

Abstract
The integral membrane protein DsbB in Escherichia coli is responsible for oxidizing the periplasmic protein DsbA, which forms disulfide bonds in substrate proteins. We have developed a high-resolution structural model by combining experimental X-ray and solid-state NMR with molecular dynamics (MD) simulations. We embedded the high-resolution DsbB structure, derived from the joint calculation with X-ray reflections and solid-state NMR restraints, into the lipid bilayer and performed MD simulations to provide a mechanistic view of DsbB function in the membrane. Further, we revealed the membrane topology of DsbB by selective proton spin diffusion experiments, which directly probe the correlations of DsbB with water and lipid acyl chains. NMR data also support the model of a flexible periplasmic loop and an interhelical hydrogen bond between Glu26 and Tyr153.
Keywords: disulfide bond generation, DsbB, membrane protein, solid-state NMR, molecular dynamics simulation
Introduction
De novo disulfide bond formation in Escherichia coli (E. coli) is catalyzed by a protein complex consisting of the periplasmic protein DsbA (21 kDa) and the membrane protein DsbB (20 kDa)1. DsbA contains a thioredoxin-like domain with the CXXC motif (Cys30-Pro31-His32-Cys33) to oxidize the reduced cysteines in substrate proteins, forming disulfide bonds for proper folding2. DsbB is an integral membrane protein with four transmembrane helices (TM1 to TM4)3, and is responsible for reoxidizing DsbA through disulfide bond rearrangements among four conserved cysteines in DsbB (Cys41–Cys44 and Cys104–Cys130). The electrons are transferred from DsbA cysteines to the cofactor ubiquinone (UQ-8)4, then to the respiratory chain5. Mutagenesis studies have shown that the disulfide bond exchange occur through the sequence of DsbA Cys30–Cys33, DsbB Cys104–Cys130 and Cys41–Cys44 with intermediate formation of disulfide bonds DsbA Cys30 – DsbB Cys104 and DsbB Cys130–Cys416 (Fig. S1). DsbB Cys44 interacts with UQ through a charge-transfer complex corresponding to a 500 nm absorption band in the UV-Vis spectrum7. The DsbA/DsbB system plays an important role in the folding of virulence factors in several pathogenic bacteria8, and thus it can be a potential target for antibiotics. Therefore, the elucidation of structure and membrane interaction can provide a better understanding of the mechanism of disulfide bond formation and the basis for future therapeutic design.
Several structures of various DsbB mutants mimicking the intermediate states in the electron transfer pathway have been solved utilizing X-ray crystallography and solution NMR spectroscopy, including DsbA(Cys33Ala)-DsbB(Cys130Ser) (3.7 Å)9, DsbA(Cys33Ala)-DsbB (3.7 Å)10, DsbB(Cys41Ser)-Fab (3.4 Å)11, and DsbB(Cys44Ser, Cys104Ser) (solution NMR)12. An amphipathic horizontal helix in the DsbB periplasmic loop was elucidated in these structures, which was proposed to control the selectivity of DsbB towards DsbA oxidation13. Based on a comparison between the disulfide bond generating systems in prokaryotic and eukaryotic cells, a similar function of the shuttle disulfide in the flexible loop region was revealed to facilitate the electron transfer from the disulfide bond donor to the electron-accepting cofactor14. Kinetic studies of the DsbB(UQ)-DsbA reaction indicated it to be highly concerted and identified the quinone reduction as the rate-limiting step15. However, the atomic-level details of disulfide bond rearrangements in the periplasmic loop and UQ binding remain elusive, due to the low electron density in the loop region of the crystal structures. Wild-type DsbB yields low-quality solution NMR spectra in DPC micelles, whereas the mutant DsbB(Cys44Ser, Cys104Ser) possesses an inter-loop disulfide bond Cys41–Cys130 and yields high-quality spectra, suggesting that conformational exchange in the loop regions is significant12. Further, the effects from the membranous environment on the DsbB structure and dynamics have not yet been investigated at high resolution, and such information would be missing in crystalline or detergent-solubilized forms.
Solid-state NMR spectroscopy (SSNMR) has been demonstrated as a high-resolution and versatile technique for probing membrane protein structure, topology and dynamics in the membranous environment16; 17; 18; 19; 20; 21; 22; 23; 24. As a powerful computational approach, molecular dynamics (MD) simulations can provide the understanding of interactions between membrane proteins and the lipid bilayers25; 26; 27. Combining these methods, we set out to determine the structure of membrane-embedded DsbB. A high-resolution structure of DsbB(Cys41Ser) was obtained by the joint calculation with X-ray reflections and SSNMR dihedral angle and distance restraints. The structure was then embedded in a 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoethanolamine (POPE) bilayer, mimicking the bacterial membrane, through MD simulations to generate a structural model of DsbB in the membrane. The model was experimentally validated not only by NMR chemical shifts and signal intensities, but also by the experiments that specifically report on the membrane topology. In 2D selective proton spin diffusion spectra, we observed specific correlations from water or lipid acyl chains to DsbB residues, which are indeed close to water or lipid acyl chains in our structural model. In the structural model, the “horizontal helix” residues adapt an extended coil conformation on the membrane surface and a hydrogen bond between Glu26 and Tyr153 is formed to offset the energy cost of the negative-charge insertion. These findings provide insights into DsbB membrane topology and the mechanism of DsbB activity.
Results
High-resolution structure of the integral membrane protein DsbB
We have chosen to study the Cys41Ser mutant of DsbB, which represents a transient intermediate state in the proposed reaction pathway7. Chemical shift assignments of the majority of the helical residues of DsbB(Cys41Ser) were obtained by 3D and 4D site-specific sequential SSNMR experiments at low temperature (Tsample = ~ −35 °C)28, but many of the resonances corresponding to the residues in the loop regions were missing. We attribute this to broadened linewidths caused by heterogeneous disorder of these regions at low temperature. Further, we found that the resolution in 2D SSNMR correlation spectra is improved when the sample temperature is close to the lipid phase transition temperature (~ −12 °C with the samples consisting of DsbB(Cys41Ser), native lipids and residual detergents)29. Therefore, we performed additional 3D SSNMR experiments at Tsample = ~ −12 °C to assign the previously missing residues in the loop regions and particularly in the active site (Fig. 1). These chemical shifts yielded 186 dihedral angle (φ, ψ) restraints from the improved version of the Torsion Angle Likeliness Obtained from Shift and Sequence Similarity (TALOS+)30 program.
Figure 1.

SSNMR chemical shift assignments of DsbB (Cys41Ser). (a) Strip plot of NCACX (blue) and NCOCX (red) of DsbB (Cys41Ser) acquired at −12 °C, showing the residues at the active site of DsbB. (b) Topology view of DsbB with assigned residues highlighted in blue and cyan. Residues in blue are at the active site of DsbB. The four rectangles represent the transmembrane helices predicted by secondary structure analysis based on chemical shifts.
To obtain distance restraints, we utilized selective labeling, in which [2-13C]glycerol or [1,3-13C]glycerol is used as the carbon source during protein expression31, to acquire 2D 13C-13C correlation spectra of [2-13C-glycerol,15N]DsbB(Cys41Ser) and [1,3-13C-glycerol,15N]DsbB(Cys41Ser)32. The cross peaks were analyzed using with chemical shift assignments from the 3D experiments28. Despite the excellent resolution from the glycerol labeling (typical 13C linewidths are 0.3–0.5 ppm), assignment degeneracies hinder the unambiguous interpretation of 2D spectra of such a large membrane protein. Therefore, we applied the probabilistic assignment algorithm for automated structure determination (PASD)33 in Xplor-NIH. Based on the peak lists in 13C-13C 2D spectra and the chemical shift assignments, considering different labeling patterns of 2-13C-glycerol and 1,3-13C-glycerol, 667 ambiguous distance restraints were extracted for DsbB(Cys41Ser).
We performed structure calculations of DsbB(Cys41Ser) using SSNMR dihedral angle, ambiguous distance restraints, and X-ray reflections from the RCSB Protein Databank for DsbB(Cys41Ser)-Fab (PDB code 2ZUQ), following methods that we have previously demonstrated for the DsbA-DsbB complex32. The joint calculations of DsbB(Cys41Ser)-Fab structure were carried out using the simulated annealing program Xplor-NIH34. The commonly used potential terms of bonds, angles, improper torsions, van der Waals, hydrogen bonds, NOEs (13C-13C distances) and dihedral angles (TALOS+ restraints) were included with a specific potential term for direct refinement against crystallographic structure factors (X-ray reflections). Table 1 shows the statistical comparison, based on the 10 lowest-energy structures out of 200 calculated, of the same protocol performed with X-ray reflections and SSNMR data alone and with both restraints together. The average r.m.s. deviation of backbone atoms (bbRMSD) from their mean positions was reduced from 2.36 Å (X-ray reflections only) to 1.35 Å (X-ray reflections + SSNMR restraints), a 43% improvement. In the helices of DsbB, where most NMR restraints of DsbB were observed, bbRMSD improvement is approximately 78% over X-ray data alone (0.50 Å vs. 2.32 Å). Fig. 2 shows the overlay of calculated structures of DsbB(Cys41Ser) without and with SSNMR restraints. Structure calculations using solely the X-ray data yielded the correct general fold of the protein (Fig. 2a), but the backbone precision is only 2.36 Å. In contrast, SSNMR restraints define the secondary structure elements to high resolution on top of the general fold maintained by X-ray data, improving the precision to 1.35 Å for the entire backbone (Fig. 2b and Fig. S2). Thus the X-ray and SSNMR data sets are complementary and self-consistent, with the combination yielding a much higher quality overall structure than either method alone.
Table 1.
Statistics of the joint calculation by SSNMR and X-ray for DsbB(Cys41Ser).
| DsbB(Cys41Ser) | |||
|---|---|---|---|
|
| |||
| Refinement from datasets | X-ray | SSNMR | X-ray + SSNMR |
| NMR restraints | |||
| Distance restraints | |||
| Ambiguous NOE | - | 1334a | 1334a |
| Intra-residue | - | 518 | 518 |
| Inter-residue | - | 826 | 826 |
| Sequential (|i-j| = 1) | - | 560 | 560 |
| Medium range (1 < |i-j| < 5) | - | 254 | 254 |
| Long range (|i-j| > 5) | - | 2 | 2 |
| Dihedral angle restraints | |||
| φ, ψ | - | 372a | 372a |
| X-ray data | |||
| Resolution (Å) | 3.4 | - | 3.4 |
| No. reflections | 25269 | - | 25269 |
| Structure statistics | |||
| Violations (mean and s.d.) | |||
| Distance restraints (Å) | - | 0.088 (0.001) | 0.090 (0.001) |
| Dihedral angle restraints (°) | - | 0.102 (0.044) | 0.442 (0.105) |
| Deviations from idealized geometry | |||
| Bond lengths (Å) | 0.016 (0.001) | 0.017 (0.001) | 0.017 (0.001) |
| Bond angles (°) | 1.984 (0.019) | 1.979 (0.003) | 1.985 (0.003) |
| Impropers (°) | 8.836 (0.093) | 8.829 (0.100) | 8.782 (0.088) |
| Average r.m.s. deviationb (Å) | |||
| DsbB helical backbonec | 2.32 | 2.24 | 0.50 |
| Backbone | 2.36 | 3.40 | 1.35 |
| Heavy | 3.25 | 4.16 | 2.11 |
| Ramachandran space analysisd (%) | |||
| Most favored | 80.0 | 100.0 | 96.1 |
| Additionally allowed | 20.0 | 0.0 | 3.9 |
| Generously allowed | 0.0 | 0.0 | 0.0 |
| Disallowed | 0.0 | 0.0 | 0.0 |
Numbers in parentheses represent the uncertainties.
Since the unit cell of DsbB-Fab crystal has two DsbB asymmetric units, each DsbB has the same distance and dihedral angle restraints.
Average r.m.s. deviation was calculated among 10 lowest-energy structures out of 200.
The transmembrane helices consist of residues 17–30, 43–60, 72–85, 146–161.
Performed in PROCHECK55 for the crystal structure or one jointly-refined structure in DsbB transmembrane regions.
Figure 2.

Structures of DsbB (Cys41Ser). (a) Overlay of ten lowest energy structures of DsbB (Cys41Ser) calculated with only X-ray reflections. (b) Overlay of ten lowest energy structures of DsbB (Cys41Ser) calculated with X-ray reflections and SSNMR restraints (PDB ID: 2LTQ). The colors represent the secondary structure elements (magenta: α-helix, cyan and white: coil and turn).
Membrane topology of DsbB in the lipid bilayers by solid-state NMR
The composition of the lipid bilayers and the difference in proton chemical shifts of water and lipid acyl chains provide the unique opportunity for SSNMR to probe the membrane topology of proteins by magnetization transfer through proton spin diffusion35; 36; 37; 38. We expect that water protons would transfer polarization through spin diffusion to DsbA faster than to DsbB due to the spatial proximity. Additionally, we expect the lipid acyl chain protons to spin diffuse to DsbB faster than to DsbA, since DsbA is in the periplasm and DsbB is transmembrane (Fig. S3a). In order to determine the relative positions of DsbA and DsbB to the lipid bilayer qualitatively, we prepared two covalently-bonded DsbA/DsbB complex samples: one with uniformly 13C, 15N-labeled DsbB (DsbA/DsbB*), and the other with uniformly 13C, 15N-labeled DsbA (DsbA*/DsbB). In both cases the samples were prepared in native phospholipid bilayer environments. The buildup rates of DsbB and DsbA from selective proton spin diffusion experiments reflect the proximity of these isotopically enriched protein to water or lipid acyl chain protons. As expected, DsbB signals build up more slowly when water protons are selected and faster when lipid acyl chain protons are selected, while DsbA signals build up faster when water protons are selected and slower when lipid acyl chain protons are selected (Fig. S3 b, c, and d). These observations also provide evidence that substantial quantities of native lipids are retained around DsbB in SSNMR samples.
To obtain site-specific information about the membrane topology of DsbB, we utilized 13C-13C 2D correlation experiments with selective proton diffusion from water or lipid acyl chains, allowing us to assign specific residues that are close to water or lipid acyl chains. A short proton mixing time was chosen to enhance the contrast of crosspeak intensities from different proton sources. In the lipid-to-DsbB spin diffusion spectrum, residues of Ala, Val, Ile and Leu with the α-helical secondary structure were identified, corresponding to the transmembrane helices of DsbB that are close to the lipid acyl chains (Fig. 3a). On the other hand, in the water-to-DsbB spin diffusion spectrum, the residues with coil or turn conformations are observed, including Pro residues in the loop regions and Ile45 in the active site with ubiquinone (Fig. 3b). The overlaid expansion of the Ala Cα-Cβ region demonstrates the secondary chemical shift distribution of Ala residues, in which water correlates mostly with Ala residues of coil or turn conformations and lipid acyl chains correlate mostly with helical Ala residues (Fig. 3d). In these 13C-13C 2D correlation spectra, we assigned 29 residues close to water and 44 residues close to lipid acyl chains (Table S1).
Figure 3.
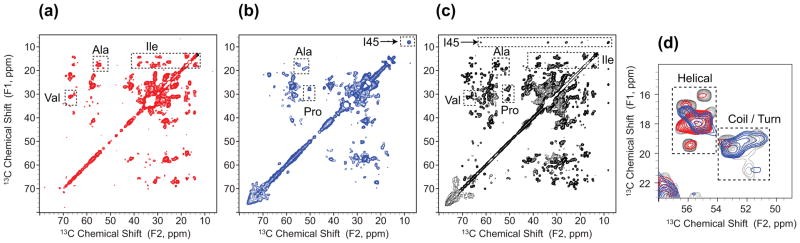
Site-specific lipid or water correlations to DsbB. (a) 13C-13C 2D correlation spectrum with proton spin diffusion from lipid acyl chains to DsbB, (b) 13C-13C 2D correlation spectrum with proton spin diffusion from water to DsbB and (c) 13C-13C 2D correlation spectrum of DsbB. (d) Overlay of expansions of Ala region in 2D spectra of lipid-DsbB (red), water-DsbB (blue) and DsbB (black).
Structural features of membrane-embedded DsbB
To investigate the influence of the membranous environment upon DsbB structure, we carried out MD simulations for DsbB(Cys41Ser) embedded in a lipid bilayer in a transmembrane orientation. A structural model of DsbB(Cys41Ser) in a POPE bilayer was generated by embedding the lowest-energy structure from the joint X-ray/SSNMR calculation into the bilayer through solvation, energy minimization and equilibrium. As shown in Fig. 4, DsbB residues correlated with water (blue) or lipid acyl chain (orange) protons in the proton spin diffusion experiments are indeed close to water or lipid acyl chains in the structural model, respectively. There are water molecules permeating the membrane through the protein and close to the active site with ubiquinone, which might provide protons for the reduction of ubiquinone to ubiquinol. This is similar to the hydration of membrane bilayers with the voltage-sensing domains of ion channels27. Interestingly, a hydrogen-bond between Glu26 in TM1 and Tyr153 in TM4 is found in the center of the membrane, which is responsible for stabilizing the negative charge on Glu26 in the membrane (Fig. 5a). These two residues are also close to each other, within 3.5 Å in the structure from joint X-ray/SSNMR calculations, but they are apart (> 8 Å) in the crystal and solution NMR structures (Fig. 5b). To confirm the proximity of Glu26 and Tyr153, a 13C–13C 2D correlation spectrum with 500 ms DARR39 mixing was acquired on [U-13C, 15N]DsbB(Cys41Ser), and 15N REDOR40 dephasing was applied to remove the interferences from the sidechains of Arg, Asn, Gln, His and Trp29. The spectrum indeed shows the crosspeaks between the sidechains of Glu26 and Tyr153 (Fig. 5c). Another charged residue, Arg83, in the middle of TM3 also has a phospholipid headgroup nearby for stabilization through hydrogen bonding and electrostatic interaction between the guanidinium and phosphate (Fig. S4).
Figure 4.

Structural model of DsbB in the membrane. The structure of DsbB (purple) was calculated jointly with X-ray and SSNMR restraints, and then put into the lipid bilayers through solvation, energy minimization and equilibrium. The highlighted residues are those that correlate with water (blue) and lipids (orange), as observed in selective proton spin diffusion experiments. They are consistent with the structural model in terms of their proximity to water (cyan) and lipid (gray) molecules. Some lipid and water molecules are not displayed to reveal DsbB more clearly.
Figure 5.
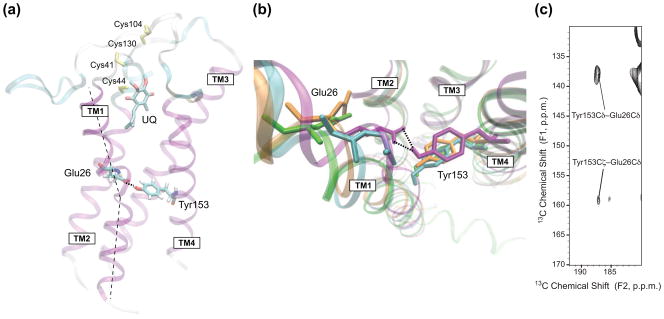
Glu26 and Tyr153 hydrogen bond in DsbB. (a) Overview of DsbB structural model from the membrane plane. The hydrogen bond between Glu26 and Tyr153, the ubiquinone cofactor (UQ) and the sidechains of the four significant Cys residues (yellow) are highlighted. The dashed line indicates the kink in TM1 due to Glu26 and Tyr153 hydrogen bond. (b) Overlay of DsbB models viewed from the periplasmic side. The structure from joint calculation (cyan) shows that Glu26 in TM1 and Tyr153 in TM4 are in close proximity. The model from MD (purple) is consistent with that Glu26 and Tyr153 are linked through hydrogen bonding. The crystal structure (orange) and solution NMR structure (green) have Glu26 relatively distant from Tyr153 (> 8 Å). (c) 13C-13C 2D correlation spectrum with 15N REDOR dephasing and 500 ms DARR mixing of [U-13C, 15N]DsbB(Cys41Ser), showing the crosspeaks between the sidechains of Glu26 and Tyr153.
Discussion
We obtained a high-resolution structure of DsbB by a joint calculation, using X-ray reflections and SSNMR restraints. The precision has improved significantly compared to the calculation solely with X-ray reflections, indicating the capability of SSNMR to yield high-resolution structural restraints for defining the detailed local structures, in addition to the essential framework for the general fold of proteins provided by X-ray reflections. The two structural approaches complement each other very well in studying structural biology of membrane proteins. In terms of structural differences, the Cα coordinates of the calculated structures show a RMSD of 2.16 Å to the crystal structure, and the deviations are mostly in the loop and turn regions where the resolution is low in the electron density map. Additionally, the sample conditions for different techniques can be a contributing factor to structural differences. The crystal structure was obtained with DsbB(Cys41Ser) bound to the monoclonal Fab antibody fragment11, while SSNMR samples consist of DsbB(Cys41Ser), native lipids and small quantities of residual detergent28; 29. The proton spin diffusion experiments also provide direct evidence that there are lipids surrounding the membrane protein DsbB in our SSNMR samples (Fig. 3 and Fig. S3).
To gain insight into the structure of DsbB in the membrane and the disulfide bond rearrangement in the periplasmic loops, the structural model of DsbB in a POPE bilayer from MD simulations was compared with the structures from X-ray (DsbB(Cys41Ser)-Fab, PDB ID: 2ZUQ), joint X-ray/SSNMR (DsbB(Cys41Ser), PDB ID: 2LTQ) and solution NMR (DsbB(Cys44Ser, Cys104Ser), PDB ID: 2K74). The loop regions in the MD model are generally lifted up for 6~8 Å relative to those structures due to the interactions between the lipids and DsbB polar residues (Fig. S5). There is an amphipathic horizontal helix (residue 116 to 120) in the long loop between TM3 and TM4 in the crystal structures of DsbB(Cys41Ser)-Fab and DsbB(Cys130Ser)-DsbA(Cys33Ala), which is associated with the membrane and important for DsbB activity to oxidize DsbA effectively11. The solution NMR structure of DsbB(Cys44Ser, Cys104Ser) also has similar horizontal helix, but it is submerged more deeply into the membrane12. Interestingly, in the structural model from MD simulations, the residues of the horizontal helix adapt a flexible coil conformation due to the interactions with lipid molecules. The feasible explanation for these structural differences is that the DsbB periplasmic loop undergoes conformational changes upon disulfide-bond rearrangements: When DsbB is alone in the membrane, the “horizontal helix” residues adapt the extended coil conformation to accommodate the Cys104–Cys130 disulfide bond (Fig. S6a). When the Cys104–Cys130 disulfide bond breaks during the disulfide-bond rearrangement, the “horizontal helix” residues adapt the compact helical conformation to accommodate the disulfide bond between DsbA Cys30 and DsbB Cys104 (Fig. S6b) or between DsbB Cys41 and Cys130 (Fig. S6c). The flexibility and amphiphilicity of the horizontal helix is likely to be critical to the ability of Cys130 to move between Cys41 and Cys104, and thus important to the electron transfer from DsbA to DsbB (Fig. S6d). As expected, the mutations on the horizontal helix to many charged or helix-breaking residues have a severe effect on the redox state in the E. coli periplasm, as demonstrated in the mutagenesis study by the presence of a large population of reduced DsbA11. As for the horizontal helix in DsbB(Cys41Ser)-Fab, the Fab molecules might stabilize the periplasmic loop to form the short helix. A similar motif is also observed in the crystal structure of a bacterial homologue of vitamin K epoxide reductase (VKOR)41, a protein evolutionarily unrelated to DsbB but which serves to oxidize DsbA in bacterial species lacking DsbB42. In this VKOR homologue from Synechococcus sp., a small amphipathic helix is next to the loop cysteines (Cys50 and Cys56) to shuttle Cys56 for reaction with the cysteines at the ubiquinone binding site.
The hydrogen bond between Glu26 and Tyr153 is intriguing, since it is not observed in any of the crystal or solution NMR structures (Fig. 5b). The hydrophobic environment in the center of the lipid bilayers may promote the hydrogen bonding to stabilize the negative charge on Glu26. According to the biological hydrophobicity scale43, the free energy for the insertion of a transmembrane helix with a Glu residue in the middle is 2.68 kcal/mol, which is very unfavorable for membrane insertion and has to be compensated by a sufficient number of hydrophobic residues, hydrogen bonds or electrostatic interactions. The hydrogen bond between Glu26 in TM1 and Tyr153 in TM4 not only offsets the energy cost for Glu insertion, but brings TM1 and TM4 together to facilitate the disulfide bond exchange between Cys41–Cys44 and Cys104–Cys130 as well (Fig. 5a). A kink in TM1 around the position of Glu26 due to the hydrogen bond is also observed in the structural model (Fig. 5a), which would open up the space in the helix bundle for UQ binding. A similar hydrogen bond between Arg135 (H3) and Tyr223 (H5) was found in rhodopsin, which is responsible for helix H5 rotation and subsequent extracellular loop EL2 displacement to stabilize the active conformation of the receptor44. Based on the relative position of the hydrogen bond between Glu25 and Tyr153 to DsbB active site and the fact that these amino acids are not conserved, the hydrogen bond is not essential to a general reaction mechanism. The structural stability imparted by this hydrogen bond and the resulting kink in TM1 may be replicated by other sets of residues in E. coli DsbB orthologues.
In summary, the structural model of membrane-embedded DsbB, generated by MD simulations combining the results from X-ray crystallography and solid-state NMR, provides insights into the mechanism of disulfide bond formation in E. coli. The flexible DsbB periplasmic loop may adapt different conformations (coil/helix) upon disulfide-bond rearrangement, and the hydrogen bond between Glu26 and Tyr153 may stabilize the membrane-embedded charge and the helix bundle. The membrane topology and structural differences reflect the influence of the membranous environment on DsbB structure compared to its crystalline or detergent soluble forms, and demonstrate the applications of this integrative approach to elucidate the structure-function relationships of membrane proteins in the lipid bilayers.
Materials and Methods
Sample Preparation
His-tagged DsbB(Cys41Ser) containing mutations of nonessential cysteines (Cys8Ala, Cys49Val) were obtained from K. Inaba and K. Ito7; 45. The expressions of [U-13C, 15N]DsbB(Cys41Ser), [2-13C-glycerol,15N]DsbB(Cys41Ser) and [1,3-13C-glycerol,15N]DsbB(Cys41Ser) were described previously28; 32. Briefly, freshly transformed E. coli C43 (DE3)/pREP4 cells were grown in LB medium containing 60 μg/ml ampicillin, 25 μg/ml kanamycin, 50 mM Na2HPO4, 50 mM KH2PO4, 5 mM Na2SO4, 2 mM MgSO4, 10 μg/ml thiamine, 10 μg/ml biotin, trace elements46, 0.3% 15N-NH4Cl, 0.2% 2-13C glycerol and 9 mM Na13CO3 for 2-DsbB or 1,3-13C glycerol and 9mM Na12CO3 for 1,3-DsbB. Flasks (2 L), with a culture volume of 250 ml, were shaken at 250 rpm at 37 °C for 9~10 h until A600 = 0.8. At this point, expression was induced with 0.2 mM isopropyl-β-D-thiogalactopyranoside (IPTG), and growth continued at 25 °C for 20 h until harvest. Membrane isolation, solubilization of DsbB in 1% dodecylmaltoside (DDM), and purification of the His-tagged DsbB were described previously28. Fractions containing DsbB were pooled and concentrated, followed by dialysis against 25 mM Tris pH 8.0 to remove dodecylmaltoside and imidazole. Centrifugation of the dialyzed suspension in a fixed-angle rotor (1 h, 100,000 × g) produced a hard pellet consisting of contaminating protein and lipid. Further centrifugation in a swinging bucket rotor (22 h, 100,000 × g) separated the resulting magenta-colored supernatant into a viscous deep red pellet (DsbB Cys41Ser) and a colorless solution. The pellet was then packed into a 3.2 mm thin wall rotor with an active volume of ~36 ml using several low speed centrifugations (5–10 min, 3,000 × g). U-13C-glucose, 2-13C-glycerol, 1,3-13C-glycerol and 15N-NH4Cl were obtained from Cambridge Isotope Laboratories, Andover, MA. The preparation of DsbB-DsbA complex was described previously47. All the membrane samples contain endogenous lipids from the plasma membrane of E. coli, and the lipid to protein ratio is approximately 24:128.
NMR spectroscopy
All SSNMR experiments are summarized in Table S2. SSNMR experiments were performed on a 750 MHz (1H frequency) three-channel Varian Inova spectrometer, a 600 MHz (1H frequency) three-channel Varian Infinity Plus spectrometer and a 500 MHz (1H frequency) three-channel Varian VNMRS spectrometer. The spectrometers were equipped with 3.2 mm 1H-13C-15N Balun MAS probes. The typical π/2 pulse widths were 2~2.5 μs on 1H, 2.7 μs on 13C and 3.7 μs on 15N. The flow rate of sample cooling gas was maintained at 100 scfh, with the gas temperature of −20 °C measured at the output of the delivery stack. The sample temperature is ~ −12°C at MAS speed of 12.5 kHz measured by ethylene glycol48. DARR39 was employed for the 13C-13C mixing, and 1H decoupling at ~75 kHz during indirect evolution and acquisition periods was used. A 90° soft pulse (270 μs) was used in the 1H spin diffusion experiments to select either water protons (4.7 ppm) or lipid acyl chain protons (1.3 ppm) before the 1H spin diffusion and magnetization transfer from 1H to 13C on the protein. The selectivity of the soft pulse was confirmed by 1H 1D spectra with the excitation soft pulse to observe only the peak from either water or lipid acyl chain protons. A short T2 filter (0.54 ms) was used before the 1H mixing to remove the proton magnetization of the protein, which was confirmed by 13C 1D spectra with the T2 filter and no 1H mixing to show no signal from the protein.
NMR Data were processed with NMRPipe49, employing zero filling and Lorentzian-to-Gaussian apodization (30~50 Hz) for each dimension before Fourier transformation. Back linear prediction and polynomial baseline correction (frequency domain) were applied to the direct dimension. Chemical shifts were referenced externally with adamantane, with the downfield 13C resonance referenced to 40.48 ppm50. Peak picking and assignment were performed with Sparky (T. D. Goddard and D. G. Kneller, SPARKY 3, University of California, San Francisco: http://www.cgl.ucsf.edu/home/sparky/). Ambiguous assignments were made using a PASD33 protocol consisting of three successive passes of simulated annealing calculations and updated to be able to treat 2D spectra with modified parameters appropriate for SSNMR 13C-13C peak lists.
Structure calculations
Xplor-NIH version 2.25 was used for the structure calculations34. A starting structure of DsbB-Fab with protons was generated from the crystal structures (PDB code 2ZUQ). For each set of experimental restraints, 200 structures were produced in a protocol with initial MD for 10 ps at 3,000 K, followed by simulated annealing with slow cooling from 3,000 to 25 K in 12.5 K decrements. The energy terms used in structure calculations included those for the 13C-13C distance restraints, TALOS-derived dihedral restraints, a term for refinement against X-ray crystal scattering data, the hydrogen bond database potential51 and the torsion angle potential of mean force52, in addition to the standard nonbonded and covalent terms. The force constant was ramped from 20 to 300 kcal/mol/Å2 for the PASD-determined restraints, the torsion angle database (0.002 to 1 kcal/mol), the nonbonded (0.004 to 4 kcal/mol/Å4), the angle (0.4 to 1 kcal/mol/radians2) and improper dihedral (0.1 to 1 kcal/mol/radians2) energy terms. The force constant for the dihedral angle restraint term was 50 kcal/mol/radian2 during high temperature dynamics and 1,000 kcal/mol/radian2 during simulated annealing. The force constant for X-ray term was 5 kcal/mol/reflection. The structures in figures were rendered by VMD-XPLOR53 and VMD54. The structural quality was analyzed by PROCHECK55.
MD simulations
The lowest energy structure of DsbB bound to UQ1 was solvated using the program SOLVATE56, and a 30 Å layer of water molecules was stripped from the transmembrane helices of DsbB. Aided by the VMD membrane plug-in54, a 100 × 100 Å2 POPE bilayer was generated with its membrane normal along the z-axis. DsbB-UQ1 was then embedded in the POPE bilayer and all lipid molecules falling within a 0.6 Å boundary of the protein were removed. The bilayer was fully hydrated with two 15 Å slabs of water with the VMD solvate plug-in54, and 23 K+ and 22 Cl− ions were added with the VMD autoionize plug-in54 to neutralize the system and to generate a 130 mM salt concentration. The system was minimized for 1000 steps, at which point the lipid tails were relaxed in a 500-ps constant temperature (310 K) and constant volume (NVT) simulation where only the lipid tails were permitted to move. The system underwent a second minimization for another 1000 steps, and yet another 500-ps simulation was performed under constant temperature (310 K) and constant pressure (1 atm) conditions (NPT), with DsbB constrained using harmonic potentials (k = 7.2 kcal/mol/Å2) and UQ1 fixed. Upon releasing the constraints for DsbB and adding constraints for UQ1 (k = 7.2 kcal/mol/Å2), the system was further simulated under the same NPT conditions for an additional 10 ns. For all MD simulations, the program NAMD 2.757 and the protein component of the CHARMM27 parameter set58, CHARMM36 lipid parameters59 and UQ1 parameters60 were used. Langevin dynamics kept the temperature constant (310 K) with a damping coefficient of 5 ps. For NPT simulations, a Langevin piston61 was employed to maintain the pressure at 1 atm. Assuming periodic boundary conditions, the particle mesh Ewald (PME) method62 was employed for computation of long-range electrostatic forces with a grid density of at least 1/Å. All simulations were performed with time steps of 1, 2, and 4 fs for calculation of bonded, nonbonded, and PME interactions, respectively.
Supplementary Material
Highlights.
The integral membrane protein DsbB in E. coli oxidizes the periplasmic protein DsbA.
High-resolution structure of DsbB in the lipid bilayer.
Selective proton spin diffusion experiments reveal membrane topology of DsbB.
Molecular dynamics simulations show a flexible periplasmic loop.
Solid-state NMR confirms an interhelical hydrogen bond between Glu26 and Tyr153.
Acknowledgments
This research was supported by the National Institutes of Health (R01GM075937, S10RR025037, R01GM103999 and R01GM073770 ARRA supplement to C.M.R., and NRSA F32GM095344 to A.E.N), the Molecular Biophysics Training Grant (PHS 5 T32 GM008276) and Ullyot Fellowship to L.J.S., and the NIH Intramural Research Program of CIT to C.D.S. The authors thank the School of Chemical Sciences NMR Facility at the University of Illinois at Urbana-Champaign for assistance with data acquisition, Mike Hallock for helpful assistance with structure calculations, and Dr. Taras Pogorelov for helpful discussion with MD simulations.
Abbreviations
- SSNMR
solid-state NMR spectroscopy
- MD
molecular dynamics
- POPE
1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoethanolamine
- UQ
ubiquinone
- DARR
dipolar assisted rotational resonance
- MAS
magic-angle spinning
Footnotes
The authors declare no conflict of interest.
Accession numbers. Biological Magnetic Resonance Bank: Chemical shifts have been deposited with accession code 18493. Protein Data Bank: Atomic coordinates and NMR restraints for DsbB(Cys41Ser) have been deposited with accession code 2LTQ.
Supplemental figures and tables are provided in the online version.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Inaba K, Ito K. Structure and mechanisms of the DsbB-DsbA disulfide bond generation machine. Biochim Biophys Acta, Mol Cell Res. 2008;1783:520–529. doi: 10.1016/j.bbamcr.2007.11.006. [DOI] [PubMed] [Google Scholar]
- 2.Martin JL, Bardwell JCA, Kuriyan J. Crystal-Structure of the Dsba Protein Required for Disulfide Bond Formation in vivo. Nature. 1993;365:464–468. doi: 10.1038/365464a0. [DOI] [PubMed] [Google Scholar]
- 3.Bardwell JC, Lee JO, Jander G, Martin N, Belin D, Beckwith J. A pathway for disulfide bond formation in vivo. Proc Natl Acad Sci U S A. 1993;90:1038–42. doi: 10.1073/pnas.90.3.1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bader MW, Xie T, Yu CA, Bardwell JC. Disulfide bonds are generated by quinone reduction. J Biol Chem. 2000;275:26082–8. doi: 10.1074/jbc.M003850200. [DOI] [PubMed] [Google Scholar]
- 5.Kobayashi T, Kishigami S, Sone M, Inokuchi H, Mogi T, Ito K. Respiratory chain is required to maintain oxidized states of the DsbA-DsbB disulfide bond formation system in aerobically growing Escherichia coli cells. Proc Natl Acad Sci U S A. 1997;94:11857–62. doi: 10.1073/pnas.94.22.11857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kadokura H, Beckwith J. Four cysteines of the membrane protein DsbB act in concert to oxidize its substrate DsbA. EMBO J. 2002;21:2354–63. doi: 10.1093/emboj/21.10.2354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Inaba K, Takahashi YH, Fujieda N, Kano K, Miyoshi H, Ito K. DsbB elicits a red-shift of bound ubiquinone during the catalysis of DsbA oxidation. J Biol Chem. 2004;279:6761–8. doi: 10.1074/jbc.M310765200. [DOI] [PubMed] [Google Scholar]
- 8.Łasica AM, Jagusztyn-Krynicka EK. The role of Dsb proteins of Gram-negative bacteria in the process of pathogenesis. FEMS Microbiol Rev. 2007;31:626–636. doi: 10.1111/j.1574-6976.2007.00081.x. [DOI] [PubMed] [Google Scholar]
- 9.Inaba K, Murakami S, Suzuki M, Nakagawa A, Yamashita E, Okada K, Ito K. Crystal structure of the DsbB-DsbA complex reveals a mechanism of disulfide bond generation. Cell. 2006;127:789–801. doi: 10.1016/j.cell.2006.10.034. [DOI] [PubMed] [Google Scholar]
- 10.Malojčić G, Owen RL, Grimshaw JPA, Glockshuber R. Preparation and structure of the charge-transfer intermediate of the transmembrane redox catalyst DsbB. FEBS Lett. 2008;582:3301–3307. doi: 10.1016/j.febslet.2008.07.063. [DOI] [PubMed] [Google Scholar]
- 11.Inaba K, Murakami S, Nakagawa A, Iida H, Kinjo M, Ito K, Suzuki M. Dynamic nature of disulphide bond formation catalysts revealed by crystal structures of DsbB. EMBO J. 2009;28:779–791. doi: 10.1038/emboj.2009.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhou YP, Cierpicki T, Jimenez RHF, Lukasik SM, Ellena JF, Cafiso DS, Kadokura H, Beckwith J, Bushweller JH. NMR solution structure of the integral membrane enzyme DsbB: Functional insights into DsbB-catalyzed disulfide bond formation. Mol Cell. 2008;31:896–908. doi: 10.1016/j.molcel.2008.08.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pan JL, Sliskovic I, Bardwell JC. Mutants in DsbB that appear to redirect oxidation through the disulfide isomerization pathway. J Mol Biol. 2008;377:1433–42. doi: 10.1016/j.jmb.2008.01.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Inaba K. Structural basis of protein disulfide bond generation in the cell. Genes Cells. 2010;15:935–43. doi: 10.1111/j.1365-2443.2010.01434.x. [DOI] [PubMed] [Google Scholar]
- 15.Tapley TL, Eichner T, Gleiter S, Ballou DP, Bardwell JCA. Kinetic characterization of the disulfide bond-forming enzyme DsbB. J Biol Chem. 2007;282:10263–10271. doi: 10.1074/jbc.M611541200. [DOI] [PubMed] [Google Scholar]
- 16.McDermott A. Structure and Dynamics of Membrane Proteins by Magic Angle Spinning Solid-State NMR. Annu Rev Biophys. 2009;38:385–403. doi: 10.1146/annurev.biophys.050708.133719. [DOI] [PubMed] [Google Scholar]
- 17.Franks WT, Linden AH, Kunert B, van Rossum BJ, Oschkinat H. Solid-state magic-angle spinning NMR of membrane proteins and protein-ligand interactions. Eur J Cell Biol. 2012;91:340–8. doi: 10.1016/j.ejcb.2011.09.002. [DOI] [PubMed] [Google Scholar]
- 18.Judge PJ, Watts A. Recent contributions from solid-state NMR to the understanding of membrane protein structure and function. Curr Opin Chem Biol. 2011;15:690–5. doi: 10.1016/j.cbpa.2011.07.021. [DOI] [PubMed] [Google Scholar]
- 19.McDermott A, Polenova T. Solid state NMR: new tools for insight into enzyme function. Curr Opin Struct Biol. 2007;17:617–22. doi: 10.1016/j.sbi.2007.10.001. [DOI] [PubMed] [Google Scholar]
- 20.Bajaj VS, Mak-Jurkauskas ML, Belenky M, Herzfeld J, Griffin RG. Functional and shunt states of bacteriorhodopsin resolved by 250 GHz dynamic nuclear polarization-enhanced solid-state NMR. Proc Natl Acad Sci U S A. 2009;106:9244–9. doi: 10.1073/pnas.0900908106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Qiang W, Sun Y, Weliky DP. A strong correlation between fusogenicity and membrane insertion depth of the HIV fusion peptide. Proc Natl Acad Sci U S A. 2009;106:15314–9. doi: 10.1073/pnas.0907360106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Renault M, Bos MP, Tommassen J, Baldus M. Solid-state NMR on a large multidomain integral membrane protein: the outer membrane protein assembly factor BamA. J Am Chem Soc. 2011;133:4175–7. doi: 10.1021/ja109469c. [DOI] [PubMed] [Google Scholar]
- 23.Shi L, Kawamura I, Jung KH, Brown LS, Ladizhansky V. Conformation of a seven-helical transmembrane photosensor in the lipid environment. Angew Chem Int Ed Engl. 2011;50:1302–5. doi: 10.1002/anie.201004422. [DOI] [PubMed] [Google Scholar]
- 24.Higman VA, Varga K, Aslimovska L, Judge PJ, Sperling LJ, Rienstra CM, Watts A. The Conformation of Bacteriorhodopsin Loops in Purple Membranes Resolved by Solid-State MAS NMR Spectroscopy. Angew Chem Int Ed Engl. 2011;50:8432–8435. doi: 10.1002/anie.201100730. [DOI] [PubMed] [Google Scholar]
- 25.Lindahl E, Sansom MS. Membrane proteins: molecular dynamics simulations. Curr Opin Struct Biol. 2008;18:425–31. doi: 10.1016/j.sbi.2008.02.003. [DOI] [PubMed] [Google Scholar]
- 26.Khalili-Araghi F, Gumbart J, Wen PC, Sotomayor M, Tajkhorshid E, Schulten K. Molecular dynamics simulations of membrane channels and transporters. Curr Opin Struct Biol. 2009;19:128–37. doi: 10.1016/j.sbi.2009.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Krepkiy D, Mihailescu M, Freites JA, Schow EV, Worcester DL, Gawrisch K, Tobias DJ, White SH, Swartz KJ. Structure and hydration of membranes embedded with voltage-sensing domains. Nature. 2009;462:473–9. doi: 10.1038/nature08542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li Y, Berthold DA, Gennis RB, Rienstra CM. Chemical shift assignment of the transmembrane helices of DsbB, a 20-kDa integral membrane enzyme, by 3D magic-angle spinning NMR spectroscopy. Protein Sci. 2008;17:199–204. doi: 10.1110/ps.073225008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tang M, Sperling LJ, Berthold DA, Nesbitt AE, Gennis RB, Rienstra CM. Solid-State NMR Study of the Charge-Transfer Complex between Ubiquinone-8 and Disulfide Bond Generating Membrane Protein DsbB. J Am Chem Soc. 2011;133:4359–66. doi: 10.1021/ja107775w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shen Y, Delaglio F, Cornilescu G, Bax A. TALOS plus: a hybrid method for predicting protein backbone torsion angles from NMR chemical shifts. J Biomol NMR. 2009;44:213–223. doi: 10.1007/s10858-009-9333-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Castellani F, van Rossum B, Diehl A, Schubert M, Rehbein K, Oschkinat H. Structure of a protein determined by solid-state magic-angle- spinning NMR spectroscopy. Nature. 2002;420:98–102. doi: 10.1038/nature01070. [DOI] [PubMed] [Google Scholar]
- 32.Tang M, Sperling LJ, Berthold DA, Schwieters CD, Nesbitt AE, Nieuwkoop AJ, Gennis RB, Rienstra CM. High-resolution membrane protein structure by joint calculations with solid-state NMR and X-ray experimental data. J Biomol NMR. 2011;51:227–33. doi: 10.1007/s10858-011-9565-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kuszewski J, Schwieters CD, Garrett DS, Byrd RA, Tjandra N, Clore GM. Completely automated, highly error-tolerant macromolecular structure determination from multidimensional nuclear overhauser enhancement spectra and chemical shift assignments. J Am Chem Soc. 2004;126:6258–6273. doi: 10.1021/ja049786h. [DOI] [PubMed] [Google Scholar]
- 34.Schwieters CD, Kuszewski JJ, Tjandra N, Clore GM. The Xplor-NIH NMR molecular structure determination package. J Magn Reson. 2003;160:65–73. doi: 10.1016/s1090-7807(02)00014-9. [DOI] [PubMed] [Google Scholar]
- 35.Etzkorn M, Martell S, Andronesi OC, Seidel K, Engelhard M, Baldus M. Secondary structure, dynamics, and topology of a seven-helix receptor in native membranes, studied by solid-state NMR spectroscopy. Angew Chem Int Ed. 2007;46:459–62. doi: 10.1002/anie.200602139. [DOI] [PubMed] [Google Scholar]
- 36.Huster D, Yao X, Hong M. Membrane protein topology probed by (1)H spin diffusion from lipids using solid-state NMR spectroscopy. J Am Chem Soc. 2002;124:874–83. doi: 10.1021/ja017001r. [DOI] [PubMed] [Google Scholar]
- 37.Kijac A, Shih AY, Nieuwkoop AJ, Schulten K, Sligar SG, Rienstra CM. Lipid-protein correlations in nanoscale phospholipid bilayers determined by solid-state nuclear magnetic resonance. Biochemistry. 2010;49:9190–8. doi: 10.1021/bi1013722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ader C, Schneider R, Seidel K, Etzkorn M, Becker S, Baldus M. Structural rearrangements of membrane proteins probed by water-edited solid-state NMR spectroscopy. J Am Chem Soc. 2009;131:170–6. doi: 10.1021/ja806306e. [DOI] [PubMed] [Google Scholar]
- 39.Takegoshi K, Nakamura S, Terao T. 13C-1H dipolar-assisted rotational resonance in magic-angle spinning NMR. Chem Phys Lett. 2001;344:631–637. [Google Scholar]
- 40.Gullion T, Schaefer J. Rotational-echo double-resonance NMR. J Magn Reson. 1989;81:196–200. doi: 10.1016/j.jmr.2011.09.003. [DOI] [PubMed] [Google Scholar]
- 41.Li W, Schulman S, Dutton RJ, Boyd D, Beckwith J, Rapoport TA. Structure of a bacterial homologue of vitamin K epoxide reductase. Nature. 2010;463:507–12. doi: 10.1038/nature08720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dutton RJ, Boyd D, Berkmen M, Beckwith J. Bacterial species exhibit diversity in their mechanisms and capacity for protein disulfide bond formation. Proc Natl Acad Sci U S A. 2008;105:11933–8. doi: 10.1073/pnas.0804621105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hessa T, Kim H, Bihlmaier K, Lundin C, Boekel J, Andersson H, Nilsson I, White SH, von Heijne G. Recognition of transmembrane helices by the endoplasmic reticulum translocon. Nature. 2005;433:377–81. doi: 10.1038/nature03216. [DOI] [PubMed] [Google Scholar]
- 44.Ahuja S, Hornak V, Yan EC, Syrett N, Goncalves JA, Hirshfeld A, Ziliox M, Sakmar TP, Sheves M, Reeves PJ, Smith SO, Eilers M. Helix movement is coupled to displacement of the second extracellular loop in rhodopsin activation. Nat Struct Mol Biol. 2009;16:168–75. doi: 10.1038/nsmb.1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Inaba K, Ito K. Paradoxical redox properties of DsbB and DsbA in the protein disulfide-introducing reaction cascade. EMBO J. 2002;21:2646–54. doi: 10.1093/emboj/21.11.2646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Studier FW. Protein production by auto-induction in high density shaking cultures. Protein Expr Purif. 2005;41:207–34. doi: 10.1016/j.pep.2005.01.016. [DOI] [PubMed] [Google Scholar]
- 47.Sperling LJ, Berthold DA, Sasser TL, Jeisy-Scott V, Rienstra CM. Assignment strategies for large proteins by magic-angle spinning NMR: the 21-kDa disulfide bond forming enzyme DsbA. J Mol Biol. 2010;399:268–282. doi: 10.1016/j.jmb.2010.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Raiford DS, Fisk CL, Becker ED. Calibration of methanol and ethylene glycol nuclear magnetic resonance thermometers. Anal Chem. 1979;51:2050–2051. [Google Scholar]
- 49.Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A. Nmrpipe: a Multidimensional Spectral Processing System Based On Unix Pipes. J Biomol NMR. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- 50.Morcombe CR, Zilm KW. Chemical shift referencing in MAS solid state NMR. J Magn Reson. 2003;162:479–486. doi: 10.1016/s1090-7807(03)00082-x. [DOI] [PubMed] [Google Scholar]
- 51.Grishaev A, Bax A. An empirical backbone-backbone hydrogen-bonding potential in proteins and its applications to NMR structure refinement and validation. J Am Chem Soc. 2004;126:7281–7292. doi: 10.1021/ja0319994. [DOI] [PubMed] [Google Scholar]
- 52.Clore GM, Kuszewski J. χ1 Rotamer populations and angles of mobile surface side chains are accurately predicted by a torsion angle database potential of mean force. J Am Chem Soc. 2002;124:2866–2867. doi: 10.1021/ja017712p. [DOI] [PubMed] [Google Scholar]
- 53.Schwieters CD, Clore GM. The VMD-XPLOR visualization package or NMR structure refinement. J Magn Reson. 2001;149:239–244. doi: 10.1006/jmre.2001.2300. [DOI] [PubMed] [Google Scholar]
- 54.Humphrey W, Dalke A, Schulten K. VMD: Visual molecular dynamics. J Mol Graph. 1996;14:33. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 55.Laskowski RA, Macarthur MW, Moss DS, Thornton JM. Procheck - a Program to Check the Stereochemical Quality of Protein Structures. J Appl Crystallogr. 1993;26:283–291. [Google Scholar]
- 56.Grubmuller H, Heymann B, Tavan P. Ligand binding: molecular mechanics calculation of the streptavidin-biotin rupture force. Science. 1996;271:997–9. doi: 10.1126/science.271.5251.997. [DOI] [PubMed] [Google Scholar]
- 57.Phillips JC, Braun R, Wang W, Gumbart J, Tajkhorshid E, Villa E, Chipot C, Skeel RD, Kale L, Schulten K. Scalable molecular dynamics with NAMD. J Comput Chem. 2005;26:1781–802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.MacKerell AD, Bashford D, Bellott, Dunbrack RL, Evanseck JD, Field MJ, Fischer S, Gao J, Guo H, Ha S, Joseph-McCarthy D, Kuchnir L, Kuczera K, Lau FTK, Mattos C, Michnick S, Ngo T, Nguyen DT, Prodhom B, Reiher WE, Roux B, Schlenkrich M, Smith JC, Stote R, Straub J, Watanabe M, Wiórkiewicz-Kuczera J, Yin D, Karplus M. All-Atom Empirical Potential for Molecular Modeling and Dynamics Studies of Proteins. J Phys Chem B. 1998;102:3586–3616. doi: 10.1021/jp973084f. [DOI] [PubMed] [Google Scholar]
- 59.Klauda JB, Venable RM, Freites JA, O’Connor JW, Tobias DJ, Mondragon-Ramirez C, Vorobyov I, MacKerell AD, Jr, Pastor RW. Update of the CHARMM all-atom additive force field for lipids: validation on six lipid types. J Phys Chem B. 2010;114:7830–43. doi: 10.1021/jp101759q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Autenrieth F, Tajkhorshid E, Schulten K, Luthey-Schulten Z. Role of Water in Transient Cytochrome c2 Docking. J Phys Chem B. 2004;108:20376–20387. [Google Scholar]
- 61.Feller SE, Zhang Y, Pastor RW, Brooks BR. Constant pressure molecular dynamics simulation: The Langevin piston method. J Chem Phys. 1995;103:4613–4621. [Google Scholar]
- 62.Darden T, York D, Pedersen L. Particle mesh Ewald: An N-log(N) method for Ewald sums in large systems. J Chem Phys. 1993;98:10089–10092. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.