

Abstract
Factor VIII functions as a cofactor for Factor IXa in a membrane-bound enzyme complex. Membrane binding accelerates the activity of the Factor VIIIa–Factor IXa complex approx. 100000-fold, and the major phospholipid-binding motif of Factor VIII is thought to be on the C2 domain. In the present study, we prepared an fVIII-C2 (Factor VIII C2 domain) construct from Escherichia coli, and confirmed its structural integrity through binding of three distinct monoclonal antibodies. Solution-phase assays, performed with flow cytometry and FRET (fluorescence resonance energy transfer), revealed that fVIII-C2 membrane affinity was approx. 40-fold lower than intact Factor VIII. In contrast with the similarly structured C2 domain of lactadherin, fVIII-C2 membrane binding was inhibited by physiological NaCl. fVIII-C2 binding was also not specific for phosphatidylserine over other negatively charged phospholipids, whereas a Factor VIII construct lacking the C2 domain retained phosphatidyl-L-serine specificity. fVIII-C2 slightly enhanced the cleavage of Factor X by Factor IXa, but did not compete with Factor VIII for membrane-binding sites or inhibit the Factor Xase complex. Our results indicate that the C2 domain in isolation does not recapitulate the characteristic membrane binding of Factor VIII, emphasizing that its role is cooperative with other domains of the intact Factor VIII molecule.
Keywords: blood coagulation, C2 domain, Factor VIII, lactadherin, phosphatidylserine
INTRODUCTION
Factor VIII is an essential cofactor in the blood-clotting cascade. Its importance is illustrated by the bleeding disorder haemophilia A, which is caused by reduced or absent Factor VIII function. Factor VIII acts as a membrane-bound cofactor for Factor IXa, a serine protease that catalyses the activation of Factor X to Factor Xa [1]. Binding of Factor VIII to phospholipid membranes increases the efficiency of Factor X cleavage over 100000-fold [1], making this interaction vital for Factor VIII function. The binding of Factor VIII to phospholipid membranes is a multistep process in which a rapid initial attachment is followed by a slower step yielding higher affinity [2]. Available results indicate that membrane binding is mediated by both hydrophobic and ionic interactions [3], although the exact sequence of binding events has not been elucidated. Factor VIII binds specifically and with high affinity to membranes containing PS (phosphatidylserine) [4,5].
The sequence of Factor VIII contains three ceruloplasmin-like A domains, a unique B domain and two discoidin-like C domains [6,7]. Factor VIII is cleaved prior to secretion to yield a heterodimeric protein consisting of a heavy chain (A1-A2-B) and a light chain (A3-C1-C2). Membrane binding is mediated by the light chain [8], and particularly by the C domains. The importance of the interaction between the fVIII-C2 (Factor VIII C2 domain) and phospholipid membranes is illustrated by severe bleeding in patients who develop antibodies against the C2 domain [9].
The Factor VIII C domains share similar sequence and structure with the C domains of Factor V [6,7,10] (another membrane-bound cofactor of the clotting cascade) and lactadherin [11] (a milk fat globule membrane protein). The crystal structure of the C2 domains of all three proteins show a β-barrel core and three hairpin loops forming hydrophobic spikes that probably penetrate the membrane surface during binding [12–14]. As with Factor VIII, both Factor V and lactadherin preferentially bind to PS-containing membranes [15,16]. The membrane-binding role of the protruding hydrophobic amino acids of the fVIII-C2 [17], Factor V C2 domain [18,19] and Lact-C2 (lactadherin C2 domain) [14] has been confirmed by site-directed mutagenesis. In addition, antibodies directed to the C2 domain in both Factor VIII [20–22] and Factor V [23] inhibit phospholipid membrane binding. However, the available results are insufficient to determine the fraction of Factor VIII membrane-binding affinity that is derived from the C2 domain, whether the stereospecific recognition of PS resides within this motif, and whether the C2 domain mediates the initial adherence of Factor VIII with a phospholipid membrane.
To form the Factor Xase complex, Factor IXa interacts with Factor VIII primarily through the A2 and A3 domains [24,25]. In addition, the crystal structures of intact Factor VIII [26,27] have added to the biochemical data [28–30] in supporting a model of Factor VIII cofactor function in which the C1 and/or C2 domains also make contact with the Gla domains of Factor IXa and/or Factor X. Indeed, a recent study of the interaction between Factor IXa and the fVIII-C2 assigns a Factor VIII C2 peptide to a specific interaction with Factor IXa [31]. Thus there is motivation to determine the apparent affinity of the isolated C2 domain for Factor IXa and/or Factor X and its impact on function.
In the present study, we have characterized the binding of the fVIII-C2 to PLVs (phospholipid vesicles) and supported membranes in solution and compared this binding to that of the Lact-C2 and intact Factor VIII. We have also investigated the role of the C2 domain in the protein–protein interactions of the Factor Xase complex. Our results show a more complex role for the fVIII-C2 in phospholipid binding than expected, and suggest that other domains play a role, affecting the interaction of the C2 domain with the membrane or making initial contact with the membrane surface.
EXPERIMENTAL
Materials
Porcine brain PS, egg PE (phosphatidylethanolamine), egg PC (phosphatidylcholine), bovine liver PI (phosphatidylinositol), biotin-PE [1,2-dihexadecanoyl-sn-glycero-3-phosphoethanol-amine-N-(cap biotinyl)], dansyl-PE [1,2-dioleoyl-sn-glycero-3-phosphoethanolamine-N-(5-dimethylaminonaphthalenesulfonyl)], Ptd-D-Ser (1,2-dioleoyl-sn-glycero-3-phospho-D-serine) and Ptd-L-Ser (1,2-dioleoyl-sn-glycero-3-phospho-L-serine) were purchased from Avanti Polar Lipids. DPC (n-dodecyl phosphocholine) was purchased from Anatrace. Glass microspheres (1.6 μm nominal diameter) were from Duke Scientific. Factor VIII was a gift from Baxter. The Lact-C2 construct was prepared as described previously [14]. ESH4 and ESH8 antibodies were purchased from American Diagnostica. B02C11 was generously provided by Professor Marc Jacquemin (Centre for Molecular and Vascular Biology, University of Leuven, Leuven, Belgium). The anti-(Factor VIII A2) antibody (GMA-8021) was purchased from Green Mountain antibodies, and was labelled with FITC using a standard protocol. Human Factor IXa, Factor X and thrombin were purchased from Enzyme Research Laboratories. The fVIII-C2Pichia (fVIII-C2 prepared in Pichia pastoris) was generously provided by Dr Kathleen Pratt (Division of Hematology, University of Washington School of Medicine, Seattle, WA, U.S.A.) [13].
PLV preparation
Chloroform-dissolved phospholipids were mixed in the indicated molar ratios. Methylene chloride was added to the phospholipid mix and evaporated under argon four times to remove the chloroform. The dried phospholipids were then resuspended in TBS (Tris-buffered saline) [50 mM Tris/HCl (pH 7.85) and 150 mM NaCl] and sonicated in a bath sonicator for at least 20 min until visually clear [4]. For vesicles used in FRET (fluorescence resonance energy transfer) experiments, the phospholipid suspension was ultracentrifuged at 200000 rev./min (TLA-120.2 rotor; Beckman) for 2 h at 4°C to remove any remaining large vesicles [32]. The phospholipid concentration of the centrifuged preparations was determined using an elemental phosphorus assay [33]. Aliquots were flash-frozen in liquid nitrogen and stored at − 80°C. Thawed aliquots were stored at 4°C and used within 8 h.
Purification of fVIII-C2
The C2 domain of human Factor VIII was cloned from a plasmid containing the full Factor VIII sequence. The forward primer 5′-CTCGGATCCCATCACCATCACCATCACCTGGTGCCAC-GCGGTAGTTGCAGCATGCCATTGGGAAT-3′ and reverse primer 5′-CTCCTCGAGTCAGTAGAGGTCCTGTGCCT-3′ were used to amplify the sequence and to add an N-terminal thrombin-cleavage site and a His6 tag. The insert was cut using BamHI and XhoI and ligated into a modified pET21 ampicillin-resistant plasmid that included an N-terminal Trx (thioredoxin) sequence to increase construct solubility. The resulting plasmid was transformed into BL21(DE3) Escherichia coli cells (Novagen). Cells were grown in 400 ml of LB (Luria–Bertani) broth to a D600 of approx. 0.7 and then induced with 50 μM IPTG (isopropyl β-D-thiogalactopyranoside) for 5 h. Cells were pelleted and then lysed using 15 ml of B-PER (bacterial protein extraction reagent; Pierce Chemical) according to the manufacturer’s instructions. The lysate was loaded on to a 10 ml Ni-NTA (Ni2+-nitrilotriacetate) column (Qiagen) and washed with 15 ml of B-PER. The protein was eluted with a five-step elution of 4, 8, 30, 60 and 200 mM imidazole in 150 mM NaCl, 20 mM Tris/HCl (pH 7.5). Fractions were analysed on a LabChip BioAnalyzer (Agilent Technologies). Positive fractions were pooled and 0.0175 mg of thrombin/mg of fVIII-C2 was added to cleave the His–Trx tag. After being incubated at 37°C for 4 h, the thrombin was inactivated with a 3:1 molar ratio of PPACK (D-Phe-Pro-Arg-chloromethylketone). The product was concentrated and buffer-exchanged to 25 mM NaCl in 5 mM MES (pH 5.5) using an Amicon Ultra-15, 10000 Mr cut-off centrifugal concentrator (Millipore). The thrombin and cleaved tag were removed by cation-exchange chromatography using a 1 ml HiTrap SP HP column (GE Healthcare). The column was equilibrated with 25 mM NaCl in 5 mM MES (pH 5.5), and the fVIII-C2 was eluted using 0.2 mM NaCl in 20 mM Hepes (pH 8.0). Fractions were monitored for protein by measuring the absorbance at 280 nm. Positive fractions were analysed with a LabChip to detect fVIII-C2 and to determine purity (>98%) and concentration (see Supplementary Figure S1). fVIII-C2 aliquots were flash-frozen in liquid nitrogen and stored at − 80°C. To ensure formation of the disulfide bond, the construct was tested for the presence of free thiols using Ellman’s reagent.
Mutant fVIII-C2 construct and fluorescein labelling
Since the fVIII-C2 construct does not contain a free thiol for fluorescence labelling, a mutant in which Val2223 was replaced with a cysteine residue (fVIII-C2C) was produced through PCR mutagenesis using primers 5′-GCCTGGAG-ACCTCAGTGTAATAATCCAAAAGAGTGGCTGCAAGTG-3′ and 5′-CACTTGCAGCCACTCTTTTGGATTATTACACTGA-GGTCTCCAGGC-3′. The mutant was then purified as described above. The presence of a single free thiol was confirmed using Ellman’s reagent. fVIII-C2C was diluted in labelling buffer (0.1 M betaine, 2 mM DPC and 20 mM Hepes) and incubated with 0.1 mM fluorescein-5-maleimide (Molecular Probes) overnight at 4°C. The buffer pH was adjusted to 7.2 using 1 M sodium bicarbonate. Excess fluorescein was removed using gel filtration over a Sephadex G50 matrix (Sigma). The protein was eluted with 25 mM NaCl, 2 mM DPC and 5 mM MES buffer (pH 5.5). The DPC was removed using cation-exchange chromatography as described previously for fVIII-C2. Fractions were monitored using 280 and 490 nm absorbance readings. The protein concentration of the positive fractions was determined using a microBCA assay.
Testing the conformation of fVIII-C2 with mAbs (monoclonal antibodies)
We selected three antibodies to the C2 domain of Factor VIII: ESH4, ESH8 and B02C11. ESH4 and ESH8 were coupled to cyanogen bromide-activated cross-linked Sepharose beads (Superose; GE Healthcare), whereas B02C11 was attached to Protein G-linked agarose beads (Sigma). The beads were then used to measure the affinity of the antibodies for Factor VIII-Fl (fluorescein-labelled Factor VIII). The average fluorescence per bead was read using a CSCaliber flow cytometer (BD Biosciences). The resulting data sets were fitted to a standard one-site binding model to calculate the Kd and Fmax (maximum fluorescence) for each antibody. To test the affinity of the antibodies for fVIII-C2, unlabelled fVIII-C2 was used to compete with the Factor VIII-Fl for the antibody-binding sites. The concentration of Factor VIII-Fl used in the competition experiments was 3 nM for ESH4, 0.8 nM for ESH8 and 0.3 nM for B02C11. The binding competition was modelled to eqn (1) for single binding-site competition:
| (1) |
where F is the measured fluorescence, [fVIIIfl] is the concentration of Factor VIII-Fl, and [fVIII-C2] is the concentration of fVIII-C2. The Kd and Fmax values calculated previously were substituted into the equation, and Ki and Fres (residual background fluorescence) were fitted to the data using Graphpad Prism software.
Binding of vesicles to immobilized fVIII-C2
The integrity of the phospholipid-binding motif of the fVIII-C2 was tested using an ELISA assay. Microtitre wells were coated with mAb ESH8 (5 μg/ml) for 4 h. After blocking with 2%(w/v) BSA, Factor VIII or fVIII-C2 (10 nM) was incubated for 1 h to allow capture by the immobilized antibodies. Sonicated biotin-labelled PLVs (PS/biotin-PE/PE/PC: 15:1:19:65 molar ratio) were pre-incubated for 1 h with streptavidin–HRP (horseradish peroxidase) and then added to the bound protein in the microtitre wells. Excess vesicles were washed away with 0.5%Tween 80 in TBS and bound vesicles were detected using o-phenylenediamine dihydrochloride (Sigma). Colour generation was stopped using 3 M HCl and then read at 490 nm on a Vmax microplate reader (Molecular Devices). The experiment was repeated with different concentrations of Factor VIII and fVIII-C2 to check for cooperativity.
C2-deleted Factor VIII
ΔC2 FVIII (Factor VIII lacking the C2 domain) was prepared as a B-domainless Factor VIII using methods described previously [34]. Recombinant ΔC2 FVIII (lacking >98%of the C2 domain, residues 2173–2332) was stably expressed in BHK (baby-hamster kidney) cells and purified as described previously [34]. Protein yields for the variant ranged from >10 to approx. 100 μg from two 750 cm2 culture flasks, with a purity from approx. 85% to >95%as judged by SDS/PAGE.
Liposphere-binding assays
Glass microspheres were cleaned, size-restricted and coated with phospholipid as described previously [4]. The resulting lipospheres were washed in TBS containing 0.1% BSA and 10μM PC-sonicated vesicles to remove excess phospholipid. Lipospheres were stored on ice and used within 8 h of preparation. For binding curves, fVIII-C2C-Fluor (flourescein-labelled fVIII-C2C) or Factor VIII-Fl was incubated for 15 min with 6×105 lipospheres/ml (PS/PE/PC: 1:1:3 molar ratio) in 0.1% BSA and 20 mM Tris/HCl (pH 7) with the indicated concentration of NaCl. For antibody-inhibition experiments, various concentrations of ESH8, ESH4 or B02C11 were incubated with 60 nM fVIII-C2C-Fluor for 1 h at room temperature (25 °C) prior to the addition of lipospheres.
To characterize fVIII-C2 binding to different phospholipids, free vesicles of various PS and PI compositions were added to compete with the lipospheres for binding to 60 nM fVIII-C2C-Fluor. Factor VIII-Fl (10 nM) and ΔC2 FVIII (2 nM) were tested using free vesicles containing either 1:24 molar ratio of Ptd-L-Ser or 1:24 molar ratio of Ptd-D-Ser. To detect binding of ΔC2 FVIII, 40 nM ΔC2 FVIII was incubated for 1 h with a 2-fold molar excess of FITC-labelled anti-(Factor VIII A2) antibody prior to incubation with the lipospheres/free vesicle mixtures. Experiments with ΔC2 FVIII were carried out in TBS containing 0.1% BSA. The samples were read using flow cytometry. Competition curves were modelled using the following equation:
| (2) |
where [PL] is the concentration of free phospholipid and [fVIII-C2]bound is the measured fluorescence per liposphere.
Fluorescence resonance experiments
Resonance energy transfer, from native tryptophan residues to dansyl-PE incorporated into vesicles, was measured as described previously [14,35]. fVIII-C2, Lact-C2 or Factor VIII stock solution was titrated into a 3 μM solution of dansyl-labelled sonicated PLVs (dansyl-PE/PS/PC: 1:4:15 molar ratio) in 20 mM Tris/HCl (pH 7). Additions were made each minute and readings were taken for 40 s after mixing. Control readings were taken of the PLV solution with buffer additions at the same volume as the protein solution to account for sample dilution and photobleaching over the course of the experiment. A buffer control was subtracted from the experiment and PLV control measurements. The increase in dansyl fluorescence was calculated using eqn (3):
| (3) |
where F is the dansyl fluorescence measured from the protein sample, Fb is the baseline fluorescence from the PLV control, and Fc is the corrected fluorescence [35]. Measurements were made with a Spex Fluorolog II fluorometer with excitation and emission at 280 and 520 nm respectively. To reduce scatter, a PerkinElmer UV-35 filter and a 450 nm high-pass filter were used in the excitation and emission pathways respectively.
Factor IXa activity assays
Factor Xase activity and Factor IXa activity were measured with two-step amidolytic substrate assays. Components were mixed into reaction buffer [20 mM Tris/HCl (pH 7.0), 0.2%BSA and 1.5 mM Ca2+] to a final volume of 40 μl. Final reaction concentrations for the Factor Xase assay were 1 unit/ml Factor VIII, 0.25 μM sonicated PLV (PS/PE/PC: 3:4:13 molar ratio), 2 nM Factor IXa, 0.8 unit/ml thrombin and 40 nM Factor X. The reaction was incubated for 5 min at room temperature. Unless otherwise indicated, the final reaction concentrations used in the Factor IXa activity assays were 25 nM Factor IXa, 20 μM sonicated PLV and 230 nM Factor X. To increase signal intensity in the absence of Factor VIII, the Factor IXa reactions were incubated for 1 h at room temperature. For both protein assays, the reactions were stopped by adding 30 μl of 20 mM Tris/HCl (pH 7.0), 0.2% BSA and 16 mM EDTA to each well. A total of 4 μl of 2 mM S-2765 Xa chromogenic substrate (Diapharma) was added to each well and the speed of colour generation was measured at 405 nm using a Vmax microplate reader (Molecular Devices) in kinetic mode. Measurements in ΔAbs/min (absorbance change/min) were converted into nM Factor Xa generated using a standard curve of known Factor Xa concentrations.
RESULTS
Isolated fVIII-C2 was purified from E. coli cytosol using metal-ion chromatography, followed by cation-exchange chromatography (Supplementary Figure S1 at http://www.BiochemJ.org/bj/435/bj4350187add). Purified fVIII-C2 had the anticipated molecular mass and showed less than 2%residual free thiol, indicating formation of the disulfide bond between Cys2174 and Cys2326. The folding of the construct was evaluated through binding to three well-characterized mAbs: ESH4, ESH8 and B02C11 [9] (Table 1). Competition experiments between Factor VIII-Fl and fVIII-C2 for antibodies linked to Superose beads were performed. For all three antibodies, their Fres was less than 2 % of the Fmax value. The implied binding affinities of fVIII-C2 for the three antibodies were not significantly different than those of Factor VIII, indicating that fVIII-C2 is folded correctly. Since ESH4 [22] and B02C11 [21] both block phospholipid binding of Factor VIII, the results also suggest that the membrane-binding epitope(s) is/are intact.
Table 1. Antibody binding to fVIII-C2 compared with Factor VIII.
Antibodies ESH4, ESH8 and B02C11 were covalently linked to Superose beads and the affinity of each antibody for Factor VIII-Fl was determined using flow cytometry. Unlabeled fVIII-C2 was used to compete with Factor VIII-Fl for bead-bound antibody to calculate a Ki (eqn 1). The values shown are means ± S.E.M for the curve fits.
| Antibody | Factor VIII Kd (nM) | fVIII-C2 Ki (nM) |
|---|---|---|
| ESH4 | 38 ± 8 | 26 ± 5 |
| ESH8 | 19 ± 2 | 18 ± 6 |
| B02C11 | 0.20 ± 0.07 | 0.27 ± 0.05 |
We then used a novel solid-phase assay to further evaluate the integrity of the fVIII-C2 phospholipid-binding site [36]. fVIII-C2 or Factor VIII was immobilized in a microtitre well using ESH8. Biotin-labelled PLVs were incubated with the immobilized protein and bound vesicles were detected with streptavidin–HRP. Vesicles bound to fVIII-C2 with an implied affinity similar to Factor VIII (Figure 1A). To test whether the apparent high affinity of fVIII-C2 might arise from engagement of a vesicle by multiple fVIII-C2 modules, we repeated the experiment using various concentrations of fVIII-C2 (or Factor VIII) to load the microtitre well (Figure 1B). Unlike the Factor VIII findings, the resulting fVIII-C2 data had a sigmoidal shape, consistent with an avidity effect. The data were fitted to the Hill equation, giving a Hill constant of 2.4 ± 0.5 for fVIII-C2. Addition of B02C11 completely blocked vesicle binding to fVIII-C2 (Figure 1C). Thus the apparent high affinity of vesicles to immobilized fVIII-C2 is largely attributable to avidity and is mediated by the known membrane-binding motif.
Figure 1. Immobilized fVIII-C2 binds PLVs via the established membrane-binding motif.

(A) fVIII-C2 or intact Factor VIII was immobilized in microtitre wells coated with mAb ESH8. PLVs labelled with biotin-PE (PS/PE-biotin/PE/PC: 15:1:19:65 molar ratio) were then incubated with the immobilized proteins for 1 h. Bound vesicles were detected using streptavidin–HRP and o-phenylenediamine dihydrochloride. (B) The experiment was repeated using various concentrations of Factor VIII or fVIII-C2 binding to 5 μM phospholipid. The resulting phospholipid-binding data were fitted to a Hill-type binding model giving a Hill constant of 2.4 ± 0.5 for fVIII-C2. The plot shows means ± S.E.M. of two experiments each performed in triplicate. (C) Immobilized fVIII-C2 was incubated for 1 h with various concentrations of mAb B02C11 prior to incubation with 1.3 μM PLVs. Binding of vesicles to fVIII-C2 was inhibited by mAb B02C11. Unless otherwise indicated, plots show experiments performed in duplicate, representative of two experiments. Background absorbance from control wells lacking fVIII-C2 or Factor VIII (<0.15) was subtracted from experimental wells.
We measured the binding of solution-phase fVIII-C2 to phospholipid bilayers supported by glass microspheres (lipospheres) (Figure 2A). For these experiments, a mutant, Val2223Cys (fVIII-C2C) was produced and labelled with fluorescein (fVIII-C2C-Fluor). This mutation site was chosen because the similar mutation Val2223Ala causes no loss of phospholipid binding or activity in intact Factor VIII [17]. Flow cytometry measurements were used to record the binding of fVIII-C2C-Fluor to lipospheres with a membrane composition molar ratio of 1:1:3. PS/PE/PC. In the presence of 150 mM NaCl, fVIII-C2C-Fluor showed little binding above background. The lack of binding persisted even when the incubation time was varied to a maximum of 2 h (results not shown). In contrast, when the buffer did not contain NaCl, fVIII-C2C-Fluor had saturable binding. Under these conditions, fVIII-C2C-Fluor showed an intermediate affinity for membrane-binding sites (Kd = 150 ± 40 nM), approx. 40-fold lower than reported previously for intact Factor VIII [35].
Figure 2. Solution-phase binding of fVIII-C2 and Lact-C2 to phospholipid membranes.

(A) fVIII-C2C-Fluor was incubated with lipospheres containing 20 % PS at the indicated concentrations for 15 min. Bound fVIII-C2C-Fluor was then measured using flow cytometry. When no NaCl (●) was added to the buffer, fVIII-C2C-Fluor bound saturably. Using buffer containing 150 mM NaCl (▼) decreased fVIII-C2C-Fluor binding over 90 %. Non-linear least squares curve fitting indicated a Kd value of 150 ± 40 nM without NaCl. Results with 150 mM NaCl did not lead to a convergent fit. (B) Three mAbs were tested for their ability to inhibit fVIII-C2C-Fluor binding to 20 % PS lipospheres in the absence of NaCl. Both B02C11 and ESH4 were able to inhibit membrane binding, whereas ESH8 had little effect. (C) FRET measurements were utilized to detect the binding of unlabelled fVIII-C2 to vesicles of a composition of 5:1:15 molar ratio of PS/dansyl-PE/PC. Similar to the liposphere binding, fVIII-C2 showed at least 90 % reduction in binding with 150 mM NaCl (▼). The Kd values were 260 ± 40, 250 ± 40 and 390 ± 130 nM in 0 mM (●), 20 mM (▲) and 50 mM NaCl (■) respectively. (B) Membrane binding of Lact-C2 was measured for comparison with fVIII-C2. In contrast with fVIII-C2, Lact-C2 bound saturably to vesicles in 150 mM NaCl with a Kd value of 290 ± 30 nM. In the absence of NaCl, there was an increase in binding sites for Lact-C2, but a decrease in overall affinity. Each condition is the means ± S.D. for at least two experiments.
Because binding of fVIII-C2C-Fluor to lipospheres differed from the solid-phase binding assay (Figure 1), we performed additional experiments. To evaluate the impact of mAb ESH8, binding of fVIII-C2C-Fluor to lipospheres was measured in the presence of saturating concentrations of antibody. Saline continued to inhibit binding to a similar degree, indicating that the effects of ESH8 do not explain membrane binding in the presence of saline (results not shown). We further investigated fVIII-C2C-Fluor binding in the absence of NaCl by testing the effect of the known inhibitor antibodies B02C11 and ESH4, as well as ESH8 (Figure 2B). B02C11 and ESH4 were both able to fully inhibit fVIII-C2 liposphere binding, whereas ESH8 had little effect (~ 11% inhibition with a 1:1 ratio of antibody to fVIII-C2), indicating the participation of the known phospholipid-binding epitope(s) in the absence of NaCl.
To confirm that the inhibitory effect of NaCl and the membrane-binding affinity were not influenced by the Val2223Cys mutation, we used FRET to measure the binding of unlabelled fVIII-C2 to dansyl-labelled PLVs (Figure 2C). fVIII-C2 showed an increase in binding as the concentration of NaCl decreased. The binding affinity of fVIII-C2 for vesicles at 0 mM NaCl was similar to that of fVIII-C2C-Fluor for lipospheres (Kd = 260 ± 40 nM). To be certain that E. coli expression did not alter fVIII-C2 function, we performed an identical experiment using fVIII-C2Pichia, the benchmark fVIII-C2 used in the crystal structure [13]. fVIII-C2Pichia also showed no phospholipid binding in the presence of 150 mM NaCl and similar affinity binding (Kd = 240 ± 60 nM) as the bacterial construct when there was no NaCl (Supplementary Figure S2 at http://www.BiochemJ.org/bj/435/bj4350187add.htm).
For comparison of fVIII-C2 with a similar protein, we repeated the FRET-binding experiments with Lact-C2 (Figure 2D). In contrast with fVIII-C2, Lact-C2 was able to bind to PLVs in the presence of physiological saline (Kd = 290 ± 30 nM). In the absence of NaCl, the number of Lact-C2-binding sites increased (Bmax = 2.80 ± 0.09 with and 5.4 ± 0.1 without NaCl), while its overall affinity for the membrane decreased (Kd = 620 ± 40 nM without NaCl). Our results confirm that the binding of fVIII-C2 differs from the similar Lact-C2 with respect to the influence of salt.
To test whether the determinants for Ptd-L-Ser specificity reside in the C2 domain, sonicated PLVs of varying phospholipid compositions were used to compete with lipospheres for fVIII-C2C-Fluor binding (Figure 3A). Curves were fitted with a non-logarithmic competition binding model [eqn (2)] and Ki values for the different phospholipids were compared. fVIII-C2C-Fluor showed an increase in the Ki value as the relative concentration of PS in the vesicles decreased, with a 70-fold increase between 20% PS and 4% PS vesicles. There was no difference in the competition curves when PI or phosphatidic acid (results not shown) was used instead of PS. In contrast, Factor VIII showed marked specificity for Ptd-L-Ser over Ptd-D-Ser even in the absence of NaCl (Figure 3B), demonstrating that this specificity is not NaCl-dependent. Furthermore, ΔC2 FVIII retained Ptd-L-Ser specificity in the presence of NaCl (Figure 3C). Thus our findings show that the determinants of PS specificity do not lie within the C2 domain.
Figure 3. Effect of phospholipid structure and membrane charge on fVIII-C2 binding.

(A) Sonicated PLV of either 100 % PC or various PS or PI composition were used to compete with lipospheres for binding of 60 nM fVIII-C2C-Fluor. Vesicles with PS or PI also contained 20 % PE with the balance as PC. Experiments were performed in 0 mM NaCl and 20 mM Tris/HCl (pH 7). The inhibition constant for each vesicle type was calculated using a competition binding model [eqn (2)]. The inset shows four of the competition curves with the associated curve fits. fVIII-C2 exhibited a gradual increase in affinity as the PS concentration increased and showed no specificity for PS when compared with PI. (B) In contrast with the isolated C2 domain, Factor VIII-Fl exhibited specificity for vesicles containing Ptd-L-Ser over vesicles containing Ptd-D-Ser (Ptd-L/D-Ser/PE/PC: 1:5:19 molar ratio) in the absence of NaCl. (C) ΔC2 FVIII [2 nM; detected using FITC-labelled anti-(Factor VIII A2) antibody] also showed specificity for Ptd-L-Ser vesicles in the presence of NaCl. Results are means ± S.E.M. from non-linear least-squares fitted curves. Curves show means ± S.D. for at least two experiments for each vesicle type.
Because membrane binding of fVIII-C2 is reduced in physiological NaCl, we asked whether the concentration of NaCl affects membrane binding of intact Factor VIII. Both liposphere binding of Factor VIII-Fl and FRET between Factor VIII and dansyl-labelled sonicated vesicles were used to measure Factor VIII binding without and with 150 mM NaCl (Figure 4). In buffer with no added NaCl, the number of Factor VIII-binding sites increased. The additional binding sites had a lower affinity than those available with NaCl present, thus increasing the overall Kd. The increase in binding in the absence of NaCl is consistent with the enhanced binding of fVIII-C2 in the absence of NaCl.
Figure 4. Effect of decreased NaCl on membrane binding of intact Factor VIII.

(A) Factor VIII-Fl was incubated with lipospheres containing 20 % PS at the indicated concentrations for 15 min. Bound Factor VIII-Fl was then measured using flow cytometry. In the absence of NaCl, the number of binding sites increased (B max =5000 ± 100 fluorescence units with and 9000 ± 300 fluorescence units without NaCl), whereas the overall binding affinity of Factor VIII decreased (Kd = 3.1 ± 0.3 nM with and 8.4 ± 0.7 nM without NaCl). (B) Binding of Factor VIII to PLVs was measured with FRET. The vesicle composition was a 4:1:15 molar ratio of PS/dansyl-PE/PC. The number of Factor VIII-binding sites more than doubled when NaCl was omitted from the buffer (F max =1.22 ± 0.05 with compared with 0.41 ± 0.05 without NaCl), whereas the overall affinity decreased. Values are means ± range for two experiments.
We performed competitive-binding experiments to determine whether fVIII-C2 binds to the membrane sites recognized by intact Factor VIII (Figure 5). fVIII-C2 did not compete with Factor VIII at concentrations up to 1 μM (Figure 5A) in the presence or absence of 150 mM NaCl. However, fVIII-C2 did compete successfully with fVIII-C2C-Fluor, confirming the specificity of membrane-binding sites for fVIII-C2C-Fluor. Intact Factor VIII also competed with fVIII-C2C-Fluor for binding sites (results not shown), indicating that Factor VIII is able to occupy fVIII-C2 phospholipid-binding sites. The Factor Xase assay was used to determine whether fVIII-C2 could displace Factor IXa or Factor X from Factor VIII in the Factor Xase complex. At concentrations up to 6 μM fVIII-C2, there was no loss of Factor Xase activity (Figure 5B). These results indicate that fVIII-C2 engages membrane-binding sites distinct from those of intact Factor VIII. They also indicate that fVIII-C2 does not bind Factor IXa or Factor X with high affinity.
Figure 5. fVIII-C2 competition for Factor VIII membrane-binding sites.
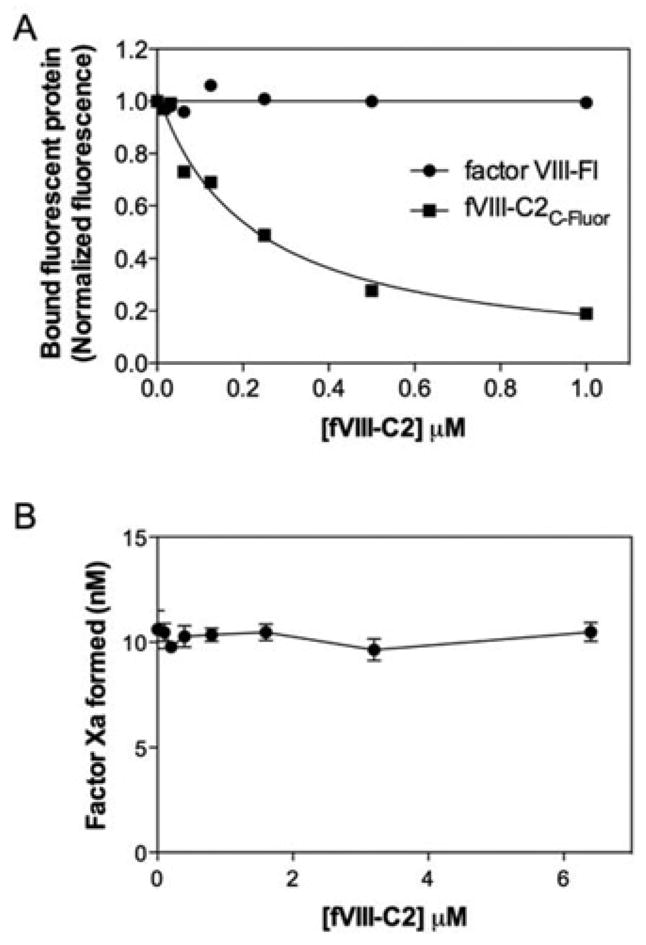
(A) Increasing concentrations of fVIII-C2 were mixed with 4 nM Factor VIII-Fl or 100 nM fVIII-C2C-Fluor prior to the addition of lipospheres. After 15 min, bound Factor VIII-Fl or fVIII-C2C-Fluor was measured by flow cytometry. fVIII-C2 competed for the binding sites of fVIII-C2C-Fluor but not intact Factor VIII. Liposphere membrane content was 3:4:13, by vol. PS/PE/PC. (B) Increasing concentrations of fVIII-C2 were mixed with Factor VIII prior to addition of PLV, Factor IXa and Factor X in a buffer without added NaCl. The reaction was started by addition of Ca2+ and thrombin. After 5 min, the reaction was stopped using EDTA and Factor Xa was measured using the S-2765 chromogenic substrate. fVIII-C2 had no effect on the Factor Xase assay. Values are means (range) from two experiments performed in duplicate.
Because a recent report [31] has identified a putative Factor IXa-binding motif on fVIII-C2, the activity of Factor IXa was evaluated in the presence of various concentrations of fVIII-C2. The experiment indicated that 6.4 μM fVIII-C2 increased Factor Xa generation by 50% (Figure 6A). We further investigated the role of fVIII-C2 by varying the phospholipid and Factor X concentrations, in turn, without and with 3 μM fVIII-C2. Addition of fVIII-C2 had no effect on the apparent affinity of Factor IXa for PLVs (Figure 6B) (Kd = 8 ± 1 μM without and 8.3 ± 0.9 μM with fVIII-C2), but increased the Vmax of the curve by approx. 50%. fVIII-C2 increased the apparent affinity of Factor IXa for Factor X (Figure 6C) (Km = 0.6 ± 0.09 μM without and 0.39 ± 0.08 μM with fVIII-C2), while not significantly increasing the Vmax(19.2 ± 0.8 nM/h without compared with 18.0 ± 0.8 nM/h with fVIII-C2). fVIII-C2 did not affect the cleavage of Factor X by Factor IXa when the NaCl concentration was 150 mM (results not shown), suggesting that intact phospholipid binding is required for the modest impact on cleavage of Factor X. These results are consistent with the weak interaction of fVIII-C2 with Factor X and/or Factor IXa.
Figure 6. Effect of fVIII-C2 on Factor IXa activity.

(A) Increasing concentrations of fVIII-C2 were added to Factor IXa, Factor X and PLVs. The reaction was started by the addition of 1.5 mM Ca2+ and, after 1 h at room temperature, was stopped with addition of EDTA. Addition of fVIII-C2 to the reaction increased the Factor Xa formation by approx. 50 %. (B) The Factor IXa activity assay was repeated with various phospholipid concentrations without or with 3 μM fVIII-C2. fVIII-C2 increased the Vmax of the curve by 50 %, but had no significant effect on the apparent Kd. (C) Factor IXa activity was measured without and with 3 μM fVIII-C2 and various Factor X concentrations. fVIII-C2 increased the apparent affinity for Factor X. Values are means ± S.D. from at least two experiments performed in duplicate.
We asked whether addition of fVIII-C2 might restore normal membrane-binding affinity to Factor VIII lacking the C2 domain. Accordingly, fVIII-C2 was titrated into a Factor Xase complex with fixed concentrations of ΔC2 FVIII, Factor IXa and Factor X. The concentration of vesicles was less than half that required to support full activity of ΔC2 FVIII. fVIII-C2 concentrations as high as 1 μM did not increase activity (results not shown). These results indicate that the C2 domain must be attached to the C1 domain to confer normal membrane-binding affinity and full activity to Factor VIII.
DISCUSSION
The results of our present experiments show that the isolated fVIII-C2 binds to PLVs with moderately high affinity and that membrane binding is dependent upon negatively charged phospholipids. In contrast with intact Factor VIII, binding of fVIII-C2 is not specific for Ptd-L-Ser, occurs only under low-salt conditions and does not compete with intact Factor VIII. The properties of fVIII-C2 contrasted with those of the similar Lact-C2, indicating that these properties relate to structural details of fVIII-C2. These results indicate that the role of fVIII-C2 in membrane binding of intact Factor VIII differs from current predictions, suggesting a membrane-binding mechanism that is more complex than previously anticipated.
Our present results agree with several previous studies that have investigated the binding of fVIII-C2 to immobilized phospholipid in a microtitre well [37–39]. These studies show that fVIII-C2 has the capacity to bind to PS in the presence of saline in some solid-phase assays. When we immobilized fVIII-C2 to a microtitre well using mAb ESH8, we also found that fVIII-C2 bound to PLVs in the presence of NaCl with an implied affinity similar to intact Factor VIII.
Our results may appear to be at odds with predictions from previous studies, including our own, that use peptides from the fVIII-C2. The peptides are able to compete with Factor VIII for membrane binding [40,41] and inhibit Factor VIII activity [42,43]. These studies make the assumption that the peptides are in the native conformation or fold into the native conformation upon contact with a phospholipid membrane. However, subsequent studies have indicated that the major membrane-binding peptide from fVIII-C2 folds into an amphipathic α-helix [41,44], whereas the crystal structure indicates that the native conformation includes strands of β-sheet and a hairpin turn [13]. Some of the properties of the peptide are emulated by other amphipathic helix-forming peptides with no sequence similarity to the C2 domain. Thus the interaction with Factor IXa, Factor X and probably with membranes is not via a physiological mechanism [45]. We are aware of no reports of fVIII-C2 competing with Factor VIII for membrane binding or substantially inhibiting lipid-dependent Factor Xase activity.
Our present results raise an apparent conflict with the implied solution-phase Kd values and Factor Xase inhibition reported by Takeshima et al. [39] in the presence of NaCl. Their indirect binding studies relied on fluorescence quenching of labelled annexin V by added PLVs to detect membrane binding of annexin V. Addition of fVIII-C2 led to decreased fluorescence quenching, interpreted as competition for binding by fVIII-C2. Unfortunately, control studies were not included to confirm that loss of quenching actually reflected fVIII-C2 binding to PLVs. Maximum inhibition of Factor Xase activity reached only 60% at approx. 200 μM fVIII-C2, a 1000-fold higher concentration than the inferred Kd for membrane binding. No explanation was evident to explain the discrepancy between the inferred Kd and inhibitory concentration. Control experiments were not included to demonstrate that the decreased Factor Xase activity resulted from competition for phospholipid-binding sites. Our present results may also be interpreted as conflicting with studies by Saenko et al. [46]. These authors utilized SPR (surface plasmon resonance) to detect binding of fVIII-C2, produced with a baculovirus vector, to a phospholipid monolayer immobilized on a gold substrate and to PLVs immobilized on a biotin-coated surface. They reported association and dissociation kinetics and a calculated Kd for fVIII-C2 that was comparable with intact Factor VIII in the presence of physiological NaCl. Since our present experiments produced comparable results using fVIII-C2 from two distinct expression systems (E. coli and Pichia), we believe that the differences between our results and those of Saenko et al. [46] are more likely to be due to different methodology than to differences between fVIII-C2 preparations. In our present studies, the differences between fVIII-C2 and intact Factor VIII and fVIII-C2 and Lact-C2, and the consistency of our results with different solution-phase assays, indicate that the effect of saline and the membrane affinity are not an artifact produced by an individual binding assay.
We verified the stability and conformation of fVIII-C2 in several ways. fVIII-C2 was purified from the cytosol, suggesting that this protein module was soluble and folded correctly. The molecular mass correlated with its predicted value and fVIII-C2 had no free thiols, indicating that the internal disulfide bond was formed correctly. The conformation was verified by measuring the binding of fVIII-C2 to three mAbs and comparing the affinities of the antibodies for full-length Factor VIII. In addition, we have shown that the binding properties of our fVIII-C2 from E. coli were equivalent to the properties of fVIII-C2 from the P. pastoris expression system described previously [13]. Thus the unexpected relationship between buffer NaCl and membrane binding is not the consequence of an improperly folded domain.
We have demonstrated that membrane binding of fVIII-C2 relies upon the epitopes of mAbs ESH4 and B02C11, which are also necessary for membrane binding of intact Factor VIII in the presence of NaCl [21,22]. We have shown previously [17] that Met2199/Phe2200 and Leu2251/Leu2252 are constituents of the membrane-binding motif, and Spiegel et al. [47] have shown that these residues contribute to the epitope of B02C11. Lact-C2 relies on residues that are similarly situated to mediate membrane binding [14]. Thus fVIII-C2 and Lact-C2 bind to membranes with similar structural motifs in spite of the contrasting membrane-binding properties.
Our present results show that fVIII-C2 slightly increased the activity of Factor IXa. The modest effect of fVIII-C2 correlated with an increase in the apparent affinity for Factor X. This suggests that fVIII-C2 interacts with either Factor X or Factor IXa. The Factor VIII light chain, composed of the A3, C1 and C2 domains, shows only weak association with Factor X [28], whereas cross-linking experiments [29] and FRET-binding experiments [30] show that the light chain binds to the Gla domain of Factor IXa. Recent results have shown that the C2 domain is able to bind to the Gla domain of Factor IXa and inhibit Factor Xase activity in the absence of phospholipid [31], and that the absence of the C2 domain results in a 24% decrease in cofactor activity [48], providing additional support for the role of the C2 domain in this interaction. Our present results are consistent with a model in which fVIII-C2 helps to anchor Factor VIIIa to Factor IXa in the Factor Xase complex.
We have considered three possible explanations for inhibition of fVIII-C2 but not intact Factor VIII by saline. First, charge shielding by salt may limit the attraction of positively charged fVIII-C2 to a negatively charged phospholipid membrane. For intact Factor VIII, the initial approach to a membrane may be mediated by additive charge components of the C2 and C1 domains. Secondly, Na+ or Cl− ions may interact with fVIII-C2 in a manner that causes a conformational or flexibility change that is not favourable for phospholipid binding. The C2 domain may assume a different conformation in the intact Factor VIII due to additional constraints resulting from contact with the A1 and/or C1 domain, thus limiting the effect of NaCl in the intact protein. Thirdly, under physiological conditions, the C2 domain may not mediate initial contact with the membrane. The C2 domain may engage the membrane only after it is brought into intimate contact by engagement of another motif, presumably on the C1 and/or A3 domain. We note that these explanations are not mutually exclusive so that all could contribute.
Our previous work has suggested that electrostatic interactions can influence membrane binding of intact Factor VIII. Factor VIII binds in a non-specific manner to negatively charged membranes when the content of negatively charged phospholipids exceeds 15% [5]. The same study found that reducing the negative electrostatic potential of PS-containing membranes decreases the affinity of Factor VIII for the PS-binding sites, indicating that the electrostatic potential contributes to Factor VIII-binding affinity. Measurements of PLVs eluted from a Factor VIII affinity column indicate that electrostatic interactions account for approx. 15% of binding energy [3]. Our present study indicates that the number of membrane sites for Factor VIII is approx. 2-fold higher in the absence of NaCl, consistent with the interpretation that fVIII-C2 is able to bind to additional distinct binding sites in the absence of NaCl (Figure 4). Thus the electrostatic interaction of Factor VIII with phospholipid membranes may have a limited role in the binding of intact Factor VIII and a larger role for the isolated C2 domain.
We have considered the possibility that the conformation of isolated fVIII-C2 may differ from the native conformation. The isolated C2 domain [13] compared with the C2 domain in complex with mAb B02C11 [49] and in complex with a small-molecule inhibitor of membrane binding [50] show some rotational flexibility of binding spike 3 (containing Leu2251/Leu2252) and lateral motion of binding spike 1 (containing Met2199/Phe2200). However, the overall C2 domain structure of intact Factor VIII [26,27] is very similar to that of the isolated C2 domain, thus constraining the magnitude of the possible conformational change brought about by the contact of C2 with the A1 and C1 domains.
Our results point to a more limited role for the C2 domain in membrane binding of Factor VIII than current models postulate. In particular, the lack of competition by fVIII-C2 suggests that fVIII-C2 does not recognize the primary membrane-binding sites for Factor VIII and that binding sites for the C2 domain might be induced only after another motif engages with the membrane. In addition, solid-phase binding, which is not inhibited by saline and has a higher apparent affinity than solution-phase binding, is dependent on having multiple fVIII-C2 molecules binding to the same vesicle, suggesting that the C2 domain alone cannot account for Factor VIII-binding affinity. In addition, recent results have shown only a 13%loss of binding energy with the deletion of the C2 domain [48] and our present results show that this construct retains Ptd-L-Ser specificity. Our recent studies indicate that residues Lys2092 and Phe2093 at the tip of the C1 domain participate in membrane binding [36]. Thus a membrane-binding role for the C1 domain has been established. However, these insights must be interpreted in light of reports that indicate that site-directed mutagenesis of the C2 domain diminishes membrane affinity [17,51] and numerous reports indicating that anti-C2 antibodies can block phospholipid binding and cause severe bleeding. Thus current findings indicate that both the C1 and the C2 domains are required to produce normal membrane binding and full functionality. Further studies will be required to explain the mechanism through which membrane engagement of the C2 domain enhances binding of intact Factor VIII and provides an essential component of biological functionality.
Supplementary Material
Acknowledgments
We are grateful to Dr Jialan Shi or preliminary experimental work and for helpful discussions, and to Sebastian Fain for excellent technical assistance.
FUNDING
This work was supported by the Department of Veterans Affairs, a National Hemophilia Foundation Award (to G.E.G.), and by the National Institutes of Health [grant number R01 HL76213 (to P.J.F.)].
Abbreviations used
- ΔC2 FVIII
Factor VIII lacking the C2 domain
- biotin-PE1
2-dihexadecanoyl-sn-glycero-3-phosphoethanolamine-N-(cap biotinyl)
- B-PER
bacterial protein extraction reagent
- dansyl-PE
1,2-dioleoyl-sn-glycero-3-phosphoethanolamine-N-(5-dimethylaminonaphthalenesulfonyl)
- DPC
n-dodecyl phosphocholine
- Factor VIII-Fl
fluorescein-labelled Factor VIII
- Fmax
maximum fluorescence
- Fres
the residual fluorescence
- FRET
fluorescence resonance energy transfer
- fVIII-C2
Factor VIII C2 domain
- fVIII-C2C
Val2223Cys fVIII-C2 mutant
- fVIII-C2C-Fluor
fluorescein-labelled fVIII-C2C
- fVIII-C2Pichia
fVIII-C2 prepared in Pichia pastoris
- HRP
horseradish peroxidase
- Lact-C2
lactadherin C2 domain
- mAb
monoclonal antibody
- PC
phosphatidylcholine
- PE
phosphatidylethanolamine
- PI
phosphatidylinositol
- PLV
phospholipid vesicle
- PS
phosphatidylserine
- Ptd-D-Ser
1,2-dioleoyl-sn-glycero-3-phospho-D-serine
- Ptd-L-Ser
1,2-dioleoyl-sn-glycero-3-phospho-L-serine
- TBS
Tris-buffered saline
- Trx
thioredoxin
Footnotes
AUTHOR CONTRIBUTION
Valerie Novakovic, David Cullinan and Gary Gilbert designed the experiments with contributions by James Baleja and Philip Fay. Valerie Novakovic, David Cullinan and Hironao Wakabayashi performed the experiments and analysed the data with Gary Gilbert. Valerie Novakovic drafted the manuscript with Gary Gilbert, and all of the authors revised the manuscript.
References
- 1.White GC, Gilbert GE. Coagulation Factors V and VIII: normal function and clinical disorders. In: Handin RI, Lux SE, Stossel TP, editors. Blood: Principles and Practice of Hematology. Lippincott Williams & Wilkins; Philadelphia: 2003. pp. 1195–1224. [Google Scholar]
- 2.Bardelle C, Furie B, Furie BC, Gilbert GE. Membrane binding kinetics of factor VIII indicate a complex binding process. J Biol Chem. 1993;268:8815–8824. [PubMed] [Google Scholar]
- 3.Atkins JS, Ganz PR. The association of human coagulation factors VIII, IXa and X with phospholipid vesicles involves both electrostatic and hydrophobic interactions. Mol Cell Biochem. 1992;112:61–71. doi: 10.1007/BF00229644. [DOI] [PubMed] [Google Scholar]
- 4.Gilbert GE, Drinkwater D, Barter S, Clouse SB. Specificity of phosphatidylserine-containing membrane binding sites for factor VIII. Studies with model membranes supported by glass microspheres (lipospheres) J Biol Chem. 1992;267:15861–15868. [PubMed] [Google Scholar]
- 5.Gilbert GE, Drinkwater D. Specific membrane binding of factor VIII is mediated by O-phospho-L-serine, a moiety of phosphatidylserine. Biochemistry. 1993;32:9577–9585. doi: 10.1021/bi00088a009. [DOI] [PubMed] [Google Scholar]
- 6.Gitschier J, Wood WI, Goralka TM, Wion KL, Chen EY, Eaton DH, Vehar GA, Capon DJ, Lawn RM. Characterization of the human factor VIII gene. Nature. 1984;312:326–330. doi: 10.1038/312326a0. [DOI] [PubMed] [Google Scholar]
- 7.Toole JJ, Knopf JL, Wozney JM, Sultzman LA, Buecker JL, Pittman DD, Kaufman RJ, Brown E, Shoemaker C, Orr EC, et al. Molecular cloning of a cDNA encoding human antihaemophilic factor. Nature. 1984;312:342–347. doi: 10.1038/312342a0. [DOI] [PubMed] [Google Scholar]
- 8.Kemball-Cook G, Edwards SJ, Sewerin K, Andersson LO, Barrowcliffe TW. Factor VIII procoagulant protein interacts with phospholipid vesicles via its 80 kDa light chain. Thromb Haemostasis. 1988;60:442–446. [PubMed] [Google Scholar]
- 9.Meeks SL, Healey JF, Parker ET, Barrow RT, Lollar P. Antihuman factor VIII C2 domain antibodies in hemophilia A mice recognize a functionally complex continuous spectrum of epitopes dominated by inhibitors of factor VIII activation. Blood. 2007;110:4234–4242. doi: 10.1182/blood-2007-06-096842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Church WR, Jernigan RL, Toole J, Hewick RM, Knopf J, Knutson GJ, Nesheim ME, Mann KG, Fass DN. Coagulation factors V and VIII and ceruloplasmin constitute a family of structurally related proteins. Proc Natl Acad Sci USA. 1984;81:6934–6937. doi: 10.1073/pnas.81.22.6934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Stubbs JD, Lekutis C, Singer KL, Bui A, Yuzuki D, Srinivasan U, Parry G. cDNA cloning of a mouse mammary epithelial cell surface protein reveals the existence of epidermal growth factor-like domains linked to factor VIII-like sequences. Proc Natl Acad Sci USA. 1990;87:8417–8421. doi: 10.1073/pnas.87.21.8417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Macedo-Ribeiro S, Bode W, Huber R, Quinn-Allen MA, Kim SW, Ortel TL, Bourenkov GP, Bartunik HD, Stubbs MT, Kane WH, Fuentes-Prior P. Crystal structures of the membrane-binding C2 domain of human coagulation factor V. Nature. 1999;402:434–439. doi: 10.1038/46594. [DOI] [PubMed] [Google Scholar]
- 13.Pratt KP, Shen BW, Takeshima K, Davie EW, Fujikawa K, Stoddard BL. Structure of the C2 domain of human factor VIII at 1.5 A resolution. Nature. 1999;402:439–442. doi: 10.1038/46601. [DOI] [PubMed] [Google Scholar]
- 14.Shao C, Novakovic VA, Head JF, Seaton BA, Gilbert GE. Crystal structure of lactadherin C2 domain at 1.7A resolution with mutational and computational analyses of its membrane-binding motif. J Biol Chem. 2008;283:7230–7241. doi: 10.1074/jbc.M705195200. [DOI] [PubMed] [Google Scholar]
- 15.Krishnaswamy S, Mann KG. The binding of factor Va to phospholipid vesicles. J Biol Chem. 1988;263:5714–5723. [PubMed] [Google Scholar]
- 16.Andersen MH, Graversen H, Fedosov SN, Petersen TE, Rasmussen JT. Functional analyses of two cellular binding domains of bovine lactadherin. Biochemistry. 2000;39:6200–6206. doi: 10.1021/bi992221r. [DOI] [PubMed] [Google Scholar]
- 17.Gilbert GE, Kaufman RJ, Arena AA, Miao H, Pipe SW. Four hydrophobic amino acids of the factor VIII C2 domain are constituents of both the membrane-binding and von Willebrand factor-binding motifs. J Biol Chem. 2002;277:6374–6381. doi: 10.1074/jbc.M104732200. [DOI] [PubMed] [Google Scholar]
- 18.Kim SW, Quinn-Allen MA, Camp JT, Macedo-Ribeiro S, Fuentes-Prior P, Bode W, Kane WH. Identification of functionally important amino acid residues within the C2-domain of human factor V using alanine-scanning mutagenesis. Biochemistry. 2000;39:1951–1958. doi: 10.1021/bi992256r. [DOI] [PubMed] [Google Scholar]
- 19.Ortel TL, Devore-Carter D, Quinn-Allen M, Kane WH. Deletion analysis of recombinant human factor V. Evidence for a phosphatidylserine binding site in the second C-type domain. J Biol Chem. 1992;267:4189–4198. [PubMed] [Google Scholar]
- 20.Shima M, Scandella D, Yoshioka A, Nakai H, Tanaka I, Kamisue S, Terada S, Fukui H. A factor VIII neutralizing monoclonal antibody and a human inhibitor alloantibody recognizing epitopes in the C2 domain inhibit factor VIII binding to von Willebrand factor and to phosphatidylserine. Thromb Haemostasis. 1993;69:240–246. [PubMed] [Google Scholar]
- 21.Jacquemin MG, Desqueper BG, Benhida A, Vander Elst L, Hoylaerts MF, Bakkus M, Thielemans K, Arnout J, Peerlinck K, Gilles JG, et al. Mechanism and kinetics of factor VIII inactivation: study with an IgG4 monoclonal antibody derived from a hemophilia A patient with inhibitor. Blood. 1998;92:496–506. [PubMed] [Google Scholar]
- 22.Ahmad SS, Walsh PN. Role of the C2 domain of factor VIIIa in the assembly of factor-X activating complex on the platelet membrane. Biochemistry. 2005;44:13858–13865. doi: 10.1021/bi0511033. [DOI] [PubMed] [Google Scholar]
- 23.Ortel TL, Moore KD, Quinn-Allen MA, Okamura T, Sinclair AJ, Lazarchick J, Govindan R, Carmagnol F, Kane WH. Inhibitory anti-factor V antibodies bind to the factor V C2 domain and are associated with hemorrhagic manifestations. Blood. 1998;91:4188–4196. [PubMed] [Google Scholar]
- 24.Fay PJ, Beattie T, Huggins CF, Regan LM. Factor VIIIa A2 subunit residues 558–565 represent a factor IXa interactive site. J Biol Chem. 1994;269:20522–20527. [PubMed] [Google Scholar]
- 25.Lenting PJ, van de Loo JW, Donath MJ, van Mourik JA, Mertens K. The sequence Glu1811–Lys1818 of human blood coagulation factor VIII comprises a binding site for activated factor IX. J Biol Chem. 1996;271:1935–1940. doi: 10.1074/jbc.271.4.1935. [DOI] [PubMed] [Google Scholar]
- 26.Shen BW, Spiegel PC, Chang CH, Huh JW, Lee JS, Kim J, Kim YH, Stoddard BL. The tertiary structure and domain organization of coagulation factor VIII. Blood. 2008;111:1240–1247. doi: 10.1182/blood-2007-08-109918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ngo JC, Huang M, Roth DA, Furie BC, Furie B. Crystal structure of human factor VIII: implications for the formation of the factor IXa–factor VIIIa complex. Structure. 2008;16:597–606. doi: 10.1016/j.str.2008.03.001. [DOI] [PubMed] [Google Scholar]
- 28.Lapan KA, Fay PJ. Localization of a factor X interactive site in the A1 subunit of factor VIIIa. J Biol Chem. 1997;272:2082–2088. doi: 10.1074/jbc.272.4.2082. [DOI] [PubMed] [Google Scholar]
- 29.Blostein MD, Furie BC, Rajotte I, Furie B. The Gla domain of factor IXa binds to factor VIIIa in the tenase complex. J Biol Chem. 2003;278:31297–31302. doi: 10.1074/jbc.M302840200. [DOI] [PubMed] [Google Scholar]
- 30.Jenkins PV, Dill JL, Zhou Q, Fay PJ. Contribution of factor VIIIa A2 and A3-C1-C2 subunits to the affinity for factor IXa in factor Xase. Biochemistry. 2004;43:5094–5101. doi: 10.1021/bi036289p. [DOI] [PubMed] [Google Scholar]
- 31.Soeda T, Nogami K, Nishiya K, Takeyama M, Ogiwara K, Sakata Y, Yoshioka A, Shima M. The factor VIIIa C2 domain (residues 2228–2240) interacts with the factor IXa Gla domain in the factor Xase complex. J Biol Chem. 2009;284:3379–3388. doi: 10.1074/jbc.M804955200. [DOI] [PubMed] [Google Scholar]
- 32.Barenholz Y, Gibbes D, Litman BJ, Goll J, Thompson TE, Carlson RD. A simple method for the preparation of homogeneous phospholipid vesicles. Biochemistry. 1977;16:2806–2810. doi: 10.1021/bi00631a035. [DOI] [PubMed] [Google Scholar]
- 33.Chen PS, Toribara TY, Warner H. Microdetermination of phosphorus. Anal Chem. 1956;28:1756–1758. [Google Scholar]
- 34.Wakabayashi H, Freas J, Zhou Q, Fay PJ. Residues 110–126 in the A1 domain of factor VIII contain a Ca2+ binding site required for cofactor activity. J Biol Chem. 2004;279:12677–12684. doi: 10.1074/jbc.M311042200. [DOI] [PubMed] [Google Scholar]
- 35.Gilbert GE, Furie BC, Furie B. Binding of human factor VIII to phospholipid vesicles. J Biol Chem. 1990;265:815–822. [PubMed] [Google Scholar]
- 36.Meems H, Meijer AB, Cullinan DB, Mertens K, Gilbert GE. Factor VIII C1 domain residues Lys 2092 and Phe 2093 contribute to membrane binding and cofactor activity. Blood. 2009;114:3938–3946. doi: 10.1182/blood-2009-01-197707. [DOI] [PubMed] [Google Scholar]
- 37.Spiegel PC, Kaiser SM, Simon JA, Stoddard BL. Disruption of protein-membrane binding and identification of small-molecule inhibitors of coagulation factor VIII. Chem Biol. 2004;11:1413–1422. doi: 10.1016/j.chembiol.2004.08.006. [DOI] [PubMed] [Google Scholar]
- 38.Spiegel PC, Murphy P, Stoddard BL. Surface-exposed hemophilic mutations across the factor VIII C2 domain have variable effects on stability and binding activities. J Biol Chem. 2004;279:53691–53698. doi: 10.1074/jbc.M409389200. [DOI] [PubMed] [Google Scholar]
- 39.Takeshima K, Smith C, Tait J, Fujikawa K. The preparation and phospholipid binding property of the C2 domain of human factor VIII. Thromb Haemostasis. 2003;89:788–794. [PubMed] [Google Scholar]
- 40.Foster PA, Fulcher CA, Houghten RA, Zimmerman TS. Synthetic factor VIII peptides with amino acid sequences contained within the C2 domain of factor VIII inhibit factor VIII binding to phosphatidylserine. Blood. 1990;75:1999–2004. [PubMed] [Google Scholar]
- 41.Gilbert GE, Baleja JD. Membrane-binding peptide from the C2 domain of factor VIII forms an amphipathic structure as determined by NMR spectroscopy. Biochemistry. 1995;34:3022–3031. doi: 10.1021/bi00009a033. [DOI] [PubMed] [Google Scholar]
- 42.Brinkman HJ, Mertens K, van Mourik JA. Phospholipid-binding domain of factor VIII is involved in endothelial cell-mediated activation of factor X by factor IXa. Arterioscler Thromb Vasc Biol. 2002;22:511–516. doi: 10.1161/hq0302.105359. [DOI] [PubMed] [Google Scholar]
- 43.Nogami K, Shima M, Nishiya K, Hosokawa K, Saenko EL, Giddings JC, Tanaka I, Yoshioka A. Anticoagulant effects of a synthetic peptide containing residues Thr-2253-Gln-2270 within factor VIII C2 domain that selectively inhibits factor Xa-catalysed factor VIII activation. Br J Haematol. 2002;116:868–874. doi: 10.1046/j.0007-1048.2002.03360.x. [DOI] [PubMed] [Google Scholar]
- 44.Veeraraghavan S, Baleja JD, Gilbert GE. Structure and topography of the membrane-binding C2 domain of factor VIII in the presence of dodecylphosphocholine micelles. Biochem J. 1998;332:549–555. doi: 10.1042/bj3320549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Blostein MD, Rigby AC, Furie BC, Furie B, Gilbert GE. Amphipathic helices support function of blood coagulation factor IXa. Biochemistry. 2000;39:12000–12006. doi: 10.1021/bi000655c. [DOI] [PubMed] [Google Scholar]
- 46.Saenko EL, Scandella D, Yakhyaev AV, Greco NJ. Activation of factor VIII by thrombin increases its affinity for binding to synthetic phospholipid membranes and activated platelets. J Biol Chem. 1998;273:27918–27926. doi: 10.1074/jbc.273.43.27918. [DOI] [PubMed] [Google Scholar]
- 47.Spiegel PC, Jr, Jacquemin M, Saint-Remy JM, Stoddard BL, Pratt KP. Structure of a factor VIII C2 domain-immunoglobulin G4κ Fab complex: identification of an inhibitory antibody epitope on the surface of factor VIII. Blood. 2001;98:13–19. doi: 10.1182/blood.v98.1.13. [DOI] [PubMed] [Google Scholar]
- 48.Wakabayashi H, Griffiths AE, Fay PJ. Factor VIII lacking the C2 domain retains cofactor activity in vitro. J Biol Chem. 2010;285:25176–25184. doi: 10.1074/jbc.M110.106906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pratt KP, Qian J, Ellaban E, Okita DK, Diethelm-Okita BM, Conti-Fine B, Scott DW. Immunodominant T-cell epitopes in the factor VIII C2 domain are located within an inhibitory antibody binding site. Thromb Haemostasis. 2004;92:522–528. doi: 10.1160/TH03-12-0755. [DOI] [PubMed] [Google Scholar]
- 50.Liu Z, Lin L, Yuan C, Nicolaes GA, Chen L, Meehan EJ, Furie B, Furie B, Huang M. Trp2313–His2315 of factor VIII C2 domain is involved in membrane binding: structure of a complex between the C2 domain and an inhibitor of membrane binding. J Biol Chem. 2010;285:8824–8829. doi: 10.1074/jbc.M109.080168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Schatz SM, Zimmermann K, Hasslacher M, Kerschbaumer R, Dockal M, Gritsch H, Turecek PL, Schwarz HP, Dorner F, Scheiflinger F. Mutation of the surface-exposed amino acid Trp to Ala in the FVIII C2 domain results in defective secretion of the otherwise functional protein. Br J Haematol. 2004;125:629–637. doi: 10.1111/j.1365-2141.2004.04959.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.