

Abstract
Phylogeographical analyses have become commonplace for a myriad of organisms with the advent of cheap DNA sequencing technologies. Bayesian model-based clustering is a powerful tool for detecting important patterns in such data and can be used to decipher even quite subtle signals of systematic differences in molecular variation. Here, we introduce two upgrades to the Bayesian Analysis of Population Structure (BAPS) software, which enable 1) spatially explicit modeling of variation in DNA sequences and 2) hierarchical clustering of DNA sequence data to reveal nested genetic population structures. We provide a direct interface to map the results from spatial clustering with Google Maps using the portal http://www.spatialepidemiology.net/ and illustrate this approach using sequence data from Borrelia burgdorferi. The usefulness of hierarchical clustering is demonstrated through an analysis of the metapopulation structure within a bacterial population experiencing a high level of local horizontal gene transfer. The tools that are introduced are freely available at http://www.helsinki.fi/bsg/software/BAPS/.
Keywords: genetic population structure, phylogeographics, Bayesian inference, evolutionary epidemiology
Introduction
Given the recent advances in DNA sequencing technology, phylogeographical analysis of molecular variation has become an increasingly important approach for finding clues to the interplay of ecological factors, dispersal, and evolution (Beaumont et al. 2010). Analysis of the transmission patterns and genetic population structure of pathogens within a host population are two examples of applications where both the spatial dimension of the data and the hierarchy of relatedness among strains introduce statistical challenges to the discovery of mechanisms affecting genetic isolation, dispersal, and evolution. The evolutionary patterns, genetic population structure, and links to ecological factors are notoriously difficult to decipher for some bacterial populations due to high rates of horizontal gene transfer caused by homologous recombination, which can occur between distantly related lineages and across named species. Hence, phylogenetic tools, such as BEAST (Drummond and Rambaut 2007), need to be complemented with population genetic analysis that allows for an admixture within and between lineages. We have recently successfully identified significant variation in the extent of recombination and its association with several ecological and genetic factors using the population genetic software package, BAPS (Corander and Marttinen 2006; Corander and Tang 2007; Corander et al. 2008a; Corander et al. 2008b; Tang et al. 2009; Cheng et al. 2011), on large collections of DNA sequence data from pathogen populations (Hanage et al. 2009; Castillo-Ramírez et al. 2012; Connor et al. 2012; Corander et al. 2012; Willems et al. 2012). For instance, we have showed that hospital-adapted virulent and resistant strains of the major source of nosocomial infections, Enterococcus faecium, display a marked reduction in their amount of recombination compared with commensal strains. Moreover, in contrast to the previous understanding about their evolution, we discovered that the hospital-adapted strains are linked to multiple independent introductions and that these are likely to represent different animal reservoirs of the pathogen (Willems et al. 2012). To enable the latter discovery, we applied the BAPS clustering model in a hierarchical manner and analyzed the associations with strain metadata using both the major clusters and the substructure within them.
An example of an analysis of pathogen population structure where the spatial dimension is of central importance is provided by Castillo-Ramirez et al. (2012), who studied the linkage of founder events with regional variation in recombination rates within a global clone of methicillin-resistant Staphylococcus aureus (MRSA). Using both BAPS and BEAST on a large, worldwide collection of whole-genome DNA sequence data derived from samples from hospital patients, they identified several genetically isolated lineages within the ST239 clone and estimated their times of introduction into particular geographical regions. In addition, it was shown that within a single country, geographical isolation of a hospital from other hospitals has consequences on the extent at which recombination does affect genomic evolution.
To further facilitate analyses of the type discussed above, we have implemented the spatially explicit BAPS model for clustering DNA sequence data, which was previously available only for molecular marker data and has been popular, for instance, in the analysis of variation detected at microsatellite loci (Corander et al. 2008b). In addition, to simplify the application of the hierarchical model-based clustering of DNA sequences, we have implemented a tandem version of BAPS (termed hierBAPS), which can accommodate large multiple sequence alignments and provides output directly in a hierarchically structured manner. Using DNA sequence data from Borrelia burgdorferi, the viridans group Streptococci, and a simulated bacterial metapopulation, we highlight the usefulness of these tools for the analysis of molecular variation in the contexts of evolutionary and spatial epidemiology.
Results
Lyme borreliosis, which is caused by the tick-borne bacterium B. burgdorferi, is a commonly occurring disease in North America and Europe, for which a multilocus sequence typing scheme has been introduced to enable studies of the spread dynamics and evolutionary trajectories of the population (Margos et al. 2008). Figure 1 shows the results of applying the spatially explicit BAPS clustering model to all publicly available North American sequence types (366 isolates) of B. burgdorferi containing eight housekeeping genes combined with spatial information (accessible at the database http://borrelia.mlst.net/, last accessed November 5, 2012). In this analysis, k = 12 clusters of genetically significantly distinct strains were detected and the BAPS output can be used directly to produce a geographical representation of the population structure in Google Maps with the tool available in the portal http://www.spatialepidemiology.net/, last accessed November 5, 2012. In addition, a colored tessellation representation of the output similar to genetic marker locus-based analysis is available (Corander et al. 2008b). The flexible zooming interface of Google Maps provides a way to rapidly produce a series of spatial representations of the estimated genetic population structure at different levels of resolution.
Fig. 1.

Google Maps representation of the estimated spatial genetic population structure of North American Borrelia burgdorferi produced from the BAPS output using the tool available in the portal http://www.spatialepidemiology.net/, last accessed November 5, 2012.
In modern evolutionary epidemiology, it is common for hundreds to thousands of bacterial strains to be considered within a single study, which poses challenges for statistical analysis. Phylogenetic trees are most often the tool of choice, but preferentially they need to be complemented with population genetic analyses to establish the extent to which recombination affects the estimated levels of relatedness. In addition, when large numbers of strains are jointly analyzed, it becomes increasingly difficult to specify the boundaries of separate lineages, in particular when a nonnegligible level of recombination is present in the population, because this tends to strongly affect the bootstrap support values of internal nodes. Figure 2 shows a phylogenetic tree estimated for 427 strains representing 23 species in the viridans group Streptococci based on the eMLSA typing scheme (Bishop et al. 2009). The leaf node coloring represents the clustering detected in the BAPS analysis which resulted in k = 13 groups of strains. Most clusters correspond to well-resolved clades in the tree, the notable exceptions being lineages that are represented by only a very few samples and are quite distinct in genetic terms, resembling thus the phenomenon known as “long-branch attraction.” The primary reason for such a grouping of outliers is that the statistical power to detect the outlier samples in a highly heterogeneous population is limited by the fact that cluster-specific parameters need to be estimated from a small number of sequences and their level of dissimilarity to the remaining population weighted against the increased complexity of the model where outliers were kept as separate groups.
Fig. 2.

BAPS clustering of 427 genotypes from 23 species in the viridans group Streptococci. Each leaf node of the tree is labeled with a color corresponding to a BAPS cluster.
Figure 3 illustrates the usefulness of the hierarchically applied model-based clustering approach to resolve “conservative” clusters arising from the Occam’s razor effect. The statistical power to detect the underlying population substructure is increased by the fact that in a heterogeneous population many sequence sites are variable only within a specific lineage, and hence, when focusing the cluster analysis on a single cluster detected in the first stage of the analysis, many sites that are variable outside the cluster will be monomorphic, leading to a decrease in the number of parameters to be estimated in the second stage of analysis. The data presented in figure 3 have been generated under a metapopulation model with no migration and a high rate of local within-patch recombination. Noting that every patch represents sequence data from 1,000 strains, the degree to which the underlying population structure was uncovered in this analysis is certainly encouraging. While the first-stage clustering did leave some of the underlying 25 patches undetected, i.e., several patches were merged into a single cluster, the second-stage clustering applied to the first-stage clusters did resolve the patch boundaries nearly perfectly.
Fig. 3.

Results from a hierarchical BAPS clustering of 25,000 strains of simulated bacteria from a population subdivided into 25 patches of 1,000 strains each with no between-patch migration and no patch turnover. The mutation rate of 0.0001 per locus/individual/generation was used in the simulation such that the population is subject to local recombination at a per locus rate 10 times more frequent than mutation. The tree on the left is the result from the first level of BAPS clustering, with leaf colors indicating their assignment into detected clusters. The trees on the right show cluster assignments from the second level of BAPS clustering, where two “conservative” clusters are correctly split with respect to the underlying patches used in the simulation process.
Materials and Methods
New Approaches
Several spatial models for estimating genetic population structure from molecular marker loci have been introduced in the past few years (Wasser et al. 2004; Guillot et al. 2005; Francois et al. 2006; Chen et al. 2007; Corander et al. 2008b). A common feature of these models is to introduce a spatially explicit prior for cluster structure that will combine sample locations with likelihood of the genetic data to provide improved inferences about geographical boundaries to gene flow in the underlying population. A specific feature of the model introduced by Corander et al. (2008b) is that it allows analytical integration of the parameters in both the spatial prior and the likelihood of genetic data, which enables the use of highly efficient stochastic optimization methods to estimate the posterior mode over the space of clustering solutions, in contrast to standard Markov chain Monte Carlo methods, which can be extremely tedious to use for large and complex data sets. Here, we developed an implementation of the spatial prior combined with the Markovian sequence clustering model introduced by Corander and Tang (2007) to enable spatially explicit clustering of DNA sequence data in the presence of geographical sample coordinates. This new implementation is provided by the spatial clustering module of the BAPS software version 6.0, which is freely available for research purposes at http://www.helsinki.fi/bsg/software/BAPS/, last accessed November 5, 2012. In addition to the earlier standard output from the spatial analysis, which includes both numerical and graphical representations of the estimated population structure, we have added an output format which provides a direct interface to the web portal http://www.spatialepidemiology.net/, last accessed November 5, 2012 where a user-defined Google Maps representation of the estimated clustering can be created. The zoomability of these maps provides a useful way to produce a series of spatial images at different levels of resolution.
As demonstrated in Willems et al. (2012), a hierarchical approach to model-based DNA sequence clustering, where data from a cluster at particular stage of the hierarchy are reclustered in the next stage, provides a useful way of increasing statistical power to detect separate lineages residing within the data. To preserve the internal consistency of the outputs from different BAPS modules, we implemented the hierarchical clustering approach in a separate program that can be used in tandem with BAPS. This tool, hierBAPS, is freely available for research purposes at http://www.helsinki.fi/bsg/software/BAPS/, last accessed November 5, 2012. hierBAPS accepts standard multiple sequence alignments up to whole-genome level as an input and provides access to improved imaging of the hierarchical clustering results. Distinct from the standard prior used in BAPS for nonspatial clustering, hierBAPS uses a uniform prior on the number of clusters k, such that any particular clustering solution has the prior probability proportional to 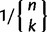 , where the denominator equals the Stirling number of the second kind and n is the number of objects to be clustered. Such a prior introduces an additional penalty for an increase in the number of clusters, because the Stirling number of the second kind increases rapidly as a function of k for a given n (until it reaches its maximum value, whereafter it decreases). Given a partition, hierBAPS uses the standard multinomial likelihood for each single-nucleotide polymorphism site in each cluster and a conjugate Dirichlet prior distribution for the frequencies of the distinct variants detected at the sequence site in question, similar to the basic clustering model in BAPS. For technical details about the distributional assumptions, see for example, Corander and Marttinen (2006).
, where the denominator equals the Stirling number of the second kind and n is the number of objects to be clustered. Such a prior introduces an additional penalty for an increase in the number of clusters, because the Stirling number of the second kind increases rapidly as a function of k for a given n (until it reaches its maximum value, whereafter it decreases). Given a partition, hierBAPS uses the standard multinomial likelihood for each single-nucleotide polymorphism site in each cluster and a conjugate Dirichlet prior distribution for the frequencies of the distinct variants detected at the sequence site in question, similar to the basic clustering model in BAPS. For technical details about the distributional assumptions, see for example, Corander and Marttinen (2006).
Real Sequence Data
The B. burgdorferi data were accessed from http://borrelia.mlst.net/ on November 27, 2012. It contains 366 multilocus sequence genotypes over 8 housekeeping loci, representing samples from North America with spatial location information available. Data on the viridans group Streptococci were taken from Bishop et al. (2009) and contain 427 multilocus sequence genotypes over 7 housekeeping genes (see also http://www.emlsa.net/, last accessed November 5, 2012). All trees presented in this work were obtained using the maximum composite likelihood method and the neighbor-joining algorithm available in the MEGA4 software (Tamura et al. 2007).
Simulated Sequence Data
Sequence data were simulated to mimic characteristics of real MLST data under a metapopulation model with no migration between patches and no patch turnover while having high recombination to mutation rate locally within each patch (r/m = 10). A population with a total of 25 patches with 1,000 bacterial strains each was generated by assuming a mutation rate of 0.0001 per locus/individual/generation, such that 7 unlinked genes with the total concatenated sequence length of 3500 bp were considered.
Acknowledgments
J.C. was supported by ERC grant no. 239784 and grant no. 251170 from the Academy of Finland and a grant from Sigrid Juselius Foundation. L.C. was supported by the Graduate School in Population Genetics.
References
- Beaumont MA, Nielsen R, Robert C, et al. (22 co-authors) In defence of model-based inference in phylogeography. Mol Ecol. 2010;19:436–446. doi: 10.1111/j.1365-294X.2009.04515.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bishop CJ, Aanensen DM, Jordan GE, Kilian M, Hanage WP, Spratt BG. Assigning strains to bacterial species via the internet. BMC Biol. 2009;7:3. doi: 10.1186/1741-7007-7-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castillo-Ramírez S, Corander J, Marttinen P, Aldeljawi M, Hanage WP, Westh H, Boye K, Gulay Z, Holden M, Feil EJ. Linking founder events with regional variation in recombination rates within a global clone of Methicillin Resistant Staphylococcus aureus (MRSA) Genome Biol. Forthcoming 2012;13:R126. doi: 10.1186/gb-2012-13-12-r126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C, Durand E, Forbes F, Francois O. Bayesian clustering algorithms ascertaining spatial population structure: a new computer program and a comparison study. Mol Ecol Notes. 2007;7:747–756. [Google Scholar]
- Cheng L, Connor TR, Aanensen DM, Spratt BG, Corander J. Bayesian semi-supervised classification of bacterial samples using MLST databases. BMC Bioinformatics. 2011;12:302. doi: 10.1186/1471-2105-12-302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connor TR, Corander J, Hanage WP. Population subdivision and the detection of recombination in non-typable Haemophilus influenzae. Microbiology. 2012;158:2958–2964. doi: 10.1099/mic.0.063073-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corander J, Connor TR, O’Dwyer CA, Kroll JS, Hanage WP. Population structure in the Neisseria, and the biological significance of fuzzy species. J R Soc Interface. 2012;9:1208–1215. doi: 10.1098/rsif.2011.0601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corander J, Marttinen P. Bayesian identification of admixture events using multi-locus molecular markers. Mol Ecol. 2006;15:2833–2843. doi: 10.1111/j.1365-294X.2006.02994.x. [DOI] [PubMed] [Google Scholar]
- Corander J, Marttinen P, Sirén J, Tang J. Enhanced Bayesian modelling in BAPS software for learning genetic structures of populations. BMC Bioinformatics. 2008a;9:539. doi: 10.1186/1471-2105-9-539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corander J, Sirén J, Arjas E. Bayesian spatial modelling of genetic population structure. Comp Stat. 2008b;23:111–129. [Google Scholar]
- Corander J, Tang J. Bayesian analysis of population structure based on linked molecular information. Math Biosci. 2007;205:19–31. doi: 10.1016/j.mbs.2006.09.015. [DOI] [PubMed] [Google Scholar]
- Drummond AJ, Rambaut A. BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol Biol. 2007;7:214. doi: 10.1186/1471-2148-7-214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francois O, Ancelet S, Guillot G. Bayesian clustering using hidden Markov random fields in spatial population genetics. Genetics. 2006;174:805–816. doi: 10.1534/genetics.106.059923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guillot G, Estoup A, Mortier F, Cosson JF. A spatial statistical model for landscape genetics. Genetics. 2005;170:1261–1280. doi: 10.1534/genetics.104.033803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanage WP, Fraser C, Tang J, Connor T, Corander J. Hyper-recombination, diversity and antibiotic resistance in the pneumococcus. Science. 2009;324:1454–1457. doi: 10.1126/science.1171908. [DOI] [PubMed] [Google Scholar]
- Margos G, Gatewood AG, Aanensen DM, et al. (17 co-authors) MLST of housekeeping genes captures geographic population structure and suggests a European origin of Borrelia burgdorferi. Proc Natl Acad Sci U S A. 2008;105:8730–8735. doi: 10.1073/pnas.0800323105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura K, Dudley J, Nei M, Kumar S. MEGA4: Molecular Evolutionary Genetics Analysis (MEGA) software version 4.0. Mol Biol Evol. 2007;24:1596–1599. doi: 10.1093/molbev/msm092. [DOI] [PubMed] [Google Scholar]
- Tang J, Hanage WP, Fraser C, Corander J. Identifying currents in the gene pool for bacterial populations using an integrative approach. PLoS Comput Biol. 2009;5(8):e1000455. doi: 10.1371/journal.pcbi.1000455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wasser SK, Shedlock AM, Comstock K, Ostrander EA, Mutayoba B, Stephens M. Assigning African elephant DNA to geographic region of origin: applications to the ivory trade. Proc Natl Acad Sci U S A. 2004;101:14847–14852. doi: 10.1073/pnas.0403170101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willems RJL, Top J, van Schaik W, Leavis H, Bonten M, Sirén J, Hanage WP, Corander J. Restricted gene flow among hospital subpopulations of Enterococcus faecium. mBio. 2012;3:e00151-12. doi: 10.1128/mBio.00151-12. [DOI] [PMC free article] [PubMed] [Google Scholar]