

Abstract
Across eukaryotes, Hsp70-based chaperone machineries display an underlying unity in their sequence, structure, and biochemical mechanism of action, while working in a myriad of cellular processes. In good part, this extraordinary functional versatility is derived from the ability of a single Hsp70 to interact with an array of J-protein cochaperones to form a functional chaperone network. Among J-proteins, the DnaJ-type is the most prevalent, being present in all three kingdoms and in several different compartments of eukaryotic cells. However, because these ancient DnaJ-type proteins diverged at the base of the eukaryotic phylogeny, little is understood about the evolutionary basis of their diversification and thus the functional expansion of the chaperone network. Here, we report results of evolutionary and experimental analyses of two more recent members of the cytosolic DnaJ family of Saccharomyces cerevisiae, Xdj1 and Apj1, which emerged by sequential duplications of the ancient YDJ1 in Ascomycota. Sequence comparison and molecular modeling revealed that both Xdj1 and Apj1 maintained a domain organization similar to that of multifunctional Ydj1. However, despite these similarities, both Xdj1 and Apj1 evolved highly specialized functions. Xdj1 plays a unique role in the translocation of proteins from the cytosol into mitochondria. Apj1’s specialized role is related to degradation of sumolyated proteins. Together these data provide the first clear example of cochaperone duplicates that evolved specialized functions, allowing expansion of the chaperone functional network, while maintaining the overall structural organization of their parental gene.
Keywords: J-protein, Hsp70, gene duplication, yeast, Hsp40, divergent evolution
Introduction
All Hsp70-based molecular chaperone machineries use the same fundamental biochemical mechanism of action, cycles of interaction with client proteins driven by ATP binding, and hydrolysis to function in many essential cellular processes (Bukau et al. 2006; Mayer 2010). These processes include folding of nascent chains as they emerge from ribosomes, driving protein translocation across membranes, protecting cells from heat stress, and facilitating biogenesis of Fe/S clusters (Kampinga and Craig 2010; Hartl et al. 2011). In good part, this functional versatility is due to the fact that Hsp70s interact with an array of J-protein cochaperones to form a functional chaperone network (Craig et al. 2006). All members of the J-protein superfamily possess a J-domain that binds Hsp70 and is responsible for stimulation of Hsp70’s ATPase activity, an obligatory step for stabilizing Hsp70’s interaction with client protein. However, outside their J-domains, J-proteins vary widely in sequence and structure (Kampinga and Craig 2010). Even the placement of the J-domain in their overall topology can differ. Often these “extra-J-domain” regions interact with client proteins, targeting them to Hsp70. Thus, binding of a client to a J-protein prioritizes its interaction with Hsp70 (fig. 1). The cycle of interaction of clients with Hsp70 initiated by its binding to a J-protein, is then followed by transfer to Hsp70, an interaction which is stabilized upon ATP hydrolysis. The cycle culminates upon Hsp70’s interaction with a nucleotide exchange factor, which effects ADP release, resulting in exchange of ATP for ADP and dissociation of the client protein from Hsp70.
Fig. 1.
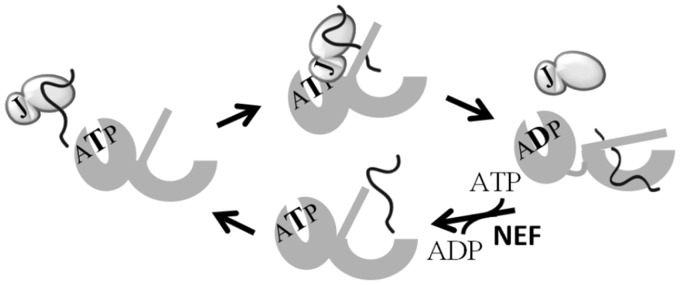
J-protein dependent cycle of interaction of Hsp70 with client protein. J-protein (light gray) binds client protein (line). Client protein is delivered to ATP-bound Hsp70, which has a fast on and off rate for client. J-domain of J-protein (J) stimulates Hsp70’s ATPase activity. As the resulting ADP-bound Hsp70 (dark gray) has a slow client off rate, the interaction of the client with Hsp70 is stabilized. Interaction of a nucleotide exchange factor (NEF) facilitates release of ADP, followed by binding of ATP, causing client dissociation and priming of Hsp70 for another cycle of interaction.
The first J-protein identified was DnaJ of Escherichia coli. The DnaJ family members (often referred to as Hsp40s) proliferated in eukaryotic cells with paralogous proteins present in the endoplasmic reticulum, cytosol/nucleus, and mitochondrium, where they function with compartment-specific Hsp70 partners (Walsh et al. 2004; Kampinga and Craig 2010). Members of the DnaJ family, which are typically homodimers, are structurally complex. They have an approximately 70-residue N-terminal J-domain, followed by a region rich in glycines and/or phenylalanines (G/F region) and a C-terminal region that is critical for client–protein interaction. Structural work established that the C-terminal region of Ydj1, an abundant member of the DnaJ family in the cytosol of the budding yeast Saccharomyces cerevisiae, is composed of two barrel topology domains, called CTD1 and CTD2, with CTD1 having a Zinc finger-like region (ZnFR) extruding from it. A hydrophobic client peptide-binding site was revealed in CTD1 when the structure of a cocrystal of the C-terminus of Ydj1 and a client peptide was solved (Li et al. 2003; Summers et al. 2009). Single amino acid alterations within this cleft affected the ability of Ydj1 to bind and facilitate refolding of client proteins such as denatured luciferase (Li and Sha 2005). The ZnFR has been suggested to play roles in client protein binding and/or interaction with Hsp70 (Linke et al. 2003; Fan et al. 2005).
The cytosol/nucleus of the budding yeast, S. cerevisiae, which contains a large functional network of 13 J-protein superfamily members, has been useful as a model system for understanding the diversity of J-protein function in cellular processes. This has particularly been the case for J-proteins structurally different from those of the DnaJ-type, such as Cwc23 (Sahi et al. 2010) and Jjj1 (Demoinet et al. 2007; Meyer et al. 2007), which are involved in mRNA splicing and ribosome biogenesis, respectively. The abundant DnaJ-type J-protein of the cytosol/nucleus, Ydj1, has also been studied extensively and shown to play general roles in de novo protein folding, protein translocation across membranes, and thermotolerance (Becker et al. 1996; Glover and Lindquist 1998; Summers et al. 2009). However, little information is available regarding the evolutionary origin and possible functional diversification and specialization of DnaJ-type J-proteins in any eukaryotic compartment. Here, we report the identification and analysis of two additional DnaJ-type proteins of the S. cerevisiae cytosol, Xdj1 and Apj1. The results of our evolutionary analysis are consistent with the idea that both proteins evolved by sequential duplications of the gene encoding Ydj1; Xdj1 in a common ancestor of Ascomycota, Apj1 more recently in a common ancestor of the Saccharomycetaceae and CTG clade (Dujon 2010) to which both S. cerevisiae and Candida albicans belong. Sequence analyses revealed that, consistent with their low abundance, both Xdj1 and Apj1 evolve significantly faster than Ydj1, and that their presence did not affect the mode or the rate of Ydj1 evolution. But, sequence analysis alone was not sufficient to predict whether Xdj1 and Apj1 had diversified functionally from Ydj1. However, our experimental work revealed that both Xdj1 and Apj1 have specialized roles, with Xdj1 being involved in mitochondrial protein import and Apj1 playing a role in degradation of sumolyated proteins.
Results
Three Members of DnaJ-Like Subfamily of S. cerevisiae Cytosol
Like Ydj1, two other J-proteins of the cytosol, Apj1 and Xdj1, have the easily discernible characteristics that historically placed them in a group called class I J-proteins (Cheetham and Caplan 1998): an N-terminal J-domain, an adjacent region rich in glycine and/or phenylalanine residues (G/F region) and four cysteine residues appropriately spaced to form a Zn finger (fig. 2A). To determine whether Xdj1 and Apj1 could be considered of the DnaJ-type (e.g., having the beta-barrel topology of CTD1 and CTD2), we extended our comparison using molecular modeling of the sequences C-terminal to the G/F region. Apj1 and Xdj1 had predicted topologies very similar to CTD1 and CTD2 of Ydj1 (fig. 2A). The Ydj1 client-binding site, revealed by cocrystallization with a client peptide (Li et al. 2003), is composed of six hydrophobic residues. Examination of the putative peptide binding clefts of Xdj1 and Apj1 revealed hydrophobic residues positioned similarly to those in the cleft of Ydj1 (fig. 2A). We conclude that the Ydj1, Apj1, and Xdj1 are members of the DnaJ family. However, we also found several differences amongst the three proteins. First, although the three hydrophobic-binding clefts were similarly positioned, the residues were not identical (fig. 2A). Second, although Xdj1 has a predicted prenylation site at its extreme C-terminus, similarly positioned to that of the experimentally determined Ydj1 site (supplementary fig. S1, Supplementary Material online), Apj1 does not contain a C-terminal cysteine, a requirement for this posttranslational modification. Third, the putative C-terminal dimerization region of Apj1 is longer than that of either Xdj1 or Ydj1. Fourth, the G/F-rich region of Apj1 is almost 2-fold longer than that of either Ydj1 or Xdj1, and although being rich in G/F residues as required of class I J-proteins, it also has serine, glutamine, and asparagine present in higher than expected proportions (supplementary fig. S1, Supplementary Material online). Herein, we refer to this segment of Apj1 G/F-S region.
Fig. 2.

Domain structures, deletion phenotypes, and cochaperone activities of DnaJ-like proteins from the cytosol/nucleolus of Saccharomyces cerevisiae. (A) (Top) Diagrams indicating domain structure of DnaJ-like proteins, drawn to scale: J-domain (J), glycine/phenylalanine-rich region (GF), beta barrel domain I with ZnFR (CTDI + ZnF), beta barrel domain II (CTDII), dimerization domain (DD). Amino acid residues indicated above each diagram indicate the boundaries of the regions. (Middle) Ribbon diagrams of CTDI/II region. Ydj1-CTDI CTDII domains cocrystallized with client peptide (magenta) (PDB:1NLT); Xdj1 and Apj1 homology models prepared using SWISS-MODEL (Kiefer et al. 2009) and the Ydj1 structure as a template. (Bottom) Space-filled diagrams of peptide binding cleft. Ydj1, based on crystal structure, with client peptide shown in magenta and Ydj1 residues involved in peptide binding shown in color, as indicated in the sequence alignment (bottom). Homology models of peptide-binding cleft of Xdj1 and Apj1. (B) WT and mutant cells, as indicated, were plated as 10-fold dilutions on rich media and incubated at indicated temperatures for 3 days. (C) Ssa1 ATPase activity under single-turnover conditions was measured in the presence of Apj1, Xdj1, and Ydj1 as indicated, or without addition (none). (Left) Hydrolysis of bound ATP over time (average of two determinations); (right) fold-change in the apparent first order rate constant for ATP hydrolysis determined using the data from left panel.
As our goal was to better understand both the functional and evolutionary relationships among these three J-proteins, we carried out three experimental comparisons. First, to assess their cellular abundance, we determined their levels in whole cell extracts derived from wild-type (WT) cells, using purified proteins as standards and antibodies specific for each protein. Our results indicate that WT cells have approximately 90,000 Ydj1 molecules/cell (supplementary fig. S2, Supplementary Material online). Xdj1 and Apj1 are significantly less abundant, at 500 and 100 molecules/cell, respectively. Second, we tested side-by-side the affect of deletion of the individual genes on cell growth. Consistent with previous reports (Caplan and Douglas 1991; Caplan et al. 1992), the absence of Ydj1 resulted in severe growth defects (fig. 2B). However, no obvious growth defects were observed for apj1Δ or xdj1Δ under a variety of conditions. Third, we set up systems to purify the three proteins to test their ability to stimulate Hsp70’s ATPase activity. Xdj1 and Ydj1 were readily purified, whereas Apj1 was prone to aggregation. However, sufficient quantities of Apj1 were obtained to assess the ability to stimulate the ATPase activity of Ssa1, the abundant cytosolic/nuclear Hsp70. Like previously observed for Ydj1, Xdj1, and Apj1 stimulated Ssa1 efficiently (fig. 2C). We conclude that Ydj1, Apj1, and Xdj1 are members of DnaJ-like family and are able to function with the Ssa class of Hsp70 in the cytosol/nucleus of S. cerevisiae.
Ydj1 and Xdj1 Are Found throughout the Ascomycetes Species
To begin to understand the evolutionary relationships amongst cytosolic DnaJ-like proteins, we first focused on Xdj1 because, as described earlier, some features of Xdj1 are more similar to those of Ydj1 (i.e., the size and amino acid composition of the G/F region and the putative prenylation site) than are those of Apj1. We searched for Ydj1 and Xdj1 orthologs in 43 Ascomycetes species (supplementary fig. S3, Supplementary Material online) using the S. cerevisiae sequences as a query for BLAST analysis. All surveyed species exhibited two significant BLAST hits (fig. 3), which we could readily identify as orthologs of Ydj1 and Xdj1. Reciprocal BLAST confirmed our predictions.
Fig. 3.

Phylogenetic distributions of YDJ1, XDJ1, and APJ1. The phylogenetic distributions of genes are presented in a simplified cladogram of the fungal phylum Ascomycota, which consists of three subphyla Taphrinomycotina, Pezizomycotina, and Saccharomycotina (Dujon 2010). The species of the Taphrinomycotina and Pezizomycotina clades are represented as separate triangles. The Saccharomycotina species were separated into three clades: Yarrowia, CTG, and Saccharomycetaceae. The Saccharomycetaceae consisting primary of species from the genera Saccharomyces and Kluyveromyces was further divided into the lineage that underwent the whole genome duplication (post-WGD) and the one that did not (pre-WGD). Shading in the post-WGD triangle indicates five closely relate species belonging to Saccharomyces sensu stricto. One representative species of each clade is named for clarity, with the total number of surveyed species indicated in parentheses. The presence (+) or absence (−) of the orthologs is indicated. The phylogenetic position of the putative origin of Xdj1 is marked with an open circle and those of Apj1 with a closed circle. For the list of additionally surveyed eukaryotic species, see supplementary figure S4, Supplementary Material online.
Next, we asked whether Ydj1 and Xdj1 orthologs were also present in well-annotated genomes of other fungi, metazoa, plantae, and protozoa. The BLAST search using the sequence of S. cerevisiae YDJ1 and XDJ1 as query provided one or more hits in each of the 32 surveyed species (supplementary fig. S4, Supplementary Material online). However, in each case, the YDJ1 hits had higher scores than those of XDJ1 hits. Moreover, except for 12 cases, reciprocal BLAST of the top three putative XDJ1 orthologs returned YDJ1 as a best hit in S. cerevisiae. Of the 12 questionable cases, none returned XDJ1, but rather other S. cerevisiae J-proteins (SCJ1, JEM1, and SIS1). That even the best XDJ1 hits are more similar to YDJ1 than to XDJ1 supports the hypothesis that XDJ1 orthologs are not present in the surveyed species. Thus, the birth of XDJ1 can be traced to the common ancestor of the Ascomycota clade. In contrast, orthologs of YDJ1 were found in all surveyed eukaryotic genomes, consistent with the hypothesis that this ancient member of DnaJ-like subfamily is indispensable for proper functioning of the cytosol/nucleus.
Xdj1 Toxicity, Whose Peptide-Binding Specificity Differs from Ydj1, Is Relieved by Reduction in Peptide Binding
The maintenance of Xdj1 throughout Ascomycetes species, even though expressed at levels almost 200-fold less than Ydj1, points to Xdj1 having a specialized function. To understand what biological role Xdj1 plays, we began an analysis of the effects of both its absence and its overexpression. We found that increased expression of Xdj1 was detrimental. No transformants with plasmids having XDJ1 under the control of either the strong TEF or GPD promoter were recovered. However, transformants expressing Xdj1 under the control of the weaker ADH1 promoter were obtained. These cells, which expressed Xdj1 at levels approximately four times higher than that found in WT cells, grew more poorly (fig. 4A). To determine whether this toxicity was dependent on its Hsp70 cochaperone function (e.g., as a J-protein), we used a mutant encoding an H to Q alteration in the invariant HPD motif of the J-domain, which has been shown in a number of J-proteins to inactivate the functional interaction of this domain with its Hsp70 partner (Kampinga and Craig 2010). Cells expressing Xdj1H37Q from the ADH promoter grew as well as WT cells. From this observation, we concluded that toxicity was dependent on Xdj1’s cochaperone function with Hsp70 (fig. 4B). Next, we tested variants having alterations in one of five residues in the putative peptide-binding cleft (fig. 2). Alanine alterations at L151, I243, L275, and F277 alleviated toxicity, whereas F153A did not (fig. 4C).
Fig. 4.

Moderate over-expression of Xdj1 is toxic: (A) Lower panel. Ten-fold serial dilutions of WT cells transformed with empty vector (—) or pRS414-ADH-Xdj1 (ADH-Xdj1) were spotted on Trp omission media and incubated at 30 °C for 2 days. (Upper panel) Total protein isolated from xdj1Δ cells transformed with empty vector (xdj1Δ) or WT cells transformed with either empty vector (—) or pRS414-ADH-Xdj1 (ADH-Xdj1) was resolved by electrophoresis, electro-blotted, and probed with anti-Xdj1 (Xdj1) and, as loading control, anti-Ssc1 antibody (c). (B) Ten-fold serial dilutions of xdj1Δ cells transformed with empty vector (—), pRS414-ADH-Xdj1 (WT) or pRS414-ADH-Xdj1H37Q (H37Q) were spotted on Trp omission medium and incubated at 30 °C for 2 days. (C) Ten-fold serial dilution of xdj1Δ cells transformed with pRS414-ADH-Xdj1 (WT) or pRS414-ADH-Xdj1 encoding the indicated variants having single amino acid alterations in the putative peptide-binding cleft were spotted on Trp omission medium and incubated at 30 °C for 2 days. (D) Ten-fold serial dilutions of xdj1Δ cells transformed with empty vector (—, left) or pRS413-GPD-Sse1 (GPD-Sse1, right) and a second plasmid: empty (—), pRS414-ADH-Xdj1 (WT), pRS414-ADH-Xdj1H37Q (H37Q), or pRS414-ADH-Xdj1L275A (L275A) were spotted on Trp-His double omission plates and incubated at 30 °C for 2 days. (E, F) Purified Xdj1, Xdj1L275A, Ydj1, or Ydj1L135A were immobilized on 96-well plates and assessed for their ability to bind coliphage expressing on their surface various 12-mer peptides (4 × 107 phage/well) that were selected using Xdj1 (left) or Ydj1 (right) using phage-specific antibody. (E) 4 and 12 unique coliphage selected for binding to Xdj1 (left) or Ydj1 (right) in panning experiments were tested for binding to Xdj1 and Xdj1L275A or Ydj1 and Ydj1L135A, respectively. (F) Indicated coliphage whose binding to cognate J protein was significantly impaired in cleft mutants were tested for specificity of binding to either Xdj1 or Ydj1.
The results described earlier indicate that the toxicity caused by Xdj1 overexpression requires both interaction with Hsp70 and binding of client protein, suggesting to us that “inappropriate” or prolonged association of certain client proteins with Hsp70 due to targeting by Xdj1 is the cause of the toxicity. To test this idea, we overexpressed Sse1, a nucleotide exchange factor for the Hsp70 Ssa (Dragovic et al. 2006; Raviol et al. 2006), concurrently with overexpression of Xdj1. The logic behind this test was that release of ADP from the Hsp70 Ssa by the action of Sse would speed up the cycle of interaction of Hsp70 with client protein. In this way, the residency time of client proteins targeted by Xdj1 would be reduced. We found that, as hypothesized, overexpression of Sse1 partially relieved the toxicity of Xdj1 overexpression, as cells having levels of Xdj1 4-fold above normal grew better when Sse1 expression was higher than that normally found (fig. 4D).
An increase of Xdj1 levels to only 2,000 molecules/cell resulted in toxicity, whereas Ydj1 is normally present at 90,000 molecules/cell, suggested to us that Xdj1 and Ydj1 may have different peptide-binding specificities. To test this idea, we proceeded to search for peptides that bound to Xdj1 or Ydj1. Using purified Xdj1 or Ydj1, we screened a phage library expressing on their surface random 12 residue peptides. We obtained phage from the Xdj1 and Ydj1 screen having 4 and 10 distinct peptide sequences, respectively (supplementary fig. S5, Supplementary Material online). To assess whether these peptides were indeed interacting in the client-binding cleft of the subdomain CTD1 (fig. 2A), we compared phage interaction of WT proteins and variants having alterations in the cleft, Xdj1L275S and Ydj1L135A. Two of the Xdj1- and 9 of the Ydj1-selected phage showed substantial reduction in binding to the variants compared with WT protein (fig. 4E). Next, we “cross-tested” the 11 phage, comparing their ability to bind the J-protein not used for their selection with the J-protein used in the initial screening. In all cases, the phage bound more efficiently to the protein used for their selection than to the other J-protein (fig. 4F). The best cross-binder, Y6, only bound Xdj1 18% as efficiently under the conditions used for screening than it bound Ydj1, with which it was selected. Together our results are consistent with the conclusion that the peptide-binding specificities of Xdj1 and Ydj1 are obviously distinct and that a decrease in the affinity of Xdj1 for client protein reduces its toxicity.
Xdj1 Functions in Mitochondrial Import
Toxicity is a useful tool for understanding Xdj1. However, its analysis did not reveal information regarding Xdj1’s biological function. As we had observed no obvious growth phenotypes of xdj1Δ cells, we set up tests to validate synthetic genetic interactions previously noted in genomic wide screens. The most dramatic report was a lethal interaction with a temperature sensitive allele of the molecular chaperone Hsp90 (Zhao et al. 2005). However, we did not detect any genetic interaction (supplementary fig. S6, Supplementary Material online). We also noted the report in another genomic-wide screen (Costanzo et al. 2010) of a genetic interaction, between deletions of XDJ1 and PAM17, which encodes a nonessential component of the protein import apparatus of the mitochondrial inner membrane (van der Laan et al. 2005). As Xdj1, but not Ydj1, had been found in a proteomic analysis of mitochondrial outer membranes (Burri et al. 2006), we pursued this observation. Although pam17Δ cells, such as xdj1Δ cells had no obvious effect on cell growth, xdj1Δ pam17Δ cells grew somewhat more poorly than WT cells at 18 °C (fig. 5A). This synthetic growth interaction was dependent on a functional interaction with Hsp70, as xdj1H37Q pam17Δ cells also grew poorly at 18 °C (fig. 5B). Many proteins synthesized on cytosolic ribosomes, but destined for mitochondria, contain an N-terminal-targeting presequence that is cleaved after translocation to the matrix. Thus, a defect in mitochondrial import can be detected by monitoring the accumulation of the precursor form of mitochondrial proteins, including Mdj1 (Pais et al. 2011). A small amount of Mdj1 precursor accumulated in pam17Δ cells at both 23°C and 18 °C (fig. 5C). This effect was exacerbated when Xdj1 was also absent, validating the synthetic genetic growth interaction between a deletion of XDJ1 and a deletion of PAM17. We also tested whether precursor accumulated in cells overexpressing Xdj1. Approximately one-third of the Mdj1 protein was present in the precursor form in cells expressing Xdj1 at levels 4-fold above normal (fig. 5D). This accumulation was ameliorated by alterations in Xdj1 that affect J-domain function (H37Q) or client protein binding (L275A). Together our in vivo results indicate that Xdj1 specializes in facilitating import of proteins from the cytosol into mitochondria, and that the cellular toxicity caused by modest overexpression of Xdj1 is due to affects on the import process, likely by increased association of client proteins to Hsp70, that are targeted by Xdj1.
Fig. 5.

Xdj1 has a role in mitochondrial protein import. (A) Ten-fold serial dilutions of WT, xdj1Δ, pam17Δ, and xdj1Δ pam17Δ cells were spotted on rich media and incubated at 18 °C for 7 days. (B) Ten-fold serial dilutions of xdj1Δ pam17Δ cells transformed with empty vector pRS313 (—), pRS313-Xdj1 (WT) or pRS313-Xdj1H37Q (H37Q) were spotted on a His omission plate and incubated at 18 °C for 7 days. (C, D) Total protein isolated from indicated strains were resolved by electrophoresis, electro-blotted and probed with anti-Mdj1 and, as loading control, anti-Sse1 antibody (c). Precursor (p) and mature (m) forms of Mdj1 are indicated. (C) WT, xdj1Δ, pam17Δ, and xdj1Δ pam17Δ cells grown at either 23 °C or 18 °C. (D) xdj1Δ cells expressing empty vector pRS414 (—), pRS414-ADH-Xdj1 (WT), pRS414-ADH-Xdj1H37Q (H37Q), pRS414-ADH-Xdj1L153A (L153A), and pRS414-ADH-Xdj1L275A (L275A) grown at 30 °C. All samples were run on the same gel; dotted lines indicate splicing out of lanes having irrelevant samples.
APJ1 Arose via Duplication of YDJ1
Next, turning our attention to Apj1, we used BLAST analysis to search for the presence of APJ1 orthologs in Ascomycetes species, using the S. cerevisiae sequence as a query. Using reciprocal BLAST, we could confidently identify the candidate sequence as an APJ1 ortholog within species belonging to the Saccharomycetaceae. However, the same query in the sister CTG clade returned three hits with similar E values for each analyzed species, two of which we had previously identified by means of reciprocal BLAST search, phylogeny (discussed later) and the presence of the C-terminal prenylation motif (CXXQ) as orthologs of YDJ1 and XDJ1. More in-depth analysis of the domain structure and presence of conserved sequence motifs such as a longer and an S-, Q-, and N-rich G/F-S region, as well as protein phylogeny, allowed us to annotate the remaining hits from the CTG clade as putative APJ1 orthologs. When the sequence of the putative APJ1 ortholog from Candida albicans, a member of the CTG clade, was used as reciprocal BLAST query within the CTG clade, best results corresponded to the annotated APJ1 sequences. However, when a similar analysis was carried using orthologs from species belonging to the Saccharomycetaceae, similar E values were returned for previously identified APJ1, YDJ1, and XDJ1 (data not shown), indicating that APJ1 orthologs exhibited a high level of clade-specific sequence divergence.
Outside the Saccharomycetaceae and CTG clade, only two high score BLAST hits were obtained for each species, which we identified as orthologs of YDJ1 and XDJ1 (fig. 3). Moreover, reciprocal BLAST analysis confirmed the annotation of these two sequences in each genome tested. No Apj1 ortholog was found in species outside the Saccharomycetaceae and CTG clade. This phylogenetic distribution of APJ1 exclusively in species forming a monophyletic group suggested to us that APJ1 arose in a common ancestor of the Saccharomycetaceae and the CTG clade, most likely via a gene duplication event. To identify the parental APJ1 gene, we analyzed phylogenetic relationships among APJ1, YDJ1, and XDJ1, using as an out group two sequences encoding DnaJ-like proteins functioning in other cellular compartments: SCJ1 of the endoplasmic reticulum and MDJ1 of the mitochondrium. Within this large maximum likelihood (ML) reconstruction of the amino acid–based phylogeny, the branching order of individual gene trees recapitulated the accepted species tree (Fitzpatrick et al. 2006) with the main phylogenetic groups retrieved with high support (fig. 6 and supplementary fig. S7, Supplementary Material online). However, placement of individual species within the major groups was somewhat variable, although no significant deviations from the accepted taxonomy were observed. The discrepancies can likely be explained by loss of the protein-specific phylogenetic signal in the alignment of all five DnaJ-like proteins used for tree inference. Inspection of the protein phylogeny indicated that APJ1, YDJ1, and XDJ1 formed a strongly supported monophyletic clade, suggesting that DnaJ-like proteins present in the cytosol/nucleus are closely related (fig. 6 and supplementary fig. S7, Supplementary Material online). Moreover, APJ1 sequences formed a sister clade to the YDJ1 sequences, indicating that APJ1 is most likely related to YDJ1 by a gene duplication event that took place in a progenitor of the Saccharomycetaceae and the CTG clade.
Fig. 6.

Phylogeny of Ydj1, Xdj1, and Apj1. ML-inferred phylogeny of amino acid sequences of the Ydj1, Xdj1, and Apj1, rooted using Scj1 and Mdj1 sequences from the set of surveyed fungi species. Scale is in substitutions per amino acid site. Bootstrap support values out of 100 pseudoreplicates are indicated. The complete phylogenetic tree, including fully resolved Scj1 and Mdj1 clades is provided as supplementary figure S7, Supplementary Material online.
Apj1’s Specialized Function Is Related to Sumo-Targeted Protein Degradation
APJ1’s retention in all postduplication species combined with its low expression levels in S. cerevisiae suggests that it has functionally diverged from its parental gene. To gain a better understanding of the functional differences between Apj1 and Ydj1, we searched for a phenotype of apj1Δ cells. As we observed no growth defect of apj1Δ cells under a variety of conditions, we extended our analysis to include synthetic genetic interactions. Noting apj1Δ listed amongst candidates in a global screen for synthetic genetic interactions with a deletion of SLX5, which encodes a subunit of a SUMO-targeted ubiquitin ligase (Pan et al. 2006), we compared the growth of slx5 and slx5Δ apj1Δ cells (fig. 7A). Although slx5Δ cells grew more slowly than WT cells at all temperatures tested, slx5Δ apj1Δ cells grew even more poorly and were unable to form individual colonies at 37 °C. To verify the genetic interaction between SLX5 and APJ1, we compared accumulation of SUMO-modified proteins by immunoblot analysis, using SUMO-specific antibodies. As expected (Mullen and Brill 2008), a wide range of proteins having SUMO modifications was observed in WT cells, with slx5Δ cells having higher levels. This greater amount of sumolyated proteins in the absence of Slx5 is attributed to impairment of the ubiquitination of these proteins and thus their degradation (fig. 7B). The level of sumolyated proteins in the slx5Δ apj1Δ double mutant was higher than that in the slx5Δ single mutant, consistent with Apj1 playing a role in the proteolysis of proteins targeted for degradation by sumoylation, and validating the genetic interaction between a deletion of APJ1 and a deletion of SLX5. To test whether the synthetic genetic interaction was dependent on the ability of Apj1 to function as a J-protein, we made use of an H to Q alteration in the J-domain, analogous to the Xdj1 variant described earlier. apj1H34Q was not able to substitute for WT APJ1 to allow growth at 37 °C (fig. 7C), indicating that Apj1 functions in protein degradation as part of a larger Hsp70-based chaperone machinery.
Fig. 7.

apj1Δ and slx5Δ have a synthetic genetic interaction related to Sumo-mediated degradation. (A) Ten-fold serial dilutions of WT, apj1Δ, slx5Δ, and apj1Δ slx5Δ cells were spotted on rich media and incubated at indicated temperatures for 2 days. All strains in upper and lower panels were spotted on the same plate; dotted line indicates that irrelevant strains were spliced out of the figure. (B) Extracts of total protein isolated from WT, apj1Δ, slx5Δ, or apj1Δ slx5Δ cells were resolved by electrophoresis, electroblotted and probed with antibodies specific for Smt3, Saccharomyces cerevisiae sumo protein and, as a control, anti-Ssc1 antibodies (c). (C, D, E) Ten-fold serial dilutions of apj1Δ slx5Δ carrying the plasmids indicated below were spotted on media lacking tryptophan and incubated at indicated temperatures for 2 days. (C) pRS314-Apj1 (WT) or pRS314-Apj1H34Q (H34Q). (D) Empty vector (—), pRS314-Apj1 (WT), pRS314-Apj1Δ79-161 (ΔGFS), pRS314-Apj1Δ206-262 (ΔZn) or pRS314-Apj1Ydj1-143-206 (Zn swap). (E) pRS314-Apj1 (WT), pRS314-Apj1I179S (I179S), pRS314-Apj1L198S (L198S), pRS314-Apj1L200S (L200S), pRS314-Apj1L277S (L277S), pRS314-Apj1V316S (V316S), pRS314-Apj1I318S (I318S).
We took a genetic approach to define regions of Apj1 critical for its specialized function. To assess possible roles of the G/F-S region and ZnFR in functional specificity, we created internal deletion mutations removing DNA segments encoding these regions. We were particularly interested in Apj1’s G/F-S region. However, apj1ΔG/F-S slx5Δ consistently grew nearly as well as slx5Δ cells, even at 37 °C, indicating that the G/F-S region, though distinct from the G/F region of Ydj1, is not critical for function. On the other hand, deletion of the Zn-binding region had deleterious effects on Apj1 function, as apj1ΔZnFR slx5Δ cells grew nearly as poorly as apj1Δ slx5Δ. However, substitution of Apj1’s ZnFR with Ydj1’s restored function, indicating that the while the ZnFR is critical, it does not provide functional specificity for Apj1. These results suggest that the specificity for Apj1 function lies elsewhere in the C-terminal segment. To address whether peptide binding is critical for function, six variants, each having one of the hydrophobic cleft residues changed to a serine, were expressed in apj1Δ slx5Δ cells, and their ability to substitute for WT Apj1 tested. Three (I179S, L200S, and L277S) were unable to substitute for WT Apj1, as indicated by failure of the double mutants to form colonies at 37 °C. Therefore, we concluded that client binding ability is critical for Apj1’s function in protein degradation.
Apj1 and Xdj1 Are Rapidly Evolving
Evident functional differences among the three cytosolic members of DnaJ-subfamily prompted us to compare the rates of evolution of their sequences. Pair-wise comparisons of all Ydj1 amino acid sequences revealed a high level of conservation (∼60% identity), whereas the sequence conservation was much lower for Xdj1 (∼38% identity) and Apj1 (∼34% identity) (table 1). Consistent with their high sequence variability, the branches of both the amino acid–based tree (fig. 6) and the nucleotide-based tree (data not shown) are longer for XDJ1 and APJ1 compared with YDJ1. Furthermore, analysis of codon evolution indicated that nonsynonymous rates of evolution are significantly higher for XDJ1 and APJ1 (dN = 0.08 ± 0.07 and 0.1 ± 0.09, respectively) than for YDJ1 (dN = 0.04 ± 0.03) (table 1). We also noticed that the dN values determined only for YDJ1 sequences from the species in which YDJ1 coexists with APJ1 (YDJ1post-Apj1) or only for the YDJ1 sequences from species lacking APJ1 (YDJ1pre-APJ1) were identical, indicating that the coexistence of APJ1 with YDJ1 had no effect on the rate of nonsynonymous substitutions in YDJ1.
Table 1.
Summary of Sequence Analysis.
| Gene | Sequence Statistics |
Codon Usage Bias |
Evolutionary Rates |
||||||
|---|---|---|---|---|---|---|---|---|---|
| Lengtha | Identityb | Similarityc | ENC | CAI | dSall | dNall | dNpre-Apj1 | dNpost-Apj1 | |
| YDJ1 | 411 ± 6 | 59.53 ± 11.7 | 73.18 ± 9.14 | 41.27 ± 5.81 | 0.36 ± 0.05 | 1.18 ± 1.04 | 0.04 ± 0.03 | 0.04 ± 0.04 | 0.04 ± 0.04 |
| XDJ1 | 436 ± 22 | 37.78 ± 12.4* | 55.26 ± 10.73* | 50.89 ± 6.25* | 0.13 ± 0.01* | 1.58 ± 1.35 | 0.08 ± 0.07* | 0.08 ± 0.08* | 0.09 ± 0.08* |
| APJ1 | 513 ± 38 | 34.30 ± 15.2* | 49.27 ± 14.54* | 52.68 ± 4.62* | 0.14 ± 0.01* | 2.51 ± 2.34* | 0.10 ± 0.09* | — | — |
Note.—All values are shown as mean ± SD.
aMean number of residues in the amino acid sequence.
bMean pairwise amino acid sequence identity.
cMean pairwise amino acid sequence similarity (see Materials and Methods for details).
*Significantly different from YDJ1 at the P = 0.01 level.
As the per site frequency of synonymous substitutions was saturated (dS > 1) for each analyzed gene, preventing us from accurate estimation of the dN/dS ratio, we proceeded to analyze amino acid sequence evolution further. We searched for two types of divergence (Gu 1999, 2001): type I, sites highly conserved in one group of paralogous sequences that are highly variable in another group, and often interpreted as indication of relaxation of functional constraints and site-specific shift of evolutionary rate; and type II, sites conserved within each group of paralogs, but differ radically in their biochemical properties, and often interpreted as associated with putative functional changes. Using the DIVERGE software (Gu and Vander Velden 2002) to compare all Xdj1 to all Ydj1 sequences, we obtained a moderately high coefficient of type I sequence divergence ΘI (0.48 ± 0.05) (table 2). A similar ΘI value (0.39 ± 0.06) was obtained from comparison of Apj1 sequences with Ydj1post-Apj1. However, a lower ΘI value (0.26 ± 0.05) was obtained from comparison between Ydj1post-Apj1 and Ydj1pre-Apj1, suggesting that most of the type I divergence was associated with Xdj1 and Apj1 sequences. To further verify this prediction, we inspected multisequence alignments, searching for sites in strict agreement with the type I divergence definition—invariant in one group of compared sequences but variable in another group. The Xdj1:Ydj1 comparison revealed 50 type I sites, of which 48 were variable in the Xdj1 sequence (table 2). In the case of the Apj1:Ydj1postApj1 comparison, we found 69 type I sites, with 67 of them being variable in Apj1. In striking contrast, the Ydj1postApj1: Ydj1preApj1 comparison identified 56 type I sites, but variable sites were distributed equally, 34 and 22, respectively, between two groups of sequences. We conclude that both Xdj1 and Apj1 exhibit elevated rates of sequence divergence, whereas that of Ydj1 was rather limited. Moreover, the divergence of Ydj1 was not affected by the emergence of Apj1 and thus, the coexistence of these two proteins. However, in all cases, type I sites were distributed across the entire alignment. Therefore, we were unable to identify specific domains as being responsible for the increase of the sequence divergence; rather the entire sequences of both Xdj1 and Apj1 were more variable than those of Ydj1 (data not shown). Interestingly, despite relatively high level of type I sequence divergence, we did not identify any type II divergent sites in any of the sequence comparisons discussed earlier.
Table 2.
Amino Acid Sequence Divergence.
| Group A | Group B | Residuesa | Divergence Coefficients |
Type I Sitesd |
||
|---|---|---|---|---|---|---|
| ΘIb ± SE | ΘIIc ± SE | Invariant in A | Invariant in B | |||
| Ydj1 | Xdj1 | 317 | 0.48 ± 0.05 | −0.46 ± 0.24 | 48 | 2 |
| Ydj1post-Apj1 | Apj1 | 325 | 0.39 ± 0.06 | −0.37 ± 0.18 | 67 | 2 |
| Ydj1post-Apj1 | Ydj1pre-Apj1 | 363 | 0.26 ± 0.05 | −0.11 ± 0.10 | 34 | 22 |
aResidues are the number of sites in the alignment.
bΘI is the coefficient of type I divergence.
cΘII is the coefficient of type II divergence.
dType I sites are the number of sites in the alignment, which were invariant in the one group of sequences but variable in the other group (see Materials and Methods for details).
The results of our analyses of YDJ1, XDJ1, and APJ1 sequences described earlier are consistent with a scenario in which over a long evolutionary time scale both XDJ1 and APJ1 evolved faster than YDJ1. To test whether this was also the case for a shorter time evolution scale, we analyzed codon evolution for a set of five species that are closely related to S. cerevisiae, belonging to the Saccharomyces sensu stricto (Dujon 2010). As for this data set, the dS values were not saturated (dS ≪ 1, table 3), and we could analyze distribution of dN/dS ratio along the phylogeny. Specifically, for each set of orthologous sequences (XDJ1, APJ1, and YDJ1), we evaluated the hypothesis that all branches of the phylogeny exhibit the same dN/dS ratio against a set of hypotheses under which the dN/dS ratio along the external branch for a given species was different from that in the rest of the phylogeny. All the analyzed genes did not reject the null hypothesis of a uniform dN/dS ratio across the phylogeny at the P = 0.01 level. However, as expected, YDJ1 evolved more slowly than either XDJ1 or APJ1 (dN/dS values of 0.027, 0.155, and 0.080, respectively) (table 3). Finally, as the dN/dS values obtained for each analyzed gene were obviously lower than 1, we concluded that on a finer time scale YDJ1, XDJ1, and APJ1 are subject to strong purifying selection.
Table 3.
Evolutionary Rates in Saccharomyces sensu stricto.
| Gene | dN | dS | dN/dS |
|---|---|---|---|
| YDJ1 | 0.006 ± 0.004 | 0.233 ± 0.148 | 0.027 |
| XDJ1 | 0.039 ± 0.016 | 0.255 ± 0.104 | 0.155 |
| APJ1 | 0.028 ± 0.014 | 0.349 ± 0.173 | 0.080 |
Note.—Values for dN and dS are shown as mean ± SD.
Discussion
The results presented here support the idea that the DnaJ type J-proteins of the S. cerevisiae cytosol, Apj1 and Xdj1, play specialized roles in critical biological functions—proteolysis and protein translocation across membranes, respectively. The absence of Apj1 exacerbated the defect in degradation of sumolyated proteins when the cell was challenged by a reduction in sumo-dependent ubiquitin ligase activity. Translocation of proteins into mitochondria was defective when xdj1Δ cells were challenged by the absence of a component of the inner membrane protein translocase. That these phenotypes were observed is remarkable given that Ydj1, the third DnaJ-type J-protein, is present at over 200-fold higher levels than either Apj1 or Xdj1, and underscores the idea of functional specificity of Apj1 and Xdj1. The results presented here also demonstrate that Xdj1 and Apj1 play their specialized roles as components of the Hsp70 machinery. For both Xdj1 and Apj1, alteration of the J-domain, which is required for Hsp70 activation, resulted in the loss of both Apj1’s and Xdj1’s specialized activity. Moreover, the toxicity connected with modest overexpression of Xdj1 is partially suppressed by simultaneous overexpression of the nucleotide release factor Sse1, suggesting that acceleration of the Hsp70-ATPase cycle subdues the toxic effect.
How did these highly specialized J-proteins originate? Several lines of evidence suggest that the emergence of Xdj1 and Apj1 can be traced back to sequential duplications of an ancient YDJ1 gene, with both significantly predating the whole genome duplication event that occurred in the Saccharomyces lineage approximately 100 Ma (Wolfe and Shields 1997). Most likely, the first duplication that gave rise to Xdj1 took place in a common ancestor of Ascomycetes, whereas the second duplication event responsible for emergence of Apj1 was more recent and most likely took place in a common ancestor of the Saccharomycetaceae and the CTG clade to which S. cerevisiae belongs. The presence of Xdj1 orthologs in the Ascomycetes species and their absence in species outside the Ascomycota phylum, coupled with the presence of Apj1 orthologs being restricted to species belonging to the Saccharomycetaceae and the CTG clade (Dujon 2010), is consistent with this idea. In addition, the phylogenetic reconstruction show that Xdj1, Apj1, and Ydj1 form a strongly supported monophyletic group and are more closely related to each other than to other members of DnaJ-family functioning in endoplasmic reticulum (Scj1) or mitochondrium (Mdj1). Moreover, in agreement with its more restricted phylogenetic distribution, and thus, likely younger age, Apj1 forms a sister cluster to Ydj1, which strongly supports the hypothesis that they are related by a gene duplication event. We note that the topology of the protein tree does not directly support the origin of XDJ1 as a duplicate of YDJ1. However, we consider this evolutionary scenario very likely, as XDJ1 and YDJ1 coexist in all surveyed Ascomycetes species and that, based on protein phylogeny, XDJ1 and YDJ1 belong to the same clade.
The fact that XDJ1 and APJ1 were universally retained after they emerged points to the functional importance of these J-proteins and is consistent with our experimental analyses showing that in S. cerevisiae both Xdj1 and Apj1 play specialized roles in important cellular processes. Hints of what drives the specialized function of these two J-proteins can be gleaned from the experimental data presented here. Evidence indicates that client protein binding is important for both. Xdj1’s peptide binding specificity is clearly different from that of Ydj1. Moreover, in vivo tests indicated that changes within the peptide-binding cleft abolished the toxicity and defect in mitochondrial import caused by modest overexpression. Although biochemical data available for Apj1 are limited due to our inability to purify it in large amounts, the results of our genetic analyses points to the conclusion that not only is client protein binding critical for function, but also the functional specificity of Apj1 is determined by the domain containing the client-binding cleft. This idea is supported by the fact that neither the deletion of the GF-S region nor replacement of the Zn-binding region by the Ydj1 homologous sequence affected Apj1’s ability to function. Although these data support the idea of specialized client protein binding being behind the specificity, other explanations are possible. Indeed, localization of J-proteins to particular sites of action is an established mode of specialization (Kampinga and Craig 2010). That Xdj1 has been found in proteomic studies to be present in mitochondrial outer membranes supports this idea (Burri et al. 2006). Perhaps, both client-binding specificity and localization to the outer mitochondrial membrane help to drive the functional specificity of Xdj1.
How do the specialized functions of Xdj1 and Apj1 relate to those of Ydj1? The results described in this report could be interpreted according to the classical model of protein evolution purporting that after gene duplication, one copy of the duplicated gene preserves the original function (YDJ1), allowing the other (XDJ1 or APJ1) to be free to evolve new functions (Ohno 1970). At first glance, our data fits this neo-functionalization model, as indeed, the rate of YDJ1 evolution was not affected by the presence of APJ1, and both XDJ1 and APJ1 did evolve specialized functions. However, we favor the alternative subfunctionalization scenario (Lynch and Conery 2000) that Apj1 and Xdj1 took over functions already carried out by multifunctional Ydj1. We hypothesize that these functions were fine-tuned, but not strictly new. We note that the broad biological functions in which Apj1 and Xdj1 are involved, regulation of protein degradation and import of proteins into mitochondria are ancient and ones in which Ydj1 is also known to function (Becker et al. 1996; Youker et al. 2004), albeit in different aspects of these complex processes. An argument that may favor a neo-functionalization hypothesis for APJ1 and XDJ1 evolution is their rapid sequence evolution. However, it is also well documented that the rate of sequence evolution is reversely proportional to the protein expression level (Drummond et al. 2005; Drummond and Wilke 2009), and thus the low expression level in S. cerevisiae (and presumably other species) may well explain the rapid rate. Moreover, the low and steady rate of YDJ1 evolution, which was not affected by the presence of Apj1, could well be due to a combination of its very high expression level and significant functional constraints resulting from its important biological roles.
In summary, based on the results of our functional and evolutionary analysis, we propose that the specialized functions of both Xdj1 and Apj1 evolved in two steps after gene duplication: 1) at an early stage, relaxation of selection due to low expression of one of the duplicates allowed accumulation of changes that led to functional diversification and perhaps enhancement of a subset of specialized functions performed by the ancestral gene, and 2) at a later stage, purifying selection maintained the newly specialized functions. Our results have implications for the functional expansion and diversification of chaperone networks, beyond the proteins functioning in yeast cytosol. Interestingly, human cells also contain three closely related members of the DnaJ family in the cytosol/nuclear compartment (Vos et al. 2008). Similar to their yeast counterparts, they are involved in many important processes. Quite remarkably they also function in regulated proteolysis and protein import to mitochondria (Terada et al. 1997; Bhangoo et al. 2007; Fan and Young 2011). Thus, the functional diversification process based on gene duplication and subsequent specialization described here might have played an important role in formation of the taxon-specific Hsp70–Hsp40 networks functioning in critical biological processes.
Materials and Methods
Ortholog Detection and Assignment
Using amino acid sequences of S. cerevisiae YDJ1, XDJ1, APJ1, SCJ1, and MDJ1 as queries, BLAST searches (Altschul et al. 1990) were performed with fungal genomic data obtained from NCBI, BROAD Institute, SGD, Genolevures, Podospora anserina genome project, and JGI databases. A total of 43 species from the Ascomycetes, which were further subdivided into three clades (Taphrinomycotina, Pezizomycotina, and Saccharomycotina) were analyzed (supplementary fig. S3, Supplementary Material online, for details).
To assess the presence or absence of particular DnaJ-like orthologs, BLASTP and TBLASTN were used to identify putative homologs within databases of annotated open reading frames (ORFs) and raw contig data for each species. The genomic locations of the best hit ORFs obtained with BLASTP were verified to correspond with the location of the best matches from the raw genomic data obtained with TBLASTN. If a high scoring hit could not be identified within the annotated ORFs, putative ORFs were identified directly from the genomic region containing the best TBLASTN hit using ORF finder (http://www.ncbi.nlm.nih.gov/gorf/gorf.html). Hits with a resulting E value of less than 10−10 were further inspected for the presence or absence of specific sequence motifs characteristic of the reference sequences from S. cerevisiae. Annotated results from each BLAST search were then used as query sequences in a reciprocal BLASTP search against the S. cerevisiae protein database. We used reciprocal-best-BLAST sequences as our operational definition of orthologs.
Phylogenetic Tree Construction
Nucleotide and amino acid sequence data of YDJ1, XDJ1, APJ1, SCJ1, and MDJ1 orthologs were used to construct multiple sequence alignments for phylogenetic reconstruction. To reduce method-specific bias, we used MAFFT v6.849b (Katoh et al. 2002) and Prank v121002 (Loytynoja and Goldman 2005) with default settings. The obtained alignments were used to infer the phylogenetic trees using ML and Bayesian Inference (BI) methods. The ML analyses were performed using RaxML v.7.3.5 (Stamatakis 2006) with 100 bootstrap replicates, under the LG (Le and Gascuel 2008) model of amino acid substitution, taking into account the empirical amino acid frequencies and using four categories derived from the gamma (γ) distribution to estimate rate heterogeneity (LG + G). This model was estimated as the best fitting substitution model by ProtTest v3.2 (Abascal et al. 2005). The BI analyses were performed using MrBayes v3.2 (Ronquist and Huelsenbeck 2003) under the WAG model (Whelan and Goldman 2001) of amino acid substitution and assuming gamma distribution rate variation across sites (WAG + G model). This model was estimated by ProtTest as the best fitting substitution model other than the LG model, which was not implemented in that version MrBayes. The analyses were performed with two independent runs, each with four chains, running for 4,000,000 generations and sampling every 100 generations. The first 25% of sampled trees were discarded as burn-in before estimation of posterior probabilities for branch supports.
Sequence Analyses
Sequence identity and similarity scores were calculated as an arithmetic mean of all pairwise sequence identity/similarity comparisons in a given multiple sequence alignment. Similarity scores were calculated as an arithmetic mean of all pairwise sequence similarity comparisons, using BLOSUM62 for YDJ1 and BLOSUM35 for XDJ1 and APJ1. Two residues were counted as similar if their substitution score in the corresponding BLOSUM matrix was greater than 0.
Synonymous (dS) and nonsynonymous (dN) substitution rates were determined for the YDJ1, XDJ1, and APJ1 orthologs using the ML approach as implemented in the PAML package v4.6 (Yang 2007) using the “codeml” program. A single dN/dS value was assumed for the entire tree obtained with ML methods and mean dN and dS rates were obtained by averaging them across all branches of the tree.
Testing for lineage-specific changes in evolutionary rates of YDJ1, XDJ1, and APJ1 in species belonging to Saccharomyces sensu stricto (S. cerevisiae, S. paradoxus, S. mikatae, S. kudriavzevii, and S. bayanus) (Dujon 2010) was performed as a series of likelihood ratio tests. The likelihood of a null hypothesis of a single dN/dS ratio on the whole tree was compared against six alternative hypotheses. The first five hypotheses assumed a unique dN/dS ratio on an external branch leading to a different Saccharomyces sensu stricto species. The sixth hypothesis assumed that all branches of the Saccharomyces sensu stricto tree have evolved under a different dN/dS ratio.
In all PAML analyses, the codon frequencies were averaged from nucleotide frequencies at the three codon positions (codon frequency model F3 × 4). To avoid falling into suboptimal likelihood peaks and ensure proper convergence, the analyses were carried out multiple times, using both low and high initial settings of the estimated parameters (dN/dS ratio and transition to transversion rate kappa, κ). In each analysis, initial values of 0.1, 0.5, 1, and 5 were used for both parameters in all 16 different combinations.
Divergence Analyses
Coefficients of types I and II amino acid sequence divergence, as defined in (Gu 2001), were calculated using the DIVERGE software (Gu and Vander Velden 2002). For type I changes, we used the likelihood-based method (Gu 2001), and all analyses were bootstrapped on 100 replicates. Additional searches for sites exhibiting type I and type II divergence was performed using a custom script written in BioPerl (Stajich et al. 2002). Multiple alignments of selected sequences (Ydj1:Xdj1, Ydj1postApj1:Ydj1preApj1, or Ydj1postApj1:Apj1) were inspected for sites in strict agreement with the definition of type I divergence, that is, invariant in one group of compared sequences but variable in another group. To correct for rare, single time substitution events, the total count of type I sites was reduced by the number of cases when a residue was invariant in all but a single sequence in the data set.
Codon Usage Bias
Codon usage bias was estimated using the effective number of codons (ENC) (Wright 1990) and Codon Adaptation Index (CAI) (Sharp and Cowe 1991). Both statistics were calculated using the CodonW software (Peden 1999). ENC was calculated for all surveyed species and CAI was calculated for orthologs from five closely related Saccharomyces sensu stricto species (S. cerevisiae, S. paradoxus, S. mikatae, S. kudriavzevii, and S. bayanus) with the reference set of optimal codons from S. cerevisiae.
Statistical Analyzes
Assessment of statistical significance of differences in sequence identity/similarity values, as well as that of the dN and dS rates were performed in the R language for statistical computation (R Development Core Team 2012). Distributions of dN, dS, identity, and similarity values were first checked for nonnormality using the Shapiro–Wilk test and the level of statistical significance of difference between them was assessed using the Kolmogorov–Smirnov test. The t test for CAI values was performed as implemented in the LibreOffice Calc v3.5.4.2. All tests were done at P = 0.01 significance.
Yeast Strains and In vivo Methods
All yeast strains used in this study were of the W303 genetic background. apj1Δ, xdj1Δ, ydj1Δ, and pam17Δ were described previously (Sahi and Craig 2007; Schiller 2009). slx5Δ and slx8Δ strains were constructed by polymerase chain reaction (PCR) amplifying the KanMx gene cassette from the respective deletions strains of a knock-out library (Open Biosystems), using specific flanking primers and then transforming the gene disruption cassette into W303. apj1Δ slx5Δ and xdj1Δ pam17Δ were constructed by crossing the parental yeast strains, tetrad dissection, and selecting for spores carrying the appropriate gene disruption markers. Cells were grown in glucose-based media.
Protein Purification
WT and variant Xdj1, Ydj1, and Apj1 were expressed as Smt3 (SUMO)-His6 fusion proteins in the Rosetta E. coli strain (Roche). Induction was carried out at 18 °C for 18 h using 0.5 mM isopropyl β-d-1-thiogalactopyranoside (IPTG) in 1 l of rich media having chloramphenicol (34 µg/ml) and kanamycin (40 µg/ml). Cells were chilled on ice before pelleting. Cell pellets were dissolved in 30-ml ice-cold lysis buffer (20 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid [HEPES], 0.5 M NaCl, glycerol [5%], 10 mM ß-mercaptoethanol, 30 mM imidazole, pH 7.7) with protease inhibitor (complete Mini, ethylenediaminetetraacetic acid-free, Roche Diagnostics). Cells were lysed in a French Press, cleared by centrifugation at 14 k for 30 min. Ni–NTA His–Bind resin (2 ml) (Qiagen) equilibrated with the lysis buffer was added to the clarified cell lysate and incubated with mild agitation for 2.5 h in the cold room. Ni resin was spun down and the supernatant was discarded. Resin was transferred to a column and sequentially washed with 25 ml of ice-cold lysis buffer, 25 ml of lysis buffer with 1 M NaCl, and again with 15 ml of lysis buffer. Proteins were eluted with 10 ml 0.5 M imidazole in lysis buffer and dialyzed against 1 l of lysis buffer for 3 h. Ulp1 protease, purified as described (Malakhov et al. 2004), was added (at an approximate ratio to protein of 1:100) directly into the dialysis bag (14 kDa cut off). Dialysis was continued overnight. The protein was injected onto a pre-equilibrated HiPrep 16/60 Sephacryl S-200 HR column (GE Healthcare) and eluted with 120 ml lysis buffer at a flow rate of 0.5 ml per min, 0.2 MPa. 1.4 ml fractions were collected and analyzed on 10% sodium dodecyl sulphate (SDS)–polyacrylamide gel. Pooled protein fractions were concentrated using Amicon Ultra centrifugal filters (Ultracel, 3,000 MWCO, Millipore Corp.) and frozen in liquid nitrogen until further use.
For generating anti-Apj1 antibody, a C-terminal fragment encoding the C-terminal 105 amino acids of Apj1 was PCR amplified with primers encoding a 5′ BamHI and a 3′ SacI site and cloned into the pGEX-KG vector in frame as an N-terminal glutathionine-S-transferase fusion. For anti-Xdj1 antibody, sequence encoding the C-terminal 210 residues was PCR amplified with appropriate primers, digested with NdeI/BamHI and cloned into the same sites in protein expression vector pet3a. Apj1 and Xdj1 expression was induced with 0.5 mM IPTG for 3 h at 30 °C in C41 and Rosetta cells, respectively. Protein was purified using glutathionine affinity chromatography. Polyclonal antibodies were generated in rabbits at Harlan laboratories (Madison, WI) using purified protein as antigen. For immunoblots, antibodies were purified using HiTrap NHS-activated HP columns (GE Healthcare) and purified Apj1 and Xdj1 according to the manufacturers’ instructions.
Screening of the Phage Display Library
A phage display dodecapeptide library (Ph.D-12, New England Biolabs) was used. 100 µg of Xdj1 or Ydj1 was immobilized in individual wells of polystyrene 96-well microtiter plates overnight in 0.1 N NaHCO3 pH 8.6 on a rocker shaker in the cold room. The Hsp40-coated dish was then washed and blocked with a solution that contained 5 mg/ml bovine serum albumin (BSA) and 0.1 M NaHCO3 for 2 h at 4 °C. Subsequently, the plate was washed with Tris-buffered saline/Tween 20 (TBS–T) (20 mM Tris–Cl, pH 8.0, 150 mM NaCl, 0.1% Tween 20) six times. 10 µl of the Ph.D-12 library diluted to 100 µl in TBS–T was added in each Hsp40 coated well and incubated for 1 h at room temperature. The plate was then washed 10 times with TBS–T. Bound phage was eluted by addition of 100 µl of 0.2 M Glycine–HCl (pH 2.2), 1 mg/ ml BSA for 20 min. The eluate was immediately neutralized with 15 µl of 1 M Tris–HCl, pH 9.1. The eluted phage displaying the peptides binding to Xdj1 or Ydj1 were amplified by infection of a 20-ml culture of early log phase E. coli ER2537 that was incubated for 4.5 h at 37 °C with vigorous shaking. The titer of the amplified phage was determined, and the second and third rounds of biopanning were performed. Later rounds were carried out as per manufacturer’s instructions. After the third round of panning, the coliphage eluate was plated. Coliphage present in isolated plaques was purified and the DNA sequenced to determine the sequence of 18 peptides from each screening.
Plasmid Construction
The genomic clone of XDJ1 was constructed by PCR amplifying the coding sequence of Xdj1 along with 800 bp of upstream and 400 bp of downstream sequence. The PCR fragment was digested with EcoRI/BamHI and cloned in same sites in pRS313. For making pRS314-Apj1, 400-bp upstream and 400-bp downstream sequence was PCR amplified and cloned into XhoI/SacI sites in pRS314. CEN-ADH-Xdj1 plasmid was constructed by subcloning the Xdj1 coding sequence from 2µ-GPD-Xdj1 described previously (Sahi and Craig 2007) into the SpeI/XhoI sites in pRS414-ADH vector. Internal deletions, pRS314-Apj1Δ79-161 (Apj1ΔG/F-S), pRS313-Xdj1Δ77-116 (Xdj1ΔGF), and all point mutations were generated by QuikChange PCR (Stratagene). ZnFR deletion mutant pRS314-Apj1Δ206-262 (Apj1ΔZn) was generated similarly using primers that deleted the ZnFR and inserted a GGSG linker. pRS314-Apj1-Zn Swap and pRS313-Xdj1-Zn Swap were made by designing primers that were complementary at their 5′-ends to either Apj1 or Xdj1 coding sequence just outside their respective ZnFR and amplifying the ZnFR (residues 143–206) of Ydj1. The resulting PCR fragment was used as a megaprimer for a QuikChange PCR to substitute the Zn finger in Apj1 and Xdj1 with that of Ydj1. The complete coding sequences of Apj1, Xdj1, and Ydj1 were PCR amplified and digested with BamHI/XhoI and cloned into the same sites in pSmt3 (Gift from W. Hendrickson, Columbia University) to generate pSmt3-Apj1, pSmt3-Xdj1, and pSmt3-Ydj1. pRS413-GPD-Sse1 (Shaner et al. 2006) was a gift of K. Morano, University of Texas.
Other Methods
Extracts of total cellular proteins were prepared by treating cells with 0.1 N NaOH and resuspension in SDS sample buffer (62.5 mM Tris–HCl, pH 6.8, 5% glycerol, 2% SDS, 2% β-mercaptoethanol, and 0.01% bromophenol blue). Protein was detected by Enhanced Chemiluminescence kit (Amersham Pharmacia) according to manufacturer’s instructions. SUMO-conjugates were resolved on a 4–15% gradient gel (BioRad) and probed with Anti-Smt3 polyclonal antibody (Gift from P. Hieter, University of British Columbia). Quantification of protein bands was performed with ImageJ software. Single point phage ELISA were performed in 96-well polystyrene microtiter plates essentially similar to the biopanning of the 12mer Ph.D library, except 2 µg of protein was coated and a single amplified phage clone was added in each well. Bound phage was detected with horseradish peroxidase-conjugated anti-M13 monoclonal antibody (GE Healthcare). Plates were visualized in a plate reader (Biotek) at 415 nm. Single-turnover ATPase assays of Ssa1 were carried out as described previously (Lopez-Buesa et al. 1998). Isolated Hsp70-[α-32P]ATP complexes were incubated with 2-fold molar excess of Apj1, Xdj1, or Ydj1 at 25 °C, and samples were removed for detection of ATP and ADP. The fractions of ATP hydrolyzed to ADP over the time courses were quantified by PhosphorImager analysis (Molecular Dynamics) and the data used to determine apparent rate constants for hydrolysis of bound ATP.
Supplementary Material
Supplementary figures S1–S7 are available at Molecular Biology and Evolution online (http://www.mbe.oxfordjournals.org/).
Acknowledgments
The authors thank Sanjith Reddy Yeruva for excellent technical assistance, Chris T. Hittinger for thoughtful discussions, and Brenda Schilke for helpful comments on the manuscript. They also thank Phil Heiter, Kevin Morano, and Wayne Hendrickson for antibody or expression plasmids. This work was supported by the National Institutes of Health GM31107 to E.A.C. and TEAM/2009-3/5 to J.M. and J.K.
References
- Abascal F, Zardoya R, Posada D. ProtTest: selection of best-fit models of protein evolution. Bioinformatics. 2005;21:2104–2105. doi: 10.1093/bioinformatics/bti263. [DOI] [PubMed] [Google Scholar]
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- Becker J, Walter W, Yan W, Craig EA. Functional interaction of cytosolic hsp70 and a DnaJ-related protein, Ydj1p, in protein translocation in vivo. Mol Cell Biol. 1996;16:4378–4386. doi: 10.1128/mcb.16.8.4378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhangoo MK, Tzankov S, Fan AC, Dejgaard K, Thomas DY, Young JC. Multiple 40-kDa heat-shock protein chaperones function in Tom70-dependent mitochondrial import. Mol Biol Cell. 2007;18:3414–3428. doi: 10.1091/mbc.E07-01-0088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bukau B, Weissman J, Horwich A. Molecular chaperones and protein quality control. Cell. 2006;125:443–451. doi: 10.1016/j.cell.2006.04.014. [DOI] [PubMed] [Google Scholar]
- Burri L, Vascotto K, Gentle IE, Chan NC, Beilharz T, Stapleton DI, Ramage L, Lithgow T. Integral membrane proteins in the mitochondrial outer membrane of Saccharomyces cerevisiae. FEBS J. 2006;273:1507–1515. doi: 10.1111/j.1742-4658.2006.05171.x. [DOI] [PubMed] [Google Scholar]
- Caplan AJ, Douglas MG. Characterization of YDJ1: a yeast homologue of the bacterial dnaJ protein. J Cell Biol. 1991;114:609–621. doi: 10.1083/jcb.114.4.609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caplan AJ, Tsai J, Casey PJ, Douglas MG. Farnesylation of YDJ1p is required for function at elevated growth temperatures in Saccharomyces cerevisiae. J Biol Chem. 1992;267:18890–18895. [PubMed] [Google Scholar]
- Cheetham ME, Caplan AJ. Structure, function and evolution of DnaJ: conservation and adaptation of chaperone function. Cell Stress Chaperones. 1998;3:28–36. doi: 10.1379/1466-1268(1998)003<0028:sfaeod>2.3.co;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costanzo M, Baryshnikova A, Bellay J, et al. (53 co-authors) The genetic landscape of a cell. Science. 2010;327:425–431. doi: 10.1126/science.1180823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craig EA, Huang P, Aron R, Andrew A. The diverse roles of J-proteins, the obligate Hsp70 co-chaperone. Rev Physiol Biochem Pharmacol. 2006;156:1–21. doi: 10.1007/s10254-005-0001-0. [DOI] [PubMed] [Google Scholar]
- Demoinet E, Jacquier A, Lutfalla G, Fromont-Racine M. The Hsp40 chaperone Jjj1 is required for the nucleo-cytoplasmic recycling of preribosomal factors in Saccharomyces cerevisiae. RNA. 2007;13:1570–1581. doi: 10.1261/rna.585007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dragovic Z, Broadley SA, Shomura Y, Bracher A, Hartl FU. Molecular chaperones of the Hsp110 family act as nucleotide exchange factors of Hsp70s. EMBO J. 2006;25:2519–2528. doi: 10.1038/sj.emboj.7601138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drummond DA, Bloom JD, Adami C, Wilke CO, Arnold FH. Why highly expressed proteins evolve slowly. Proc Natl Acad Sci U S A. 2005;102:14338–14343. doi: 10.1073/pnas.0504070102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drummond DA, Wilke CO. The evolutionary consequences of erroneous protein synthesis. Nat Rev Genet. 2009;10:715–724. doi: 10.1038/nrg2662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dujon B. Yeast evolutionary genomics. Nat Rev Genet. 2010;11:512–524. doi: 10.1038/nrg2811. [DOI] [PubMed] [Google Scholar]
- Fan AC, Young JC. Function of cytosolic chaperones in Tom70-mediated mitochondrial import. Protein Pept Lett. 2011;18: 122–131. doi: 10.2174/092986611794475020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan CY, Ren HY, Lee P, Caplan AJ, Cyr DM. The type I Hsp40 zinc finger-like region is required for Hsp70 to capture non-native polypeptides from Ydj1. J Biol Chem. 2005;280:695–702. doi: 10.1074/jbc.M410645200. [DOI] [PubMed] [Google Scholar]
- Fitzpatrick DA, Logue ME, Stajich JE, Butler G. A fungal phylogeny based on 42 complete genomes derived from supertree and combined gene analysis. BMC Evol Biol. 2006;6:99. doi: 10.1186/1471-2148-6-99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glover JR, Lindquist S. Hsp104, Hsp70, and Hsp40: a novel chaperone system that rescues previously aggregated proteins. Cell. 1998;94:73–82. doi: 10.1016/s0092-8674(00)81223-4. [DOI] [PubMed] [Google Scholar]
- Gu X. Statistical methods for testing functional divergence after gene duplication. Mol Biol Evol. 1999;16:1664–1674. doi: 10.1093/oxfordjournals.molbev.a026080. [DOI] [PubMed] [Google Scholar]
- Gu X. Maximum-likelihood approach for gene family evolution under functional divergence. Mol Biol Evol. 2001;18:453–464. doi: 10.1093/oxfordjournals.molbev.a003824. [DOI] [PubMed] [Google Scholar]
- Gu X, Vander Velden K. DIVERGE: phylogeny-based analysis for functional-structural divergence of a protein family. Bioinformatics. 2002;18:500–501. doi: 10.1093/bioinformatics/18.3.500. [DOI] [PubMed] [Google Scholar]
- Hartl FU, Bracher A, Hayer-Hartl M. Molecular chaperones in protein folding and proteostasis. Nature. 2011;475:324–332. doi: 10.1038/nature10317. [DOI] [PubMed] [Google Scholar]
- Kampinga HH, Craig EA. The HSP70 chaperone machinery: J proteins as drivers of functional specificity. Nat Rev Mol Cell Biol. 2010;11:579–592. doi: 10.1038/nrm2941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh K, Misawa K, Kuma K, Miyata T. MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002;30:3059–3066. doi: 10.1093/nar/gkf436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiefer F, Arnold K, Kunzli M, Bordoli L, Schwede T. The SWISS-MODEL repository and associated resources. Nucleic Acids Res. 2009;37:D387–D392. doi: 10.1093/nar/gkn750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le SQ, Gascuel O. An improved general amino acid replacement matrix. Mol Biol Evol. 2008;25:1307–1320. doi: 10.1093/molbev/msn067. [DOI] [PubMed] [Google Scholar]
- Li J, Qian X, Sha B. The crystal structure of the yeast Hsp40 Ydj1 complexed with its peptide substrate. Structure. 2003;11:1475–1483. doi: 10.1016/j.str.2003.10.012. [DOI] [PubMed] [Google Scholar]
- Li J, Sha B. Structure-based mutagenesis studies of the peptide substrate binding fragment of type I heat-shock protein 40. Biochem J. 2005;386:453–460. doi: 10.1042/BJ20041050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linke K, Wolfram T, Bussemer J, Jakob U. The roles of the two zinc binding sites in DnaJ. J Biol Chem. 2003;278:44457–44466. doi: 10.1074/jbc.M307491200. [DOI] [PubMed] [Google Scholar]
- Lopez-Buesa P, Pfund C, Craig EA. The biochemical properties of the ATPase activity of a 70-kDa heat shock protein (Hsp70) are governed by the C-terminal domains. Proc Natl Acad Sci U S A. 1998;95:15253–15258. doi: 10.1073/pnas.95.26.15253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loytynoja A, Goldman N. An algorithm for progressive multiple alignment of sequences with insertions. Proc Natl Acad Sci U S A. 2005;102:10557–10562. doi: 10.1073/pnas.0409137102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch M, Conery JS. The evolutionary fate and consequences of duplicate genes. Science. 2000;290:1151–1155. doi: 10.1126/science.290.5494.1151. [DOI] [PubMed] [Google Scholar]
- Malakhov MP, Mattern MR, Malakhova OA, Drinker M, Weeks SD, Butt TR. SUMO fusions and SUMO-specific protease for efficient expression and purification of proteins. J Struct Funct Genomics. 2004;5:75–86. doi: 10.1023/B:JSFG.0000029237.70316.52. [DOI] [PubMed] [Google Scholar]
- Mayer MP. Gymnastics of molecular chaperones. Mol Cell. 2010;39:321–331. doi: 10.1016/j.molcel.2010.07.012. [DOI] [PubMed] [Google Scholar]
- Meyer AE, Hung NJ, Yang P, Johnson AW, Craig EA. The specialized cytosolic J-protein, Jjj1, functions in 60S ribosomal subunit biogenesis. Proc Natl Acad Sci U S A. 2007;104:1558–1563. doi: 10.1073/pnas.0610704104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullen JR, Brill SJ. Activation of the Slx5-Slx8 ubiquitin ligase by poly-small ubiquitin-like modifier conjugates. J Biol Chem. 2008;283:19912–19921. doi: 10.1074/jbc.M802690200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohno S. Evolution by gene duplication. New York: Springer; 1970. [Google Scholar]
- Pais JE, Schilke B, Craig EA. Reevaluation of the role of the Pam18:Pam16 interaction in translocation of proteins by the mitochondrial Hsp70-based import motor. Mol Biol Cell. 2011;22:4740–4749. doi: 10.1091/mbc.E11-08-0715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan X, Ye P, Yuan DS, Wang X, Bader JS, Boeke JD. A DNA integrity network in the yeast Saccharomyces cerevisiae. Cell. 2006;124:1069–1081. doi: 10.1016/j.cell.2005.12.036. [DOI] [PubMed] [Google Scholar]
- Peden JF. [Nottingham (UK)]: University of Nottingham; 1999. Analysis of codon usage [PhD thesis] [Google Scholar]
- R Development Core Team. R: a language and environment for statistical computing [Internet]. Vienna, Austria: R Foundation for Statistical Computing; 2012. R version 2.15.1 (2012-06-22). ISBN 3-900051-07-0. Available from: http://www.R-project.org [Google Scholar]
- Raviol H, Sadlish H, Rodriguez F, Mayer MP, Bukau B. Chaperone network in the yeast cytosol: Hsp110 is revealed as an Hsp70 nucleotide exchange factor. EMBO J. 2006;25:2510–2518. doi: 10.1038/sj.emboj.7601139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronquist F, Huelsenbeck JP. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics. 2003;19:1572–1574. doi: 10.1093/bioinformatics/btg180. [DOI] [PubMed] [Google Scholar]
- Sahi C, Craig EA. Network of general and specialty J protein chaperones of the yeast cytosol. Proc Natl Acad Sci U S A. 2007;104:7163–7168. doi: 10.1073/pnas.0702357104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahi C, Lee T, Inada M, Pleiss JA, Craig EA. Cwc23, an essential J protein critical for pre-mRNA splicing with a dispensable J domain. Mol Cell Biol. 2010;30:33–42. doi: 10.1128/MCB.00842-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiller D. Pam17 and Tim44 act sequentially in protein import into the mitochondrial matrix. Int J Biochem Cell Biol. 2009;41:2343–2349. doi: 10.1016/j.biocel.2009.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaner L, Sousa R, Morano KA. Characterization of Hsp70 binding and nucleotide exchange by the yeast Hsp110 chaperone Sse1. Biochemistry. 2006;45:15075–15084. doi: 10.1021/bi061279k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharp PM, Cowe E. Synonymous codon usage in Saccharomyces cerevisiae. Yeast. 1991;7:657–678. doi: 10.1002/yea.320070702. [DOI] [PubMed] [Google Scholar]
- Stajich JE, Block D, Boulez K, et al. (21 co-authors) The Bioperl toolkit: Perl modules for the life sciences. Genome Res. 2002;12:1611–1618. doi: 10.1101/gr.361602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamatakis A. RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics. 2006;22:2688–2690. doi: 10.1093/bioinformatics/btl446. [DOI] [PubMed] [Google Scholar]
- Summers DW, Douglas PM, Ren HY, Cyr DM. The type I Hsp40 Ydj1 utilizes a farnesyl moiety and zinc finger-like region to suppress prion toxicity. J Biol Chem. 2009;284:3628–3639. doi: 10.1074/jbc.M807369200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terada K, Kanazawa M, Bukau B, Mori M. The human DnaJ homologue dj2 facilitates mitochondrial protein import and luciferase refolding. J Cell Biol. 1997;139:1089–1095. doi: 10.1083/jcb.139.5.1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Laan M, Chacinska A, Lind M, Perschil I, Sickmann A, Meyer HE, Guiard B, Meisinger C, Pfanner N, Rehling P. Pam17 is required for architecture and translocation activity of the mitochondrial protein import motor. Mol Cell Biol. 2005;25:7449–7458. doi: 10.1128/MCB.25.17.7449-7458.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vos MJ, Hageman J, Carra S, Kampinga HH. Structural and functional diversities between members of the human HSPB, HSPH, HSPA, and DNAJ chaperone families. Biochemistry. 2008;47:7001–7011. doi: 10.1021/bi800639z. [DOI] [PubMed] [Google Scholar]
- Walsh P, Bursac D, Law YC, Cyr D, Lithgow T. The J-protein family: modulating protein assembly, disassembly and translocation. EMBO Rep. 2004;5:567–571. doi: 10.1038/sj.embor.7400172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whelan S, Goldman N. A general empirical model of protein evolution derived from multiple protein families using a maximum-likelihood approach. Mol Biol Evol. 2001;18:691–699. doi: 10.1093/oxfordjournals.molbev.a003851. [DOI] [PubMed] [Google Scholar]
- Wolfe KH, Shields DC. Molecular evidence for an ancient duplication of the entire yeast genome. Nature. 1997;387:708–713. doi: 10.1038/42711. [DOI] [PubMed] [Google Scholar]
- Wright F. The “effective number of codons” used in a gene. Gene. 1990;87:23–29. doi: 10.1016/0378-1119(90)90491-9. [DOI] [PubMed] [Google Scholar]
- Yang Z. PAML 4: phylogenetic analysis by maximum likelihood. Mol Biol Evol. 2007;24:1586–1591. doi: 10.1093/molbev/msm088. [DOI] [PubMed] [Google Scholar]
- Youker RT, Walsh P, Beilharz T, Lithgow T, Brodsky JL. Distinct roles for the Hsp40 and Hsp90 molecular chaperones during cystic fibrosis transmembrane conductance regulator degradation in yeast. Mol Biol Cell. 2004;15:4787–4797. doi: 10.1091/mbc.E04-07-0584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao R, Davey M, Hsu YC, Kaplanek P, Tong A, Parsons AB, Krogan N, Cagney G, Mai D, Greenblatt J, Boone C, Emili A, Houry WA. Navigating the chaperone network: an integrative map of physical and genetic interactions mediated by the hsp90 chaperone. Cell. 2005;120:715–727. doi: 10.1016/j.cell.2004.12.024. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.