber types and their proportions, which are not fixed entities but dynamic structures with an extraordinary adaptive potential. Their phenotypic profiles are affected by such factors as innervation, exercise training, mechanical loading/unloading, hormones, and aging. To date, the most useful classifications of muscle fibers are based on their two major functional characteristics: speed of contraction and the aerobic (oxidative)/anaerobic (glycolytic) production of adenosine triphosphate. In general, contractile speed is slowest in type I fibers and fastest in type IIB fibers. Fast twitch fibers (also known as type IIB or white fibers) utilize anaerobic glycolysis, are low in mitochondria and myoglobin, rich in glycogen, and are suited to short-term intense activity. Slow fibers (also known as type I or red fibers) utilize aerobic metabolism, are rich in mitochondria and myoglobin, and are suited to endurance activity. Fast fiber type IIA is an intermediate fiber that combines fast twitch capacity with aerobic fatigue, resistant metabolism, and intermediate glycogen levels [48].
Skeletal muscle is one of the primary targets of thyroid hormones [49], which have a profound influence on the relative concentrations of MHC isoforms that ultimately affect the fiber type composition and fiber type sizes. Moreover, by regulating the energy consumption and heat production associated with activity, TH is the main determinant of the resting metabolic rate of the muscle fibers [48].
In general, hypothyroidism elicits fast-to-slow muscle fiber transitions [49–51], while hyperthyroidism causes shifts in the reverse direction. Accordingly, low levels of TH result in a decrease in muscle mass and promote fast-to-slow transition in MHC isoform expression, whereas high levels cause slow-to-fast shifts [52–54].
In addition to their impact on adult muscle fiber phenotypes, TH plays an important role during muscle development and maturation. Several muscle-specific genes (myogenes) are direct targets of TH. TH–TR complexes have been demonstrated to regulate genes controlling metabolic homeostasis, such as GLUT4 [55], and energy expenditure, such as UCP3 [56], and also to modulate the activity of a number of Krebs cycle enzymes [57]. TH regulates the steady-state levels of many different contractile protein mRNAs involved in fluxes of intracellular Ca2 +, affecting the rate of contraction and relaxation of muscle fibers [58]. Moreover, the gene coding for the calcium pump of the sarcoplasmic reticulum, SERCa2, is highly T3-responsive and its regulatory region contains three thyroid hormone responsive elements (TREs) [48].
Typical examples of T3-mediated control of specific myogenes are changes in critical muscle-specific transcription factors (muscle regulatory factors, MRFs), including MyoD, the master regulator of myogenesis and of the muscle regeneration process [49,50]. The MyoD promoter contains a highly conserved TRE binding site that can bind TR in vivo [41]. Furthermore, myogenin has been indicated as a TH positively regulated gene [59]. The T3-mediated regulation of the α and β MHC chains is more complex. T3 treatment stimulates the expression of the MHCβ gene but decreases the expression of the [59] MHCα gene. In hypothyroid muscles, V3 MHC predominates, thus leading to the decreased velocity of muscle contraction. In contrast, in hyperthyroid rats, myosin is exclusively composed of V1, which leads to an accelerated contraction due to the faster turnover of the myosin moving along the thin filament [60].
Visser et al. examined the effects of l-thyroxine on the human skeletal muscle transcriptome by investigating muscle gene expression from 10 thyroidectomized patients before and 4 weeks after l-thyroxine replacement [61]. They found that a large number of TH-dependent genes involved in energy and fuel metabolism were overrepresented among the up-regulated genes. Moreover, by analyzing the miRNA expression profile in the same specimens, they showed that l-thyroxine therapy induced a large down-regulation of the primary transcripts of the noncoding microRNA pair miR-206/miR-133b, and concluded that TH plays complex role in the regulation of cellular processes in human skeletal muscle by regulating a myriad of genes, as well as microRNAs.
MicroRNA action is also critical for MHC expression as recently demonstrated by van Rooij et al. [62]. They found that the cardiac-specific miR-208 (encoded by an intron of the αMHC gene) is highly sensitive to decreases in TH induced by PTU, which affect TH-dependent transcriptional activity. The mRNA encoding TR Associated Protein 1 (THRAP1) was among the strongest predicted targets of miR-208 [63]. THRAP1 is a component of the TR-associated TRAP complex and modulates TR activity by recruiting RNA polymerase II and general initiation factors [64]. By repressing the expression of the TR coregulator THRAP1, miR-208 can exert effects on TH-dependent transcription [65]. Thus, miR-208 enhances βMHC expression and decreases αMHC expression, resulting in opposite regulation of their expression by TH [65].
2.4.2. Deiodinases and muscle stem cells
“Satellite cells” are the most relevant population of muscle stem cells; they are capable of self-renewal, and are able to differentiate into myoblasts and to form new muscle fibers. These small cells are located beneath the basal lamina of the muscle fiber and are approximately the same size as a muscle cell nucleus [66]. These cells are mitotically quiescent, but can be activated in response to muscle injury, and are required for the formation of new myofibers during postnatal and adult life, and for the myofiber repair process [67,68]. Since muscle fibers are postmitotic, postnatal muscle growth and repair after injury depend on the reservoir of satellite cells.
During embryonic muscle development, as well as during muscle regeneration, satellite cells exit their normal quiescent state to enter a defined program that includes, in temporal sequence, proliferation, differentiation, and fusion to form new myofibers (Fig. 3). At molecular level, early phases of this process are characterized by upregulation of Pax7, Myf5, and MyoD expression. Following the proliferative phase of myocyte precursor cells, myogenin and MRF4 are upregulated so driving the terminal differentiation program [69], which is completed with the withdrawal from the cell cycle, p21 upregulation, and specific MHC expression. Such a highly orchestrated process requires a hierarchy of several, time-dependent regulators.
Fig. 3.

Thyroid hormone levels and myogenesis of satellite cells. In quiescence, satellite cells are a heterogeneous population of multipotent, undifferentiated cells capable of auto-renewing. The TH signal is essential for the progression into terminal differentiation, while TH downregulation is required to retain muscle progenitor cells (mpcs) in a proliferative phase. The genes identified as T3 target during myogenesis are indicated.
Thyroid hormone has long been known to affect muscle development and differentiation. More recent is the discovery of the pivotal role of TH metabolism in muscle development. Although the presence of D2 in skeletal muscle has long been recognized, its role has been questioned given the very low specific activity and some muscle tissue-specific confounding factors. These include the complex structure of muscle causing difficulties in extracting membrane-associated proteins and the presence of a high concentration of metals that ultimately lead to peroxidation and oxidation of DTT, essential for the measurement of D2 activity in vitro [70]. Therefore, it is more appropriate to specifically quantify the labeled T3 by chromatographic methods during D2 assays, thus avoiding an overestimation of D2 activity due to an artifactual release of iodide not accompanied by T3 production [71].
Recently, using a novel, very sensitive D2-extraction protocol, we identified D2 activity in mouse neonatal and adult muscles [41,72]. Interestingly, D2 is expressed in myoblast precursor cells and potently induced during differentiation. This was demonstrated in the myogenic cell line C2C12, as well as in the more physiologically relevant primary culture of muscle precursor cells, termed “pp6” cells (Fig. 4). D2 action, by increasing intracellular T3, promotes MyoD gene expression and allows terminal differentiation. The critical role played by D2 was demonstrated by the lack of proper differentiation of pp6 cells derived from Dio2−/− mice [41].
Fig. 4.

Deiodinase expression profile during myogenesis of satellite cells. D2 and D3 show opposite expression profiles during the satellite cell differentiation program. D3 expression is sustained in the early phases of proliferation, but it declines sharply before differentiation. Conversely, D2 acts mainly in the differentiative stages, driving the TH-dependent up-regulation of several myogenic proteins.
By measuring enzymatic activity and mRNA levels, we found that the expression of the T4-activating enzyme D2 in skeletal muscle differs depending on muscle fiber composition. Although D2 activity was detectable in all hind limb muscles examined, it was higher in the oxidative, type-I, slow twitch than in type II fast-twitch muscles [72]. The role of D2 in skeletal muscle is underscored by the fact that animals lacking D2 expression (Dio2−/−) show a mildly hypothyroid pattern of muscle gene expression [41]. The consequences of this in terms of muscle physiology and function are currently under investigation.
Intracellular T3 concentration also has an impact on satellite cell proliferation as demonstrated by the finding that activated muscle precursor cells from Dio2−/− mice have a higher proliferation rate than wild-type cells. Satellite cells from Dio2−/− mice have reduced levels of MyoD, fail to fully differentiate and display a marked delay in their regeneration program (Fig. 5), similar to satellite cells from MyoD null mice [41]. This indicates that D2 activation lies upstream of MyoD in the linear cascade of myogenesis in satellite cells, which requires – for its normal sustained expression during regeneration or development – both T3 from the plasma and that generated by D2. Thus, the current hypothesis is that muscle-specific, D2-mediated T4 activation is required for the proper expression of MRFs, cell-cycle withdrawal, and subsequent fusion of myoblasts into multinucleated myotubes (Fig. 3). In vivo, increased D2 expression is required for proper muscle regeneration and circulating levels of T3 are not sufficient to sustain a full regeneration process. Such a requirement has been elegantly demonstrated by injecting 125I-T4 and 131I-T3 into wild type and Dio2−/− mice, which allowed the evaluation of the relative contributions of plasma and D2-generated T3 in regenerating muscles. The muscle production of 125I-T3 is doubled in wild type, but not in Dio2−/−, mice after skeletal muscle injury [32].
Fig. 5.
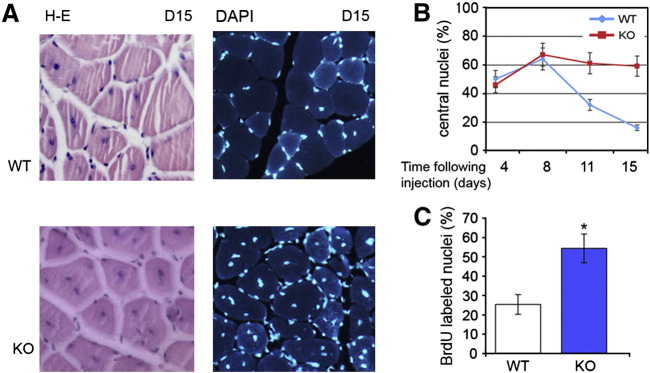
Impaired muscle regeneration in Dio2−/−mice. (A, B) Regeneration following CTX injury is significantly delayed in Dio2−/− mice compared with wild type animals, as shown by the high percentage of centrally located nuclei at day 15 following CTX injection. (C) D2 depletion enhances proliferation of muscle progenitor cells as demonstrated by BrdU incorporation during muscle regeneration.
Figure modified from [41]
How is D2 induced in differentiating myoblasts? We found that the Dio2 gene is positively regulated by FoxO3a, a forkhead transcription factor that integrates growth signals into several cellular functions, including differentiation, metabolism, and survival [73,74]. In skeletal muscle, FoxOs have been implicated in muscle differentiation and, by inducing several autophagy-related genes, FoxO3 is the main coordinator of muscle atrophy [75,76]. Acting in concert with Pax3/7 factors, FoxO3 binds to and regulates MyoD expression [77]. In this scenario, it is relevant that the Dio2 gene is a direct target of FoxO3a, and that Dio2 expression is significantly reduced in FoxO3 null mice [41]. Moreover, we demonstrated that pp6 cells from FoxO3 null mice, which fail to completely differentiate due to the absence of MyoD, can be rescued by T3, thus overcoming their differentiation defects. These data demonstrate that D2 lies between FoxO3 and MyoD expression in the intricate network that induces muscle differentiation. In the myoblasts differentiation program, D3 and D2 expressions increase at different specific times (Dentice et al., manuscript in preparation) so as to allow proliferation in advance of differentiation (Fig. 4). Such a finely regulated profile results in down regulation of TH signaling in the proliferative phase of satellite cells and a remarkable increase of intracellular TH during the differentiation phase.
2.4.3. Deiodinases and muscle regeneration
Skeletal muscle possesses substantial regenerative capacity and can undergo several rounds of repair following injury. In response to wounding, quiescent satellite cells become activated, start to proliferate, and their descendants, the mpc, undergo multiple rounds of division before fusing into new fibers. In several disease states, muscle regenerative activity is impaired — a process that leads to mass wasting and progressive muscle weakness, thereby resulting in muscular dystrophy, muscle weakness, and metabolic disorders. In Duchenne Muscular Dystrophy (DMD), a devastating inherited neuromuscular disorder caused by the absence of a functional dystrophin, the dystrophic myofibers need continuous cycles of muscle regeneration, resulting in a constant need for satellite cell proliferation. However, with time, DMD regeneration cannot compensate for the loss of muscle tissue, a process likely due to a progressive decrease in the satellite cell reservoir. Consistent with this model, several studies demonstrated that alterations in satellite cell proliferation and/or differentiation have an impact on muscular dystrophy [78–81].
Several studies have examined the role of altered TH on the onset of skeletal muscle dystrophy. They showed that alterations in TH plasma levels have a profound impact on the phenotype of mdx mice. McIntosh and Anderson demonstrated that hypothyroidism worsens the phenotype of mdx mice by enhancing mpc proliferation and delaying myotube formation [82]. McArdle and colleagues found that induction of hypothyroidism exerts an initial positive effect on the dystrophic phenotype of mdx mice by preventing necrosis in muscle and abolishing the characteristic elevation in serum Creatine Kinase. This study was the first to demonstrate that experimental manipulation of TH levels altered the onset of necrosis in mdx mice [83]. These findings were subsequently confirmed in studies demonstrating that antithyroid drugs reduce the rate of muscle degeneration in avian muscular dystrophy. King and Entrikin showed that, soon after hatching, induction of hypothyroidism improved muscle function in dystrophic chickens [84]. An opposite effect was elicited by T3 treatment, which accelerated precursor cell transition from proliferation towards fusion in regenerating mdx and control mpc, thereby limiting the number of myotubes generated during the repair process [82,85].
Taken together, these data show that gross fluctuations in TH significantly alter the muscle's response to dystrophy; hyperthyroidism worsens the phenotype of mdx dystrophy by increasing the amount of damage while hypothyroidism can, in the initial phase, ameliorate the mdx dystrophic phenotype by enhancing the proliferative potential of mpc. Indeed, using the cardiotoxin-induced muscle injury paradigm, we demonstrated that D2 is significantly up-regulated in the later stages of muscle regeneration and that repair is significantly delayed in Dio2−/− compared with wild-type mice (Fig. 5). At day 15 after injury, muscle fibers from Dio2−/− mice were still regenerating as demonstrated by the centrally located nuclei, while wild type animals showed completed repair of the myofibers; in fact, the number of laterally located nuclei was nearly double that of Dio2−/− muscle fibers (Fig. 5). In agreement with the enhanced proliferation rate of D2-deficient myoblasts, the number of nuclei in regenerating tissues of Dio2−/− mice was nearly twice that in muscles of similarly injured wild type mice, which indicates that the overall mpc proliferation rate was higher in vivo in the Dio2−/− environment than in controls. It is intriguing that a very similar phenotype is observed in MyoD-null mice in which myogenic stem cells are increased in number but impaired in their differentiation [82,86]. This correlation supports the concept of a linear cascade in which Dio2 lies upstream of MyoD expression. These studies raise the possibility that tissue-specific modulation of TH action could be used to prolong the period of myocyte precursor cell proliferation prior to transplantation.
3. Role of deiodinases in different organs
The important role of local regulation of TH levels by the deiodinases is by no means confined to the tissues described above. For example, D2-KO mice have elevated levels of both serum T4 and TSH (with a normal serum T3), suggesting that there is a resistance to the feedback mechanisms of T4 in the hypothalamus and/or anterior pituitary [87,88]. In brown adipose tissue (BAT), D2 provides an essential local source of T3 to allow adaptive thermogenesis during cold exposure. During cold exposure in D2-null mice, the animals survive only by compensatory shivering with consequent acute weight loss [89]. In the lung, an increase in lung D2 is protective against ventilator-induced lung injury, when an increase of immunoreactivity and D2 enzymatic activity was detected [90]. In the skeleton, where hypothyroidism causes delayed bone formation and linear growth retardation, important roles for Dio2 are emerging [91]. D2 activity was demonstrated in the perichondrium in the embryonic chick growth plate [22], and in murine differentiated osteoblasts [92]. Adult D2KO mice had brittle bones with impaired resistance to fracture, indicating that maintenance of adult bone mineralization and optimal bone strength requires local Dio2-mediated production of T3 [92].
Thus, this brief list of additional examples indicates the critical role of the deiodinases in conferring important local modulation of thyroid hormone responsiveness in vivo to permit the fulfillment of tissue-specific spatio-temporal requirements. One begins to sense that this pathway for control of intracellular T3 concentrations during development and regeneration may be the rule, not the exception.
4. Future directions
There is now compelling evidence that the control of TH action by deiodinases significantly affects the physiology of many organs and tissues. The data available demonstrate that thyroid hormone levels in the circulation do not faithfully reflect TH status in multiple cell types. Although our knowledge is still primitive, the results obtained to date raise the possibility of using deiodinases as therapeutic targets to modify TH action in selected tissues.
A crucial question regarding the therapeutic potential of this potent hormone is how to differentially modulate its action in a time-and tissue-specific manner in order to avoid the deleterious effects of excessive T3. In this respect, the modification of TH deiodination is a promising theoretical approach since it can increase or decrease thyroid hormone signaling irrespective of thyroid hormone serum concentrations. Thus, the spatio- and temporally-controlled regulation of TH action via deiodinases would allow for the modulation of the complex pattern of specific TH-sensitive gene expression in diverse developmental and disease states. Of course, a better understanding of thyroid hormone action at cellular level is a prerequisite for the use of deiodinases for therapeutic purposes. In this scenario, mouse models of tissue-specific knock outs and over-expression of deiodinases will reveal how these enzymes impact TH signaling in selected tissues and organs, and pave the way for their therapeutic exploitation.
Acknowledgements
This work was supported in part by AIRC grants to DS and MD; Telethon grant GGP11185 to DS and NIH grants DK044128 and DK036256 to PRL.
Footnotes
This article is part of a Special Issue entitled Thyroid hormone signalling.
References
- 1.Gereben B., Zavacki A.M., Ribich S., Kim B.W., Huang S.A., Simonides W.S., Zeold A., Bianco A.C. Cellular and molecular basis of deiodinase-regulated thyroid hormone signaling. Endocr. Rev. 2008;29:898–938. doi: 10.1210/er.2008-0019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bianco A.C., Salvatore D., Gereben B., Berry M.J., Larsen P.R. Biochemistry, cellular and molecular biology, and physiological roles of the iodothyronine selenodeiodinases. Endocr. Rev. 2002;23:38–89. doi: 10.1210/edrv.23.1.0455. [DOI] [PubMed] [Google Scholar]
- 3.Burmeister L.A., Pachucki J., St Germain D.L. Thyroid hormones inhibit type 2 iodothyronine deiodinase in the rat cerebral cortex by both pre- and posttranslational mechanisms. Endocrinology. 1997;138:5231–5237. doi: 10.1210/endo.138.12.5602. [DOI] [PubMed] [Google Scholar]
- 4.Leonard J.L., Silva J.E., Kaplan M.M., Mellen S.A., Visser T.J., Larsen P.R. Acute posttranscriptional regulation of cerebrocortical and pituitary iodothyronine 5′-deiodinases by thyroid hormone. Endocrinology. 1984;114:998–1004. doi: 10.1210/endo-114-3-998. [DOI] [PubMed] [Google Scholar]
- 5.Steinsapir J., Harney J., Larsen P.R. Type 2 iodothyronine deiodinase in rat pituitary tumor cells is inactivated in proteasomes. J. Clin. Invest. 1998;102:1895–1899. doi: 10.1172/JCI4672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hanley K., Rassner U., Elias P.M., Williams M.L., Feingold K.R. Epidermal barrier ontogenesis: maturation in serum-free media and acceleration by glucocorticoids and thyroid hormone but not selected growth factors. J. Invest. Dermatol. 1996;106:404–411. doi: 10.1111/1523-1747.ep12343405. [DOI] [PubMed] [Google Scholar]
- 7.Tomic-Canic M., Day D., Samuels H.H., Freedberg I.M., Blumenberg M. Novel regulation of keratin gene expression by thyroid hormone and retinoid receptors. J. Biol. Chem. 1996;271:1416–1423. doi: 10.1074/jbc.271.3.1416. [DOI] [PubMed] [Google Scholar]
- 8.Komuves L.G., Hanley K., Jiang Y., Elias P.M., Williams M.L., Feingold K.R. Ligands and activators of nuclear hormone receptors regulate epidermal differentiation during fetal rat skin development. J. Invest. Dermatol. 1998;111:429–433. doi: 10.1046/j.1523-1747.1998.00296.x. [DOI] [PubMed] [Google Scholar]
- 9.Slominski A., Wortsman J. Neuroendocrinology of the skin. Endocr. Rev. 2000;21:457–487. doi: 10.1210/edrv.21.5.0410. [DOI] [PubMed] [Google Scholar]
- 10.Leonhardt J.M., Heymann W.R. Thyroid disease and the skin. Dermatol. Clin. 2002;20:473–481. doi: 10.1016/s0733-8635(02)00009-8. (vii) [DOI] [PubMed] [Google Scholar]
- 11.Radoja N., Stojadinovic O., Waseem A., Tomic-Canic M., Milisavljevic V., Teebor S., Blumenberg M. Thyroid hormones and gamma interferon specifically increase K15 keratin gene transcription. Mol. Cell. Biol. 2004;24:3168–3179. doi: 10.1128/MCB.24.8.3168-3179.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Safer J.D., Crawford T.M., Holick M.F. A role for thyroid hormone in wound healing through keratin gene expression. Endocrinology. 2004;145:2357–2361. doi: 10.1210/en.2003-1696. [DOI] [PubMed] [Google Scholar]
- 13.Burman K.D., McKinley-Grant L. Dermatologic aspects of thyroid disease. Clin. Dermatol. 2006;24:247–255. doi: 10.1016/j.clindermatol.2006.04.010. [DOI] [PubMed] [Google Scholar]
- 14.Refetoff S., Matalon R., Bigazzi M. Metabolism of l-thyroxine (T4) and l-triiodothyronine (T3) by human fibroblasts in tissue culture: evidence for cellular binding proteins and conversion of T4 to T3. Endocrinology. 1972;91:934–947. doi: 10.1210/endo-91-4-934. [DOI] [PubMed] [Google Scholar]
- 15.Huang T.S., Chopra I.J., Beredo A., Solomon D.H., Chua Teco G.N. Skin is an active site for the inner ring monodeiodination of thyroxine to 3,3′,5′-triiodothyronine. Endocrinology. 1985;117:2106–2113. doi: 10.1210/endo-117-5-2106. [DOI] [PubMed] [Google Scholar]
- 16.Billoni N., Buan B., Gautier B., Gaillard O., Mahe Y.F., Bernard B.A. Thyroid hormone receptor beta1 is expressed in the human hair follicle. Br. J. Dermatol. 2000;142:645–652. doi: 10.1046/j.1365-2133.2000.03408.x. [DOI] [PubMed] [Google Scholar]
- 17.Dentice M., Luongo C., Huang S., Ambrosio R., Elefante A., Mirebeau-Prunier D., Zavacki A.M., Fenzi G., Grachtchouk M., Hutchin M., Dlugosz A.A., Bianco A.C., Missero C., Larsen P.R., Salvatore D. Sonic hedgehog-induced type 3 deiodinase blocks thyroid hormone action enhancing proliferation of normal and malignant keratinocytes. Proc. Natl. Acad. Sci. U. S. A. 2007;104:14466–14471. doi: 10.1073/pnas.0706754104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Safer J.D., Fraser L.M., Ray S., Holick M.F. Topical triiodothyronine stimulates epidermal proliferation, dermal thickening, and hair growth in mice and rats. Thyroid. 2001;11:717–724. doi: 10.1089/10507250152484547. [DOI] [PubMed] [Google Scholar]
- 19.Safer J.D., Crawford T.M., Fraser L.M., Hoa M., Ray S., Chen T.C., Persons K., Holick M.F. Thyroid hormone action on skin: diverging effects of topical versus intraperitoneal administration. Thyroid. 2003;13:159–165. doi: 10.1089/105072503321319468. [DOI] [PubMed] [Google Scholar]
- 20.Safer J.D., Crawford T.M., Holick M.F. Topical thyroid hormone accelerates wound healing in mice. Endocrinology. 2005;146:4425–4430. doi: 10.1210/en.2005-0192. [DOI] [PubMed] [Google Scholar]
- 21.Fink C.W., Ferguson J.L., Smiley J.D. Effect of hyperthyroidism and hypothyroidism on collagen metabolism. J. Lab. Clin. Med. 1967;69:950–959. [PubMed] [Google Scholar]
- 22.Dentice M., Bandyopadhyay A., Gereben B., Callebaut I., Christoffolete M.A., Kim B.W., Nissim S., Mornon J.P., Zavacki A.M., Zeold A., Capelo L.P., Curcio-Morelli C., Ribeiro R., Harney J.W., Tabin C.J., Bianco A.C. The Hedgehog-inducible ubiquitin ligase subunit WSB-1 modulates thyroid hormone activation and PTHrP secretion in the developing growth plate. Nat. Cell Biol. 2005;7:698–705. doi: 10.1038/ncb1272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Trotter W.R. The association of deafness with thyroid dysfunction. Br. Med. Bull. 1960;16:92–98. doi: 10.1093/oxfordjournals.bmb.a069828. [DOI] [PubMed] [Google Scholar]
- 24.Van Middlesworth L., Norris C.H. Audiogenic seizures and cochlear damage in rats after perinatal antithyroid treatment. Endocrinology. 1980;106:1686–1690. doi: 10.1210/endo-106-6-1686. [DOI] [PubMed] [Google Scholar]
- 25.Uziel A. Periods of sensitivity to thyroid hormone during the development of the organ of Corti. Acta Otolaryngol. Suppl. 1986;429:23–27. doi: 10.3109/00016488609122726. [DOI] [PubMed] [Google Scholar]
- 26.Forrest D., Erway L.C., Ng L., Altschuler R., Curran T. Thyroid hormone receptor beta is essential for development of auditory function. Nat. Genet. 1996;13:354–357. doi: 10.1038/ng0796-354. [DOI] [PubMed] [Google Scholar]
- 27.Rusch A., Erway L.C., Oliver D., Vennstrom B., Forrest D. Thyroid hormone receptor beta-dependent expression of a potassium conductance in inner hair cells at the onset of hearing. Proc. Natl. Acad. Sci. U. S. A. 1998;95:15758–15762. doi: 10.1073/pnas.95.26.15758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Refetoff S., Weiss R.E., Usala S.J. The syndromes of resistance to thyroid hormone. Endocr. Rev. 1993;14:348–399. doi: 10.1210/edrv-14-3-348. [DOI] [PubMed] [Google Scholar]
- 29.Ng L., Rusch A., Amma L.L., Nordstrom K., Erway L.C., Vennstrom B., Forrest D. Suppression of the deafness and thyroid dysfunction in Thrb-null mice by an independent mutation in the Thra thyroid hormone receptor alpha gene. Hum. Mol. Genet. 2001;10:2701–2708. doi: 10.1093/hmg/10.23.2701. [DOI] [PubMed] [Google Scholar]
- 30.Crantz F.R., Silva J.E., Larsen P.R. An analysis of the sources and quantity of 3,5,3′-triiodothyronine specifically bound to nuclear receptors in rat cerebral cortex and cerebellum. Endocrinology. 1982;110:367–375. doi: 10.1210/endo-110-2-367. [DOI] [PubMed] [Google Scholar]
- 31.Silva J.E., Leonard J.L., Crantz F.R., Larsen P.R. Evidence for two tissue-specific pathways for in vivo thyroxine 5′-deiodination in the rat. J. Clin. Invest. 1982;69:1176–1184. doi: 10.1172/JCI110554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Marsili A., Tang D., Harney J.W., Singh P., Zavacki A.M., Dentice M., Salvatore D., Larsen P.R. Type II iodothyronine deiodinase provides intracellular 3,5,3′-triiodothyronine to normal and regenerating mouse skeletal muscle. Am J Physiol Endocrinol Metab. 2011;301:E818–824. doi: 10.1152/ajpendo.00292.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tu H.M., Legradi G., Bartha T., Salvatore D., Lechan R.M., Larsen P.R. Regional expression of the type 3 iodothyronine deiodinase messenger ribonucleic acid in the rat central nervous system and its regulation by thyroid hormone. Endocrinology. 1999;140:784–790. doi: 10.1210/endo.140.2.6486. [DOI] [PubMed] [Google Scholar]
- 34.Bernal J. Action of thyroid hormone in brain. J. Endocrinol. Invest. 2002;25:268–288. doi: 10.1007/BF03344003. [DOI] [PubMed] [Google Scholar]
- 35.Kaplan M.M., Yaskoski K.A. Maturational patterns of iodothyronine phenolic and tyrosyl ring deiodinase activities in rat cerebrum, cerebellum, and hypothalamus. J. Clin. Invest. 1981;67:1208–1214. doi: 10.1172/JCI110136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Peeters R., Fekete C., Goncalves C., Legradi G., Tu H.M., Harney J.W., Bianco A.C., Lechan R.M., Larsen P.R. Regional physiological adaptation of the central nervous system deiodinases to iodine deficiency. Am. J. Physiol. Endocrinol. Metab. 2001;281:E54–E61. doi: 10.1152/ajpendo.2001.281.1.E54. [DOI] [PubMed] [Google Scholar]
- 37.Campos-Barros A., Amma L.L., Faris J.S., Shailam R., Kelley M.W., Forrest D. Type 2 iodothyronine deiodinase expression in the cochlea before the onset of hearing. Proc. Natl. Acad. Sci. U. S. A. 2000;97:1287–1292. doi: 10.1073/pnas.97.3.1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ng L., Goodyear R.J., Woods C.A., Schneider M.J., Diamond E., Richardson G.P., Kelley M.W., Germain D.L., Galton V.A., Forrest D. Hearing loss and retarded cochlear development in mice lacking type 2 iodothyronine deiodinase. Proc. Natl. Acad. Sci. U. S. A. 2004;101:3474–3479. doi: 10.1073/pnas.0307402101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Larsen P.R. Thyroid-pituitary interaction: feedback regulation of thyrotropin secretion by thyroid hormones. N. Engl. J. Med. 1982;306:23–32. doi: 10.1056/NEJM198201073060107. [DOI] [PubMed] [Google Scholar]
- 40.Larsen P.R., Silva J.E., Kaplan M.M. Relationships between circulating and intracellular thyroid hormones: physiological and clinical implications. Endocr. Rev. 1981;2:87–102. doi: 10.1210/edrv-2-1-87. [DOI] [PubMed] [Google Scholar]
- 41.Dentice M., Marsili A., Ambrosio R., Guardiola O., Sibilio A., Paik J.H., Minchiotti G., DePinho R.A., Fenzi G., Larsen P.R., Salvatore D. The FoxO3/type 2 deiodinase pathway is required for normal mouse myogenesis and muscle regeneration. J Clin Invest. 2010;120:4021–4030. doi: 10.1172/JCI43670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sharlin D.S., Visser T.J., Forrest D. Developmental and cell-specific expression of thyroid hormone transporters in the mouse cochlea. Endocrinology. 2011;152:5053–5064. doi: 10.1210/en.2011-1372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ng L., Hernandez A., He W., Ren T., Srinivas M., Ma M., Galton V.A., St Germain D.L., Forrest D. A protective role for type 3 deiodinase, a thyroid hormone-inactivating enzyme, in cochlear development and auditory function. Endocrinology. 2009;150:1952–1960. doi: 10.1210/en.2008-1419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Trimarchi J.M., Harpavat S., Billings N.A., Cepko C.L. Thyroid hormone components are expressed in three sequential waves during development of the chick retina. BMC Dev. Biol. 2008;8:101. doi: 10.1186/1471-213X-8-101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Marsh-Armstrong N., Huang H., Remo B.F., Liu T.T., Brown D.D. Asymmetric growth and development of the Xenopus laevis retina during metamorphosis is controlled by type III deiodinase. Neuron. 1999;24:871–878. doi: 10.1016/s0896-6273(00)81034-x. [DOI] [PubMed] [Google Scholar]
- 46.Ng L., Lyubarsky A., Nikonov S.S., Ma M., Srinivas M., Kefas B., St Germain D.L., Hernandez A., Pugh E.N., Jr., Forrest D. Type 3 deiodinase, a thyroid-hormone-inactivating enzyme, controls survival and maturation of cone photoreceptors. J Neurosci. 2010;30:3347–3357. doi: 10.1523/JNEUROSCI.5267-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Roberts M.R., Srinivas M., Forrest D., Morreale de Escobar G., Reh T.A. Making the gradient: thyroid hormone regulates cone opsin expression in the developing mouse retina. Proc. Natl. Acad. Sci. U. S. A. 2006;103:6218–6223. doi: 10.1073/pnas.0509981103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Simonides W.S., van Hardeveld C. Thyroid hormone as a determinant of metabolic and contractile phenotype of skeletal muscle. Thyroid. 2008;18:205–216. doi: 10.1089/thy.2007.0256. [DOI] [PubMed] [Google Scholar]
- 49.Ianuzzo D., Patel P., Chen V., O'Brien P., Williams C. Thyroidal trophic influence on skeletal muscle myosin. Nature. 1977;270:74–76. doi: 10.1038/270074a0. [DOI] [PubMed] [Google Scholar]
- 50.Fitts R.H., Winder W.W., Brooke M.H., Kaiser K.K., Holloszy J.O. Contractile, biochemical, and histochemical properties of thyrotoxic rat soleus muscle. Am. J. Physiol. 1980;238:C14–C20. doi: 10.1152/ajpcell.1980.238.1.C15. [DOI] [PubMed] [Google Scholar]
- 51.Nwoye L., Mommaerts W.F. The effects of thyroid status on some properties of rat fast-twitch muscle. J. Muscle Res. Cell Motil. 1981;2:307–320. doi: 10.1007/BF00713269. [DOI] [PubMed] [Google Scholar]
- 52.Caiozzo V.J., Herrick R.E., Baldwin K.M. Response of slow and fast muscle to hypothyroidism: maximal shortening velocity and myosin isoforms. Am. J. Physiol. 1992;263:C86–C94. doi: 10.1152/ajpcell.1992.263.1.C86. [DOI] [PubMed] [Google Scholar]
- 53.Canepari M., Cappelli V., Pellegrino M.A., Zanardi M.C., Reggiani C. Thyroid hormone regulation of MHC isoform composition and myofibrillar ATPase activity in rat skeletal muscles. Arch. Physiol. Biochem. 1998;106:308–315. doi: 10.1076/apab.106.4.308.4373. [DOI] [PubMed] [Google Scholar]
- 54.Izumo S., Nadal-Ginard B., Mahdavi V. All members of the MHC multigene family respond to thyroid hormone in a highly tissue-specific manner. Science. 1986;231:597–600. doi: 10.1126/science.3945800. [DOI] [PubMed] [Google Scholar]
- 55.Moreno H., Serrano A.L., Santalucia T., Guma A., Canto C., Brand N.J., Palacin M., Schiaffino S., Zorzano A. Differential regulation of the muscle-specific GLUT4 enhancer in regenerating and adult skeletal muscle. J. Biol. Chem. 2003;278:40557–40564. doi: 10.1074/jbc.M306609200. [DOI] [PubMed] [Google Scholar]
- 56.Queiroz M.S., Shao Y., Ismail-Beigi F. Effect of thyroid hormone on uncoupling protein-3 mRNA expression in rat heart and skeletal muscle. Thyroid. 2004;14:177–185. doi: 10.1089/105072504773297849. [DOI] [PubMed] [Google Scholar]
- 57.dos Santos R.A., Giannocco G., Nunes M.T. Thyroid hormone stimulates myoglobin expression in soleus and extensorum digitalis longus muscles of rats: concomitant alterations in the activities of Krebs cycle oxidative enzymes. Thyroid. 2001;11:545–550. doi: 10.1089/105072501750302840. [DOI] [PubMed] [Google Scholar]
- 58.Berchtold M.W., Brinkmeier H., Muntener M. Calcium ion in skeletal muscle: its crucial role for muscle function, plasticity, and disease. Physiol. Rev. 2000;80:1215–1265. doi: 10.1152/physrev.2000.80.3.1215. [DOI] [PubMed] [Google Scholar]
- 59.Hughes S.M., Taylor J.M., Tapscott S.J., Gurley C.M., Carter W.J., Peterson C.A. Selective accumulation of MyoD and myogenin mRNAs in fast and slow adult skeletal muscle is controlled by innervation and hormones. Development. 1993;118:1137–1147. doi: 10.1242/dev.118.4.1137. [DOI] [PubMed] [Google Scholar]
- 60.Dillmann W.H. Hormonal influences on cardiac myosin ATPase activity and myosin isoenzyme distribution. Mol. Cell. Endocrinol. 1984;34:169–181. doi: 10.1016/0303-7207(84)90173-4. [DOI] [PubMed] [Google Scholar]
- 61.Visser W.E., Heemstra K.A., Swagemakers S.M., Ozgur Z., Corssmit E.P., Burggraaf J., van Ijcken W.F., van der Spek P.J., Smit J.W., Visser T.J. Physiological thyroid hormone levels regulate numerous skeletal muscle transcripts. J. Clin. Endocrinol. Metab. 2009;94:3487–3496. doi: 10.1210/jc.2009-0782. [DOI] [PubMed] [Google Scholar]
- 62.van Rooij E., Sutherland L.B., Qi X., Richardson J.A., Hill J., Olson E.N. Control of stress-dependent cardiac growth and gene expression by a microRNA. Science. 2007;316:575–579. doi: 10.1126/science.1139089. [DOI] [PubMed] [Google Scholar]
- 63.Krek A., Grun D., Poy M.N., Wolf R., Rosenberg L., Epstein E.J., MacMenamin P., da Piedade I., Gunsalus K.C., Stoffel M., Rajewsky N. Combinatorial microRNA target predictions. Nat. Genet. 2005;37:495–500. doi: 10.1038/ng1536. [DOI] [PubMed] [Google Scholar]
- 64.Ito M., Roeder R.G. The TRAP/SMCC/Mediator complex and thyroid hormone receptor function. Trends Endocrinol. Metab. 2001;12:127–134. doi: 10.1016/s1043-2760(00)00355-6. [DOI] [PubMed] [Google Scholar]
- 65.Park S.W., Li G., Lin Y.P., Barrero M.J., Ge K., Roeder R.G., Wei L.N. Thyroid hormone-induced juxtaposition of regulatory elements/factors and chromatin remodeling of Crabp1 dependent on MED1/TRAP220. Mol. Cell. 2005;19:643–653. doi: 10.1016/j.molcel.2005.08.008. [DOI] [PubMed] [Google Scholar]
- 66.Mauro A. Satellite cell of skeletal muscle fibers. J. Biophys. Biochem. Cytol. 1961;9:493–495. doi: 10.1083/jcb.9.2.493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Grounds M.D., Yablonka-Reuveni Z. Molecular and cell biology of skeletal muscle regeneration. Mol. Cell Biol. Hum. Dis. Ser. 1993;3:210–256. doi: 10.1007/978-94-011-1528-5_9. [DOI] [PubMed] [Google Scholar]
- 68.Hawke T.J., Garry D.J. Myogenic satellite cells: physiology to molecular biology. J. Appl. Physiol. 2001;91:534–551. doi: 10.1152/jappl.2001.91.2.534. [DOI] [PubMed] [Google Scholar]
- 69.Cornelison D.D., Wold B.J. Single-cell analysis of regulatory gene expression in quiescent and activated mouse skeletal muscle satellite cells. Dev. Biol. 1997;191:270–283. doi: 10.1006/dbio.1997.8721. [DOI] [PubMed] [Google Scholar]
- 70.Ramadan W., Marsili A., Larsen P.R., Zavacki A.M., Silva J.E. Type-2 iodothyronine 5′deiodinase (D2) in skeletal muscle of C57Bl/6 mice. II. Evidence for a role of D2 in the hypermetabolism of thyroid hormone receptor alpha-deficient mice. Endocrinology. 2011;152:3093–3102. doi: 10.1210/en.2011-0139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Larsen P.R. Type 2 iodothyronine deiodinase in human skeletal muscle: new insights into its physiological role and regulation. J. Clin. Endocrinol. Metab. 2009;94:1893–1895. doi: 10.1210/jc.2009-0791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Marsili A., Ramadan W., Harney J.W., Mulcahey M., Castroneves L.A., Goemann I.M., Wajner S.M., Huang S.A., Zavacki A.M., Maia A.L., Dentice M., Salvatore D., Silva J.E., Larsen P.R. Type 2 iodothyronine deiodinase levels are higher in slow-twitch than fast-twitch mouse skeletal muscle and are increased in hypothyroidism. Endocrinology. 2010;151:5952–5960. doi: 10.1210/en.2010-0631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Accili D., Arden K.C. FoxOs at the crossroads of cellular metabolism, differentiation, and transformation. Cell. 2004;117:421–426. doi: 10.1016/s0092-8674(04)00452-0. [DOI] [PubMed] [Google Scholar]
- 74.Carter M.E., Brunet A. FOXO transcription factors. Curr. Biol. 2007;17:R113–R114. doi: 10.1016/j.cub.2007.01.008. [DOI] [PubMed] [Google Scholar]
- 75.Mammucari C., Milan G., Romanello V., Masiero E., Rudolf R., Del Piccolo P., Burden S.J., Di Lisi R., Sandri C., Zhao J., Goldberg A.L., Schiaffino S., Sandri M. FoxO3 controls autophagy in skeletal muscle in vivo. Cell Metab. 2007;6:458–471. doi: 10.1016/j.cmet.2007.11.001. [DOI] [PubMed] [Google Scholar]
- 76.Sandri M., Sandri C., Gilbert A., Skurk C., Calabria E., Picard A., Walsh K., Schiaffino S., Lecker S.H., Goldberg A.L. Foxo transcription factors induce the atrophy-related ubiquitin ligase atrogin-1 and cause skeletal muscle atrophy. Cell. 2004;117:399–412. doi: 10.1016/s0092-8674(04)00400-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Hu P., Geles K.G., Paik J.H., DePinho R.A., Tjian R. Codependent activators direct myoblast-specific MyoD transcription. Dev. Cell. 2008;15:534–546. doi: 10.1016/j.devcel.2008.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Wakayama Y., Schotland D.L., Bonilla E., Orecchio E. Quantitative ultrastructural study of muscle satellite cells in Duchenne dystrophy. Neurology. 1979;29:401–407. doi: 10.1212/wnl.29.3.401. [DOI] [PubMed] [Google Scholar]
- 79.Ishimoto S., Goto I., Ohta M., Kuroiwa Y. A quantitative study of the muscle satellite cells in various neuromuscular disorders. J. Neurol. Sci. 1983;62:303–314. doi: 10.1016/0022-510x(83)90207-1. [DOI] [PubMed] [Google Scholar]
- 80.Reimann J., Irintchev A., Wernig A. Regenerative capacity and the number of satellite cells in soleus muscles of normal and mdx mice. Neuromuscul. Disord. 2000;10:276–282. doi: 10.1016/s0960-8966(99)00118-2. [DOI] [PubMed] [Google Scholar]
- 81.Yablonka-Reuveni Z., Anderson J.E. Satellite cells from dystrophic (mdx) mice display accelerated differentiation in primary cultures and in isolated myofibers. Dev. Dyn. 2006;235:203–212. doi: 10.1002/dvdy.20602. [DOI] [PubMed] [Google Scholar]
- 82.McIntosh L.M., Pernitsky A.N., Anderson J.E. The effects of altered metabolism (hypothyroidism) on muscle repair in the mdx dystrophic mouse. Muscle Nerve. 1994;17:444–453. doi: 10.1002/mus.880170413. [DOI] [PubMed] [Google Scholar]
- 83.McIntosh L.M., Anderson J.E. Hypothyroidism prolongs and increases mdx muscle precursor proliferation and delays myotube formation in normal and dystrophic limb muscle. Biochem. Cell Biol. 1995;73:181–190. doi: 10.1139/o95-022. [DOI] [PubMed] [Google Scholar]
- 84.King D.B., Entrikin R.K. Thyroidal involvement in the expression of avian muscular dystrophy. Life Sci. 1991;48:909–916. doi: 10.1016/0024-3205(91)90038-d. [DOI] [PubMed] [Google Scholar]
- 85.Pernitsky A.N., McIntosh L.M., Anderson J.E. Hyperthyroidism impairs early repair in normal but not dystrophic mdx mouse tibialis anterior muscle. An in vivo study. Biochem. Cell Biol. 1996;74:315–324. doi: 10.1139/o96-034. [DOI] [PubMed] [Google Scholar]
- 86.Rudnicki M.A., Jaenisch R. The MyoD family of transcription factors and skeletal myogenesis. Bioessays. 1995;17:203–209. doi: 10.1002/bies.950170306. [DOI] [PubMed] [Google Scholar]
- 87.Schneider M.J., Fiering S.N., Pallud S.E., Parlow A.F., St Germain D.L., Galton V.A. Targeted disruption of the type 2 selenodeiodinase gene (DIO2) results in a phenotype of pituitary resistance to T4. Mol. Endocrinol. 2001;15:2137–2148. doi: 10.1210/mend.15.12.0740. [DOI] [PubMed] [Google Scholar]
- 88.Galton V.A., Wood E.T., St Germain E.A., Withrow C.A., Aldrich G., St Germain G.M., Clark A.S., St Germain D.L. Thyroid hormone homeostasis and action in the type 2 deiodinase-deficient rodent brain during development. Endocrinology. 2007;148:3080–3088. doi: 10.1210/en.2006-1727. [DOI] [PubMed] [Google Scholar]
- 89.de Jesus L.A., Carvalho S.D., Ribeiro M.O., Schneider M., Kim S.W., Harney J.W., Larsen P.R., Bianco A.C. The type 2 iodothyronine deiodinase is essential for adaptive thermogenesis in brown adipose tissue. J. Clin. Invest. 2001;108:1379–1385. doi: 10.1172/JCI13803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Barca-Mayo O., Liao X.H., DiCosmo C., Dumitrescu A., Moreno-Vinasco L., Wade M.S., Sammani S., Mirzapoiazova T., Garcia J.G., Refetoff S., Weiss R.E. Role of type 2 deiodinase in response to acute lung injury (ALI) in mice. Proc Natl Acad Sci U S A. 2011;108:E1321–E1329. doi: 10.1073/pnas.1109926108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Williams G.R., Bassett J.H. Deiodinases: the balance of thyroid hormone: local control of thyroid hormone action: role of type 2 deiodinase. J Endocrinol. 2011;209:261–272. doi: 10.1530/JOE-10-0448. [DOI] [PubMed] [Google Scholar]
- 92.Bassett J.H., Williams A.J., Murphy E., Boyde A., Howell P.G., Swinhoe R., Archanco M., Flamant F., Samarut J., Costagliola S., Vassart G., Weiss R.E., Refetoff S., Williams G.R. A lack of thyroid hormones rather than excess thyrotropin causes abnormal skeletal development in hypothyroidism. Mol. Endocrinol. 2008;22:501–512. doi: 10.1210/me.2007-0221. [DOI] [PMC free article] [PubMed] [Google Scholar]