

Abstract
Over a billion people are infected by Ascaris spp. intestinal parasites. To clarify functional differences among tissues of adult A. suum, we compared gene expression by various tissues of these worms by expression microarray methods.. The A. suum genome was sequenced and assembled to allow generation of microarray elements. Expression of over 40,000 60-mer elements was investigated in a variety of tissues from both male and female adult worms. Nearly 50 percent of the elements for which signal was detected exhibited differential expression among different tissues. The unique profile of transcripts identified for each tissue clarified functional distinctions among tissues, such as chitin binding in the ovary and peptidase activity in the intestines. Interestingly, hundreds of gender-specific elements were characterized in multiple non-reproductive tissues of female or male worms, with most prominence of gender differences in intestinal tissue. A. suum genes from the same family were frequently expressed differently among tissues. Transcript abundance for genes specific to A. suum, by comparison to Caenorhabditis elegans, varied to a greater extent among tissues than for genes conserved between A. suum and C. elegans. Analysis using C. elegans protein interaction data identified functional modules conserved between these two nematodes, resulting in identification of functional predictions of essential subnetworks of protein interactions and how these networks may vary among nematode tissues. A notable finding was very high module similarity between adult reproductive tissues and intestine. Our results provide the most comprehensive assessment of gene expression among tissues of a parasitic nematode to date.
Keywords: Nematode, parasite, Ascaris suum, tissue, expression
Introduction
Infections by parasitic nematodes produce substantial mortality and morbidity in humans and in animals that provide important nutrition for people, especially in tropical regions of Africa, Asia, and the Americas. The parasitic nematode species differ in their mode of parasitism, trophic ecology and clinical symptoms they cause, body morphology and size, among other features. This study takes advantage of the body size difference of the large roundworm A. suum and investigates functional differences among tissues of the adult parasite. The W.H.O. estimates that close to 2.9 billion people are infected by parasitic nematodes, of which Ascaris spp. are some of the most prevalent (Hotez 2007). Ascaris suum is a parasitic nematode with a cosmopolitan distribution that primarily infects pigs and secondarily humans (Crompton 2001), whereas the closely related species, A. lumbricoides, infects more than a billion people worldwide. Infections by Ascaris spp. cause damage to liver and lungs during somatic migrations, intestinal blockage /obstruction during the intestinal phase of infection, as well as malnutrition that might lead to physical and intellectual retardation (Bethony et al. 2006). It was also suggested that Ascaris spp. infections increase host susceptibility to other diseases, such as acquired immunodeficiency syndrome and tuberculosis (Fincham et al. 2003). Anthelmintic drugs are commonly used to treat infections by Ascaris spp. However, the high rate of re-infection by these parasites and the potential for helminthes to develop resistance against anthelmintics have stimulated heightened efforts to develop alternative treatment approaches, as has been widely discussed in platyhelminthes (Hagan et al. 2004; Capron et al. 2005). Deep understanding of the cellular and molecular functions in parasite tissues should greatly contribute to those efforts.
Nematologists have available the genome sequences of 10 nematode species and the sequencing of approximately 30 more species is in progress (Mitreva et al. 2007; Brindley et al. 2009). This burgeoning genomic sequence resource will facilitate functional delineation of genes and encoded proteins at an ever increasing rate. A critical aspect of this understanding involves the parasite stage and tissue in which gene products function. There is an increasing number of parasitic nematodes for which stage-specific expression data is becoming available (Neveu et al. 2003; Mitreva et al. 2004; Wang et al. 2010; Choi et al. 2011), including A. suum (Morimoto et al. 2003; Huang et al. 2008; Cantacessi et al. 2009). In contrast, much less information (e.g. (Ma et al. 2011) is available regarding gene expression across the diversity of tissues that comprise these pathogens. Reasons to gain insight on tissue patterns of gene expression are multifold. A basic reason is that knowledge of gene expression can resolve cellular functions specific to a given tissue that contribute to overall stasis of the parasite within the host. Tissue restricted functions/characteristics can provide effective approaches for parasite control as previously demonstrated by research on the nematode nervous system and intestine (Jasmer et al. 2000; Knox et al. 2003; Kimber et al. 2007; Shingles et al. 2007). Deep knowledge on genes expressed in nematode tissues will facilitate experimental dissection of mechanisms involved in mediating and regulating critical parasite functions in those tissues. The large size of A. suum and its close relationship to A. lumbricoides (Leles et al. 2012) at the biological and genetic levels (Dold and Holland 2011) makes A. suum an ideal model to investigate tissue biology of both parasites.
Previous research on tissue related gene expression in A. suum is limited and has relied on analysis of expressed sequence tags for transcripts and deep sequencing methods (Yin et al. 2008; Wang et al. 2010; Ma et al. 2011). The results of those types of investigations conveyed qualitative and quantitative data on genes expressed in intestine and germinal/reproductive tissues, as examples, from several parasitic nematodes (Yin et al. 2008; Li et al. 2011; Ma et al. 2011). In addition, comparisons of intestinal gene expression among nematode species identified genes that are predicted to be conserved across species that diverged about 350 million years ago (Yin et al. 2008). Hence, knowledge gained from these investigations may have application to many parasitic nematodes. Some limitations to these studies include the restricted number of genes queried (more so with EST analysis), restricted numbers of tissues interrogated, relatively little comparative data between genders, and the relative high cost and difficulty of sequencing methods used. This last consideration limits the routine use of these methods for experimental testing of hypotheses.
To explore alternative methods for transcriptome analysis, microarray elements were generated using publicly available sequences for A. suum and an in-house gene-set constructed through sequencing of the A. suum genome at a low coverage. A cDNA expression microarray approach was selected because it offers broad coverage at relatively low cost and has supported many investigations of gene expression involving experimental treatments of organisms, tissues and cells. Gene expression was analyzed among several tissues of adult male and female A. suum. The results delineated gene expression patterns that characterize and discriminate among each of the tissues investigated. Gender-specific distinctions were made for several tissues, and in many cases gene family members were parsed according to tissue-specific expression patterns. Functional predictions suggested findings of potential significance to parasite biology. Hence, multiple tissue-specific markers now exist that can be used in conjunction with the overall expression profile, to experimentally test hypotheses that can be generated from these observations. The results will also contribute to the functional annotation of the recently generated genome sequence of A. suum (Jex et al. 2011; Wang et al. 2011).
Materials and methods
Parasite material and RNA preparation
Adult worms were collected from infected pigs at an abattoir when being processed as part of the normal work of the abattoir. The fresh worm tissues including intestine, pharynx, head and reproductive tissues were dissected and snap frozen in liquid nitrogen for subsequent storage at −80 °C. Total RNA of each individual tissue was prepared using TRIzol (Invitrogen) according to the manufacturer’s instruction and treated with Ambion Turbo DNase (Ambion/Applied Biosystems, Austin, TX). RNA quality and quantity were assessed by Agilent 2100 Bioanalyzer (Agilent Technologies, Cedar Creek, Texas), and the yield determined using the NanoDrop ND-1000 UV-VIS spectrophotometer v.3.2.1 (NanoDrop Technologies, Wilmington, DE). Only RNA samples with an RNA integration number (RIN) greater than 7 passed our QC criteria. Fluorescence labeled cDNA was prepared from ~10 ug of total RNA using Superscript Plus Indirect labeling system (Invitrogen), with slight modifications. In brief, The anchored oligo(dT)20 primer is a mixture of 12 primers, each consisting of a string of 20 deoxythymidylic acid (dT) residues followed by two additional nucleotides represented by VN, where V is dA, dC, or dG, and N is dA, dC, dG, or dT. The VN “anchor” allows the primer to anneal preferentially at the 5′ end of the poly(A) tail of mRNA, ensuring high specificity, high yields, and longer lengths of cDNA. Two aminoallyl-modified nucleotides were used together with other dNTPs in reverse transcription, for incorporation into the cDNA. After RNA hydrolysis, neutralization, and column purification, the amino-modified cDNA is coupled with a monoreactive, N-hydroxysuccinimide (NHS)-ester fluorescent dye, either Alexa Fluor 555 succinimidyl ester or Alexa Fluor 647 succinimidyl ester. The Alexa dyes provide higher correlation coefficients, signal intensities, and single-to-background ratios. After the final column purification, the fluorescence labeled single-strand cDNA samples are quantified via spectrophotometer.
Genome sequencing, assembly and annotation
Genomic DNA from 32–64 cell Ascaris suum embryos was randomly sheared, end-repaired and size fractionated to enrich for 6–6.2 kb fragments. A total of 475,000 genome survey sequences (GSSs) were generated (373 Mb), 95% passed quality screening and were submitted to dbGSS division of GenBank. The 451,630 GSSs were assembled using PCAP (Huang et al. 2003). 48% of the sequences assembled into 54,184 contigs and subsequently into 33,222 supercontigs (average supercontig length was 2,801 bp; the largest supercontig was 60,001 bp). The total length of the contigs was 93.1 Mb (with a 3.5X coverage) and the 233,600 singletons contributed to 193 Mb of additional unique sequence. The repeat content and singletons in the assembly were evaluated by masking simple repeats and low-complexity repeats, and repeats were identified by generating a custom library of repeat sequences. The custom library was built using RECON (Bao and Eddy 2002) and default parameters. This library was screened for non-coding RNA and protein-coding genes using Pfam(Finn et al. 2006)and Non-Redundant GenBank (built 02/07/2007) databases, respectively, yielding 508 repeat families. RepeatMasker Open-3.0 (http://www.repeatmasker.org) was used to estimate the percentage of repeats. The total repeat content is estimated to be around 3.9 %, out of which 0.4% are simple repeats, 0.5% are low complexity repeats, and 3% are repeats identified by the custom library. The masked assembly was used to call genes. In total, 38,768 protein coding sequences were predicted using the assembly contigs through a 6-tier gene-calling pipeline(Ding et al. 2004)...
Microarray design and hybridization
Agilent 4×44K format chips were used for this genome-wide expression array, in which each slide had four arrays comprising ~ 44,000 probes. Probes of 60 mer were designed based on the data from our genome survey sequences (GSS) project in A. suum, which predicted 38,768 predicted protein coding sequences. An additional 5,176 EST contigs that were not covered by this predicted GSS gene-set were included for probe design to broaden the gene coverage. Therefore, a total of 43,944 putative gene sequences from A. suum were uploaded to eArray of Agilent for the probe selection and array design. The selected probes were searched against the currently available coding sequences for nematodes to ensure that they would not cross-react with non-coding sequences. The final array contains 42,212 customized elements, each derived from a single sequence. We also included 1,591 spike control spots of ten probes, each targeting a different Arabidopsis thaliana gene that did not have homology to known non-plant sequences currently in public databases (SpotReport@-10 Array Validation System, Stratagene).
A draft genome of A. suum was published with predicted gene models at the time of preparing our manuscript (Jex et al. 2011). We therefore mapped gene sequences used in our investigation to the new genome based gene models to maximize utility of our data by other users of the genome database. The table (Ascaris suum Gene Name Mappings) providing this information has been submitted as supplementary file to GEO under accession number GSE36690.
Microarray experiments were carried out using a two-color design with the pooled cDNA sample, which was made by combining an equal amount of cDNAs from each of the 10 tissues, as a common reference (Arbeitman et al. 2002). The pooled reference was used in one of the two channels of each array. Three biological replicates were independently generated for each tissue sample to assess biological variations. Dye effects originated from the differences between the two fluorescent dyes were corrected by dye-swap normalization.
Each cDNA sample was paired and balanced with the common reference, and applied to a single plex of the custom array. Hybridization was carried out at 65 °C for 20 hours. Washing procedures were carried out according to the Agilent protocols. Slides were scanned with an Axon 4000B scanner for the detection of fluorescent signals. Laser power was kept constant and PMT was varied for each slide to ensure optimal signal intensities while maintaining saturation below 0.1%. Images were processed using GenePix v6.1 (Axon) to generate gpr files containing raw intensities, gene information, etc. The data discussed in this publication have been deposited in the National Center for Biotechnical Information (NCBI) Gene Expression Omnibus (GEO) and are accessible through GEO Series accession number GPL15336 (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GPL15336) and GSE36690 (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE36690).
Microarray data processing and gene expression characterization
Scanned GPR files were first inspected for quality control (QC) using built-in QC tools from the Bioconductor package of R (Gentleman et al. 2004). Inspected microarrays were then analyzed using LIMMA of Bioconductor (Smyth 2004). After background correction and normalization, gene expression was detected through a linear model fitting which considers both the dye effect and the variation among replicates. Differentially expressed genes were defined based on the F test of the linear model fitting. To control the problem of multiple tests, we used false discovery rate (FDR) of 0.01 as a cutoff. The resulting differentially expressed gene set with corrected F-test and FDR was exported for further analysis.
Gene over- and under-expression was determined using the “2 fold” rule based on the linear modeling fitting results. For each gene, if the fitted effect coefficient for one tissue was more than 1 (log-ratio compared with the signal of pooled references), it was recorded as relatively over-expressed in this tissue; if the coefficient was less than −1, it was recorded as relatively under-expressed in that tissue. By this way, each tissue was assessed for its complement of over or under- expressed genes. The GO category analysis was performed using the full sequences from which array elements were derived. Gene lists that were exclusively over-expressed in a specific tissue were first generated and then examined for enriched ontology terms using FUNC (Prufer et al. 2007).
Gene conservation and conserved functional modules
The protein sequences predicted from A. suum gene sequences were searched against the C. elegans protein database using WU-BLAST and those having C. elegans homologs were classified as conserved nematode genes (CNG). Protein homologs were identified with cutoff of bit score > 50. The genes encoding proteins were further searched against the InterPro database and those lacking homology to any of the InterPro domain families were classified as A. suum-specific genes. The numbers of differentially expressed genes from these two groups were compared. Protein family membership of A. suum genes was assigned using PANTHER HMM (Thomas et al. 2003) through interproscan (Zdobnov and Apweiler 2001). Sequences hitting the same family were considered homologs (paralogs). The expression distances of genes from the same family and among families were compared. The expression distance between a gene pair was defined as the Euclidean distances of the vectors of the corresponding linear fitting coefficients and represented the difference of expression patterns for the corresponding gene pair. Similarly, Euclidean distance calculation was applied for comparing of expression profile of elements putatively derived from the same gene to elements derived from randomly selected genes.
Proteins usually interact with other proteins to form modules so that the cellular functions can be carried out in a concerted manner (Spirin and Mirny 2003). In the model nematode C. elegans, thousands of documented protein-protein interactions(Walhout et al. 2000) provide a convenient framework for analyzing gene expression data. To detect A. suum nematode functional modules that are potentially conserved with C. elegans, we first built a protein-protein interaction (PPI) network based on the available C. elegans PPI data from both BIND (Bader et al. 2003) and IntAct (Aranda et al. 2010) databases using Cytoscape (Shannon et al. 2003). Then we incorporated the A. suum tissue gene expression profile generated from our microarray study into the PPI network. An active functional module for a condition of interest is defined as a PPI subnetwork whose genes show significant coordinated changes based on the z scoring system specified in the Cytoscape jActiveModules plugin (Ideker et al. 2002). The higher the z score, the higher the significance placed on a functional module under a specific condition. The search strategy and default parameters were employed for the module analysis and the resultant subnetwork z-score cutoff was 3.0. The procedure of finding modules was repeated three times to improve the final output. All the module networks were constructed and visualized using Cytoscape. For the example active modules in all tissues, in males and females the.sif files, that can be directly uploaded and viewed in Cytoscape, has been made available through A. suum species page on Nematode.net (Martin et al. 2012) (nematode.net/Ascaris_suum.html).
PCR validation
Quantitative real-time PCR (qRT-PCR) was performed on the same set of cDNA samples in microarray analyses to independently validate tissue-specific gene expression of 36 most differentially expressed genes identified by the microarray. The remaining A. suum tissue cDNA templates of microarray assay stored in −80 C were used for qRT-PCR confirmation. The quality of qRT-PCR reactions was verified by amplification efficiency and melting curves analysis as described previously (Li et al. 2004a). Briefly, the primers (Table S1) were derived from the consensus sequences of genes selected using Primer Express software (Version1.0, PE Applied Biosystems, Foster City, CA) and synthesized from Integrated DNA Technology Inc (Coralville, IA). The qRT-PCR reactions were carried out in 96 well plates in a 25 ul reaction volume containing SYBR Green Master Mix (PE Biosystems), sequence-specific primers and equal amount of cDNAs from various tissues. We next compared results obtained with cDNA prepared from different tissue with those obtained with a reference cDNA sample made from pooled mRNA representing all of the tissue studied (the same as in the microarray assay) to measure the relative abundance of transcripts in each tissue. In contrast to using a housekeeping gene as an internal control, we chose to use a standardized RNA mix of all templates to serve as a control for quantitative assessment of expression differences among tissues. This choice of control stems from the concern that expression of individual “housekeeping” genes may vary markedly among tissues that differ vastly in function, structure and likely metabolism, as is expected for tissues analyzed here. Evidence that addresses this concern is essentially non-existent for A. suum. Therefore, the pooled control used here provides an alternative to compare relative expression levels of individual gene transcripts across multiple tissues, and it has been used for similar purposes elsewhere (e.g. (Arbeitman et al. 2002; Li et al. 2012)).
Each assay included duplicates of the tested sample, the reference cDNA sample made from pooled RNA representing all tissues assayed, and the negative controls. The expression level for a gene of interest from a specific tissue was represented by a ΔCt value which is the difference between the mean Ct value obtained from the specified tissue with the mean Ct value obtained for the reference cDNA sample. Differential expression between tissues was determined by comparing those ΔCts among all the tested tissues. The relative abundance of transcripts in each tissue was used to assess transcriptional profiles for each gene.
Results
This study used a two-color microarray to profile gene expression in different tissues of adult A. suum. Each 60-mer element was tested in 10 different tissues: head, pharynx, intestine, ovary, and uterus from females, and head, pharynx, intestine, testes, and seminal vesicle from males. Elements were called “detected” if microarray signals were present in at least two out of three replicate hybridizations after background correction. A primary goal was to identify genes and the predicted proteins responsible for functional differences among these tissues. The results also delineated genes that are commonly expressed among all the tissues, and thus may functionally contribute to all the tissues.
Resolution of genes commonly or differentially expressed among A. suum tissues
Transcript signals were detected from a total of 32,919 elements, which is approximately 78% of the 42,212 microarray elements (Table S2). Significant differences were observed in expression among tissues for 16,262 elements, accounting for nearly 50% of the total detected signals. As expected, distinct gene expression profiles were observed for each of the tissues investigated (Fig. 1). Differences in gene expression were recorded in multiple dimensions; e.g. relative number of over-expressed and under-expressed elements, elements specifically over-and under expressed in a given tissue and elements over- and under-expressed in a gender-specific manner.
Fig. 1.

Expression patterns of top 5000 differentially expressed genes across 10 different A. suum tissues. Hierarchical clustering was used to display the expression ratios of the tissue-associated genes. Rows represent genes and columns represent different tissues. Levels of expression are shown by color and density according to the key. FemPha, female pharynx; MalPha, male pharynx; FemHea, female head; MalHea, male head; FemInt, female intestine; MalInt, male intestine; SemVes, seminal vesicle; Tes, testis; Ova, ovary; Ute, uterus. Log2 fold changes relative to the pooled reference are depicted by the color key scale provided in the top left corner of the heatmap
PCR validation
qRT-PCR was successfully carried out for 36 differentially expressed genes that showed the highest level of quantitative differences in microarray assays. Correlation coefficients of expression patterns across different tissues between these two platforms for each individual gene were examined and there were 25 out of 36 of tested genes that displayed positive relatedness (Table S3). We did notice that the correlation was generally low, which was not completely unexpected if considering the 10 dimensional data we generated and the limited number of genes selected. The results also show that 26 transcripts (more than 70% of the tested genes) demonstrating higher abundance in a given tissue by microarray analysis, similarly showed higher abundance by qRT-PCR in the same tissue or functionally related tissue.
The level of agreement between qRT-PCR and microarray results is consistent with numerous other published reports (e.g. (Rajeevan et al. 2001; Ghedin et al. 2009)) and therefore was considered to meet current standards. However, as suggested in those studies qRT-PCR is more sensitive assay to detect changes in transcript levels than microarray hybridization, therefore might introduce some uncertainty regarding interpretation for a given gene in a given tissue. This impact is highest with regard to results displayed in Fig. 1. Nevertheless, results from comparisons that incorporate all ten tissues, where nine different tissues provide a similar result by comparison to one tissue, the reliability is elevated substantially. Most of our results are based on this kind of comparison.
Quantitative and qualitative differences in gene expression among tissues
For each tissue analyzed, a substantial number of over- and under expressed elements were detected, inclusive of tissue specific and non-tissue specific patterns of expression. The number of microarray elements observed to be relatively under-expressed always outnumbered the elements that were relatively over-expressed (Table 1). This disproportionate occurrence of under-expressed elements is likely explained, in part, by the tendency of many over-expressed elements to be specific to individual tissues. For instance, 10,209 over-expressed elements were found to be tissue-specific (63%), whereas less than 5% of under-expressed elements (1,526) were specific to an individual tissue (Table 2). Therefore, the under-expressed elements for any given tissue would include tissue-specific, over-expressed elements summed from all other tissues included in the comparison, leading to a higher representation of under-expressed elements in each tissue.
Table 1.
Differentially expressed genes per tissuea
| Tissue | Over-expressed genes | Under-expressed genes |
|---|---|---|
| Female Intestine | 1196 | 4738 |
| Male Intestine | 1500 | 4609 |
| Female Head | 1487 | 2738 |
| Male Head | 1672 | 2692 |
| Female Pharynx | 972 | 3222 |
| Male Pharynx | 903 | 2880 |
| Testis | 2364 | 3843 |
| Uterus | 3369 | 3602 |
| Ovary | 2283 | 4603 |
| Seminal Vesicle | 1422 | 4484 |
total of 16262 microarray expressed elements
Table 2.
Tissue exclusively over-expressed and under-expressed genes
| Tissue | Over-expressed | Under-expressed | C. elegans homologous of over-expressed genes |
|---|---|---|---|
| Female Intestine | 290 | 198 | 116 |
| Male Intestine | 629 | 124 | 190 |
| Intestine (Male + Female) | 506 | 322 | 151 |
| Female Head | 105 | 6 | 39 |
| Male Head | 133 | 6 | 47 |
| Head (Male + Female) | 716 | 0 | 278 |
| Female Pharynx | 98 | 25 | 32 |
| Male Pharynx | 15 | 7 | 5 |
| Pharynx (Male + Female) | 142 | 6 | 34 |
| Testis | 2008 | 240 | 548 |
| Uterus | 2888 | 186 | 605 |
| Ovary | 1831 | 335 | 574 |
| Seminal Vesicle | 848 | 71 | 248 |
Each tissue displayed characteristics that were specific to that tissue (Table 2), indicating that when linked to functional predictions this analysis will define many functional markers that can discriminate among these tissues, as will be analyzed below. The number of tissue specific markers also varied markedly among tissues (Table 2). Whereas a substantially higher number of over-expressed elements were observed in reproductive tissues compared to all others, numbers of over-expressed elements specific to intestine (next highest), head and pharynx (lowest) indicate numerous markers specific to those tissues. Homologous proteins were identified in the public database for many of the tissue specific A. suum elements, which may aid elucidation of functions in these tissues (Table S4). The GO associations compiled for the sequences from which all the elements on the chip were derived are shown (Table S5). GO associations that were enriched relative to molecular function and biological process for genes over-expressed by each tissue are listed in Tables 3A and 3B. Major differences in gene expression were also observed for each of the A. suum tissues in a gender specific manner. Although more numerous gender specific differences might be expected in reproductive tissues, as is the case here (Table 2), substantial differences were observed between genders for each of the other tissues, with the greatest disparity between male and female intestine, followed by head and pharynx. In addition, the uterus had significantly more over-expressed elements (28%) than the positionally similar tissue in male, the seminal vesicle (8%) (p<0.001 by χ2 test). It should be noted that no distinction was made between uterus or seminal vesicle and eggs or sperm, respectively, contained within them. The relative role that somatic tissues play in accomplishing gender specific functions in nematodes has not been investigated. Here we calculated for each of the somatic tissues the percentage of elements categorized as specific to a tissue and expressed by both male and female versus the total expressed by both genders and individual genders. In this case, 36% of elements specific to the intestine were found to be expressed by both males and females (506/290+629+506)(Table 2), whereas this representation was 75% and 56% for head and pharynx, respectively, with the lower percentage indicating higher representation of gender biased, tissue-specific functions. Hence, this variation in conjunction with the total number of elements measured would seem to reflect the degree to which a given somatic tissue contributes to gender-specific functions; the intestine demonstrating a clear prominence over the other tissues evaluated.
Table 3a.
GO molecular function enrichment of genes exclusively over-expressed in individual tissues
| Tissue | GO Identifier | GO descriptor | P-value | FDRa |
|---|---|---|---|---|
| Female intestine | ||||
| GO:0004478 | methionine adenosyltransferase activity | 1.97E-05 | 2.16E-03 | |
| GO:0004615 | phosphomannomutase activity | 2.58E-04 | 1.18E-02 | |
| GO:0005319 | lipid transporter activity | 2.81E-08 | 0 | |
| Male intestine | ||||
| GO:0004190 | aspartic-type endopeptidase activity | 7.28E-04 | 1.27E-02 | |
| GO:0004222 | metalloendopeptidase activity | 5.74E-07 | 0 | |
| GO:0004450 | isocitrate dehydrogenase (NADP+) a ct ivity | 5.68E-06 | 2.73E-04 | |
| Female and male intestine | ||||
| GO:0000062 | acyl-CoA binding | 7.07E-21 | 0 | |
| GO:0008234 | cysteine-type peptidase activity | 1.81E-05 | 3.27E-04 | |
| GO:0008237 | metallopeptidase activity | 3.40E-10 | 1.54E-04 | |
| Female head | ||||
| GO:0004499 | flavin-containing monooxygenase activity | 2.54E-07 | 0 | |
| GO:0004791 | thioredoxin-disulfide reductase activity | 8.25E-04 | 1.25E-02 | |
| GO:0050660 | FAD binding | 9.71E-08 | 0 | |
| GO:0050661 | NADP or NADPH binding | 9.79E-10 | 0 | |
| Male head | ||||
| GO:0004332 | fructose-bisphosphate aldolase activity | 7.91E-08 | 0 | |
| GO:0004365 | glyceraldehyde-3-phosphate dehydrogenase (phosphorylating) activity | 4.81E-04 | 3.66E-03 | |
| GO:0004601 | peroxidase activity | 2.14E-06 | 0 | |
| GO:0004645 | phosphorylase activity | 4.81E-04 | 3.66E-03 | |
| GO:0008890 | glycine C-acetyltransferase activity | 4.07E-05 | 4.24E-04 | |
| GO:0020037 | heme binding | 2.17E-04 | 2.46E-03 | |
| Female and male head | ||||
| GO:0003774 | motor activity | 1.20E-15 | 0 | |
| GO:0003995 | acyl-CoA dehydrogenase activity | 5.40E-06 | 2.58E-04 | |
| GO:0004148 | dihydrolipoyl dehydrogenase activity | 1.08E-06 | 1.00E-04 | |
| GO:0004645 | phosphorylase activity | 4.33E-05 | 6.88E-04 | |
| GO:0004807 | triose-phosphate isomerase activity | 6.21E-04 | 8.06E-03 | |
| GO:0005201 | extracellular matrix structural constituent | 1.54E-05 | 3.21E-04 | |
| GO:0005515 | protein binding | 4.87E-11 | 0 | |
| GO:0009055 | electron carrier activity | 3.33E-14 | 0 | |
| GO:0016153 | urocanate hydratase activity | 5.00E-04 | 4.88E-03 | |
| GO:0020037 | heme binding | 1.79E-07 | 0 | |
| GO:0042302 | structural constituent of cuticle | 2.47E-05 | 4.51E-04 | |
| GO:0050660 | FAD binding | 8.26E-05 | 1.25E-03 | |
| Testis | ||||
| GO:0004672 | protein kinase activity | 7.05E-05 | 0 | |
| GO:0004674 | protein serine/threonine kinase activity | 9.63E-04 | 6.20E-03 | |
| GO:0004713 | protein tyrosine kinase activity | 2.21E-11 | 0 | |
| GO:0004725 | protein tyrosine phosphatase activity | 1.60E-10 | 0 | |
| GO:0005524 | ATP binding | 3.35E-04 | 4.30E-03 | |
| GO:0008474 | palmitoyl-(protein) hydrolase activity | 1.95E-04 | 3.43E-03 | |
| GO:0008483 | transaminase activity | 9.02E-06 | 0 | |
| GO:0016787 | hydrolase activity | 1.03E-13 | 6.67E-05 | |
| GO:0030170 | pyridoxal phosphate binding | 8.19E-04 | 8.19E-03 | |
| GO:0031625 | ubiquitin protein ligase binding | 5.86E-05 | 1.68E-03 | |
| Uterus | ||||
| GO:0008060 | ARF GTPase activator activity | 1.20E-04 | 1.01E-02 | |
| Seminal vesicle | ||||
| GO:0003735 | structural constituent of ribosome | 9.27E-10 | 0 | |
| GO:0003827 | alpha-1,3-mannosylglycoprotein 2-beta-N-acetylglucosaminyl transferase activity | 7.45E-04 | 1.02E-02 | |
| GO:0003978 | UDP-glucose 4-epimerase activity | 4.74E-04 | 6.18E-03 | |
| GO:0005198 | structural molecule activity | 5.58E-04 | 0 | |
| GO:0015662 | ATPase activity, coupled to transmembrane movement of ions, phosphorylative mechanism | 3.25E-08 | 0 | |
| GO:0016149 | translation release factor activity, codon specific | 5.09E-06 | 1.43E-04 | |
| Ovary | ||||
| GO:0003937 | IMP cyclohydrolase activity | 5.10E-04 | 1.05E-02 | |
| GO:0004618 | phosphoglycerate kinase activity | 1.53E-04 | 4.72E-03 | |
| GO:0004643 | phosphoribosylaminoimidazolecarboxamide formyltransferase activity | 5.10E-04 | 1.05E-02 | |
| GO:0008061 | chitin binding | 4.24E-16 | 0 | |
| GO:0008430 | selenium binding | 1.53E-04 | 4.72E-03 | |
| GO:0015269 | calcium-activated potassium channel activity | 9.72E-04 | 1.60E-02 | |
FDR, false discovery rate
Table 3b.
GO biological porcess enrichment of genes exclusively over-expressed in individual tissues
| Tissue | GO Identifier | GO descriptor | P-value | FDRa |
|---|---|---|---|---|
| Female intestine | ||||
| GO:0006730 | one-carbon metabolic process | 7.34E-05 | 4.99E-03 | |
| GO:0006869 | lipid transport | 2.59E-08 | 0 | |
| GO:0007160 | cell-matrix adhesion | 3.90E-04 | 1.08E-02 | |
| GO:0007229 | integrin-mediated signaling pathway | 5.02E-04 | 1.65E-02 | |
| GO:0019307 | mannose biosynthetic process | 2.52E-04 | 9.84E-03 | |
| Male intestine | ||||
| GO:0006102 | isocitrate metabolic process | 7.22E-06 | 3.75E-04 | |
| GO:0006508 | proteolysis | 1.07E-09 | 0 | |
| GO:0015986 | ATP synthesis coupled proton transport | 5.40E-04 | 1.77E-02 | |
| Female and male intestine | ||||
| GO:0006508 | proteolysis | 4.30E-12 | 0 | |
| Female head | ||||
| None | ||||
| Ovary | ||||
| GO:0000103 | sulfate assimilation | 4.93E-04 | 1.02E-02 | |
| GO:0006030 | chitin metabolic process | 1.09E-12 | 0 | |
| GO:0006096 | glycolysis | 4.12E-04 | 7.26E-03 | |
| GO:0007156 | homophilic cell adhesion | 1.04E-07 | 0.00E+00 | |
| Female and male head | ||||
| GO:0006548 | histidine catabolic process | 7.23E-06 | 0 | |
| GO:0008152 | metabolic process | 3.36E-09 | 6.88E-03 | |
| GO:0009060 | aerobic respiration | 8.98E-12 | 0 | |
| GO:0022904 | respiratory electron transport chain | 2.61E-08 | 0 | |
| Testis | ||||
| GO:0006094 | gluconeogenesis | 1.21E-04 | 1.61E-03 | |
| GO:0006096 | glycolysis | 8.43E-04 | 6.59E-03 | |
| GO:0006099 | tricarboxylic acid cycle | 1.74E-05 | 5.46E-04 | |
| GO:0006468 | protein amino acid phosphorylation | 5.39E-17 | 0 | |
| GO:0006470 | protein amino acid dephosphorylation | 1.05E-08 | 0 | |
| GO:0006511 | ubiquitin-dependent protein catabolic process | 4.95E-04 | 4.39E-03 | |
| GO:0006750 | glutathione biosynthetic process | 9.12E-04 | 7.13E-03 | |
| GO:0007018 | microtubule-based movement | 5.75E-04 | 4.89E-03 | |
| GO:0008202 | steroid metabolic process | 5.50E-05 | 1.03E-03 | |
| Uterus | ||||
| GO:0032312 | regulation of ARF GTPase activity | 1.54E-04 | 1.72E-02 | |
| Seminal Vesicle | ||||
| GO:0006412 | translation | 1.51E-09 | 0 | |
| GO:0006415 | translational termination | 1.22E-04 | 1.01E-03 | |
| GO:0006754 | ATP biosynthetic process | 4.44E-08 | 0 | |
| GO:0006096 | glycolysis | 6.81E-06 | 0 | |
| GO:0006979 | response to oxidative stress | 3.57E-06 | 0 | |
FDR, false discovery rate
The combinations of quantitative and qualitative expression differences among tissues discussed present a very complex picture of expression profiles as depicted on the vertical axis of Fig. 1. The complexity may be overestimated in this graphic, for reasons mentioned with qRT-PCR results, but complexity is still expected to be high. While it is anticipated that sequence elements clustered on the dendrogram will represent coordinately regulated genes, further resolution of relationships among genes with similar expression profiles will best be accomplished with sequence information that is less fragmented than that used here.
Genes with restricted phylogenetic conservation display greater restrictions on tissue expression
About 40% of the A. suum sequences from which the elements were derived (15,941 sequences) encode predicted protein sequences with homology to PANTHER (PTHR) protein families. We assume that sequences encoding predicted proteins with homology to the PTHR families represent genes that are conserved more broadly across phyla, while the others that are more restricted may represent A. suum-specific genes. Among the elements representing putative conserved genes, 3,508 sequence elements exhibited differential expression among tissues; whereas among the provisionally A. suum-specific protein-coding sequences, 12,754 were differentially expressed. Thus, only 22% of the elements representing conserved genes exhibited differential expression whereas nearly 50% of the elements representing A. suum specific genes were differentially expressed among tissues (p<0.001 by z test). These results indicate a tendency for genes of restricted phylogenetic significance to represent adaptations of relevance to restricted cell or tissue populations.
Differential tissue expression of genes encoding individual members from multimember protein families
Genes were organized according to single or multimember families of the corresponding predicted proteins. Expression analysis showed that more than 20% of the multimember families contained at least one pair of gene members for which transcript expression was significantly different among tissues, as evidenced by an expression profile distance large than 7 (the top 5% cutoff derived from all the gene expression profiles). For example, the neurotransmitter gated ion channel family (PTHR18945) has 233 protein members. Even though this family had low mean pairwise expression distance (2.26) (Table 4, Table S6), the maximum pairwise expression distance within this family ranked third among all the calculated pairwise expression distances (23.86). Table 4 lists the 20 families that have pairs of members with the largest expression distances. The results also indicate that the magnitude of variation in expression does not always positively relate to the number of sequences within a family. For example the prion-like-(Q/N-rich)-domain bearing protein (PTHR21593, 9 sequences), which had the highest variation of expression among members with an average pairwise expression distance of 13.89, while the neurotransmitter gated ion channels super-family mentioned above have very similar expression profiles. Hence, this expression analysis resolved likely differences in tissue expression, and thus functions, of gene members within multigene families (paralogues) based on tissue expression patterns.
Table 4.
Preotein families with members for which transcript levels differ among tissues
| Family ID and descriptor | Expression
|
Sequences within family group | Tissue Distributionc | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Meana | Maxb | FemHea | FemInt | FemPha | MalHea | MalInt | MalPha | Ova | SemVes | Tes | Ute | ||
| PTHR11588 TUBULIN | 4.94 | 24.84 | 67 | 1 | 1 | 1 | 0 | 1 | 2 | 7 | 7 | 11 | 7 |
| PTHR10499 COLLAGEN ALPHA CHAIN | 5.79 | 24.14 | 252 | 63 | 3 | 7 | 56 | 8 | 6 | 12 | 6 | 5 | 33 |
| PTHR18945 NEUROTRANSMITTER GATED ION CHANNEL | 2.26 | 23.86 | 233 | 7 | 0 | 7 | 7 | 1 | 4 | 13 | 7 | 3 | 20 |
| PTHR11920 ADENYLATE AND GUANYLATE CYCLASES | 3.85 | 23.82 | 65 | 0 | 4 | 0 | 2 | 5 | 1 | 8 | 4 | 1 | 11 |
| PTHR19876 COATOMER | 3.57 | 23.44 | 19 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 6 | 0 | 1 |
| PTHR19242 ATP-BINDING CASSETTE TRANSPORTER | 3.31 | 23.43 | 165 | 0 | 34 | 3 | 0 | 44 | 10 | 16 | 4 | 4 | 10 |
| PTHR11865 NUCLEAR HORMONE RECEPTOR | 2.77 | 23.28 | 69 | 0 | 13 | 0 | 0 | 11 | 0 | 5 | 0 | 1 | 5 |
| PTHR22762 ALPHA-GLUCOSIDASE | 5.62 | 23.16 | 77 | 0 | 6 | 8 | 1 | 20 | 11 | 2 | 6 | 6 | 5 |
| PTHR11403 CYTOCHROME C OXIDASE SUBUNIT III | 4.14 | 23.06 | 21 | 17 | 1 | 0 | 17 | 0 | 16 | 0 | 0 | 0 | 1 |
| PTHR21593 PRION-LIKE-(Q/N-RICH)-DOMAIN-BEARING PROTEIN PROTEIN | 13.89 | 23.06 | 9 | 3 | 1 | 4 | 5 | 0 | 5 | 0 | 1 | 0 | 2 |
| PTHR22955 RETROTRANSPOSON | 3.61 | 22.96 | 78 | 0 | 0 | 4 | 2 | 1 | 2 | 1 | 0 | 3 | 10 |
| PTHR11347 CYCLIC NUCLEOTIDE PHOSPHODIESTERASE | 4.71 | 22.84 | 21 | 0 | 1 | 1 | 0 | 1 | 1 | 1 | 0 | 0 | 3 |
| PTHR11686 GAMMA GLUTAMYL TRANSPEPTIDASES | 8.37 | 22.62 | 6 | 1 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 1 |
| PTHR11712 POLYKETIDE SYNTHASE-RELATED | 4.59 | 22.30 | 21 | 5 | 0 | 0 | 0 | 0 | 0 | 11 | 0 | 0 | 1 |
| PTHR22986 MAPKK-RELATED SERINE/THREONINE PROTEIN KINASES | 2.33 | 22.27 | 68 | 0 | 2 | 2 | 0 | 1 | 1 | 0 | 4 | 6 | 4 |
| PTHR10796 PATCHED-RELATED | 2.68 | 22.19 | 99 | 10 | 0 | 5 | 1 | 2 | 2 | 20 | 2 | 4 | 8 |
| PTHR23151 DIHYDROLIPOAMIDE ACETYL/SUCCINYL-TRANSFERASE-RELATED | 4.14 | 22.07 | 15 | 1 | 0 | 0 | 1 | 0 | 0 | 0 | 1 | 3 | 1 |
| PTHR10432 RNA-BINDING PROTEIN | 3.47 | 22.02 | 48 | 3 | 0 | 0 | 7 | 0 | 0 | 2 | 2 | 3 | 4 |
| PTHR13712 RING FINGER-CONTAINING PROTEIN-RELATED | 7.02 | 21.78 | 24 | 1 | 0 | 0 | 1 | 0 | 1 | 2 | 0 | 3 | 5 |
| PTHR23310 ACYL-COA-BINDING PROTEIN, ACBP | 6.80 | 21.74 | 27 | 0 | 16 | 0 | 0 | 15 | 0 | 0 | 0 | 0 | 2 |
Mean=Average pairwise expression profile distance
Max=Maximum pairwise expression profile distance
element was considered highly expressed when the log ratio was greater than 1
Comparison of predicted functional modules across A. suum tissues
Organization of protein expression data for A. suum tissues into predicted functional modules provides another method to assess how individual proteins might function in higher order interactions within the tissues investigated. Of the 16,262 differentially expressed A. suum elements, 4,673 encode protein sequences that had homology to 2,695 C. elegans proteins. These proteins when integrated with the C. elegans protein-protein interaction (PPI) network and A. suum tissue expression data identified one hundred and sixty two active modules, that may exist among the A. suum tissues investigated. Thirty-three active modules were shared by all the 10 tissues (Fig. 2, Table 5). The number of the modules shared by different tissues differed markedly, with the highest number of modules shared among reproductive and intestinal tissues. Visual inspection indicated that 24 of those 33 common active modules were derived from one subnetwork centered at protein GEI-4 (Cel.17149) (Fig. 3). In C. elegans, the GEI-4 protein extensively interacts with structural proteins, adaptor proteins and hormone receptors, and has been implicated in embryogenesis, fertility, and vulval morphogenesis by regulating intermediate filament dynamics (Tsuboi et al. 2002; Kamath et al. 2003). GEI-4 and over 10 of its interacting partners are predicted to be conserved in A. suum based on our results. Our microarray data showed that transcripts for the gei-4 gene homolog in A. suum are highly enriched in the uterus. Concurrently, transcripts for many proteins that interact with GEI-4 displayed significant expression differences among tissues (Table 6). GO term analysis illustrated that those differentially expressed module members are involved in diverse molecular functions related to protein binding.
Fig. 2.

Top scored active modules identified from 10 A. suum adult tissues identified by jActiveModule analysis with a z score greater than 3. Rows represent the identified active modules and columns represent the individual tissues (Hea, head; Int, intestine; Pha, pharynx; Ova, ovary; SemVes, seminal vesicle; Ute, uterus; Tes, testis). Each black filled area indicates the existence of an active module in a specific tissue. Modules are sorted according to the descending z scores
Table 5.
Number of functional modules detected in A. suum tissues
| Tissues for interaction network analysis | Active modules detected | Conserved active modules
|
||
|---|---|---|---|---|
| All tissues | Non-reproductive | Reproductive | ||
| All 10 tissues in 2 genders | 162 | 33 | 162 | 162 |
| Male tissues only | 186 | 30 | 186 | 178 |
| Female tissues only | 124 | 28 | 111 | 124 |
Fig. 3.
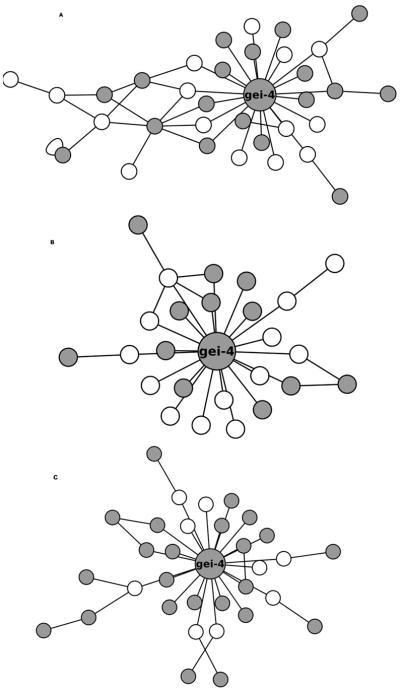
Examples of the active modules common to the all adult A. suum tissues that are centered at gene gei-4. Node indicates the protein members in the active module. Edge indicates the interaction between protein members. Conserved protein members between A. suum and C. elegans are highlighted by grey color. Interaction files for modules A, B and C are available at www.nematode.net (nematode.net/Ascaris_suum.html)
Table 6.
Genes that directly interact with gei-4 and display differential expression a across adult A. suum tissues
| Unigene ID | FemI | Mall | FemH | Mall | FemP | MalP | Ute | Tes | Ova | SemVes | Gene and Description |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Cel.18366 | nhr-31, Nuclear Hormone Receptor | ||||||||||
| Cel.15019 | bath-38, BTB and MATH domain containing | ||||||||||
| Cel.19324 | unc-54, UNCoordinated | ||||||||||
| Cel.16866 | hypothetical proteinb | ||||||||||
| Cel.18173 | rsp-6 | ||||||||||
| Cel.13744 | vab-23 | ||||||||||
| Cel.5996 | acdh-1, Acyl CoA DeHydrogenase | ||||||||||
| Cel.16930 | hypothetical protein, variant functions | ||||||||||
| Cel.16886 | lfi-1, Lin-5 Interacting protein |
green filled indicates over-expression and red filled indicated depleted-expression;
likely to be involved in eggshell synthesis and early embryonic development by interacting with other proteins.
Gender related expression patterns: Further module analyses were conducted on microarray data from female tissues only or male tissues only (Fig. S1). One hundred and eighty-two active modules were identified from male tissues and one hundred and twenty-four from female tissues. These results showed similarities between intestine and reproductive tissues, consistent with data when expression from both genders combined was evaluated. In addition, the male head had more functional modules in common with the intestine and reproductive tissues than female head. There were thirty active modules common to all male tissues and twenty-eight to all female tissues. The active modules in male tissues displayed similar convergence to the subnetwork centered at GEI-4 as those identified when the gene expression profiles from all the 10 tissues were included, but the modules in this case generally had fewer protein members and the specific members were slightly different (Fig. S2, Table S7). However, the active modules in female tissues revealed a different organization characterized by multi-centered networks and diverse hub genes (Fig. S3). In contrast to the predominance of GEI-4 centered modules in male worms, PQN-54 (Cel.2829) contributes to 18 of the 28 modules conserved among tissues and active in female (Table 5). pqn-54 encodes a Prion-like-(Q/N-rich)-domain-bearing protein and is conserved in A. suum (Scheibel and Lindquist 2001; Serio and Lindquist 2001).
Discussion
Ascaris spp. still count for a major burden of disease in humans worldwide. With the long-term goal of improved methods of parasite control, we sought to identify and compare the repertoire of genes expressed by several tissues of adult A. suum. This information can generate a systematic view of predicted gene functions at a tissue level and potential intermolecular interactions of encoded proteins. Insights were gained on putative biological and physiological functions performed by tissues in this organism and possibly other nematodes that when interrupted could be detrimental to the parasite. Our results resolved gene sequences that can be categorized as i) relatively or exclusively (tissue specific) over-expressed in each of the tissues compared, providing explicit definitions for each tissue based on gene transcript information, ii) relatively or exclusively (tissue specific) under-expressed by individual tissues, or iii) expressed by all tissues investigated. When considering all combinations of expression levels and tissues, a myriad of other categories of expression were also discerned for individual genes. Although intricate regulatory mechanism may underlie this complexity, the accuracy of lesser fluctuations in expression across tissues will require further investigation. The analysis resolved genes that are expressed in a gender-related context in each of the tissues investigated and also gene family members that displayed differential expression among tissues. Predicted protein sequences encoded by the nucleotide sequences investigated were in many cases linked to predicted functions, which will be discussed primarily in context of the overexpressed and tissue specific genes.
When initiated, the investigation did not have the benefit of a complete genome sequence, which imposes some limitations on conclusions. In particular, the fragmentation of sequences represented by the collection of microarray elements made it difficult to accurately enumerate genes interrogated in this analysis. Nevertheless, confidence in conclusions reached from this analysis is supported by the quantitative differences in expression observed across elements and among tissues, validation of microarray results by qRT-PCR (70% agreement among the two approaches), and the diversity of predicted functions based on sequence contigs from which microarray elements were derived. When the A. suum genome and gene model became available, we found that 28,380 of our 42k array elements covered 10,845 of the total 18,542 predicted A. suum genes (Jex et al. 2011). The average Euclidean expression distances for the nucleotide elements we used that derived from the same genes are significantly lower (p<0.0001, student t test) than those from different genes (data not shown), which added confidence in our findings. Accordingly, the results will have important application to deducing functions of the tissues investigated and greatly expand the annotation of the recently reported genome sequence for A. suum.
Expression of species restricted genes
Data from this investigation coupled with our previous observations support the view that species-specific genes have greater tendency to support specialized functions of tissues or life cycle stages rather than more generalized functions common to all stages and tissues. For instance, our previous transcriptome studies showed that expression of A. caninum specific genes tended to vary more across different developmental stages than genes more broadly conserved with other organisms. The current study with A. suum tissues showed that species-restricted genes were disproportionately expressed in a tissue-specific manner compared to more broadly conserved genes. Therefore, resolution of gene expression at the tissue level has identified novel adaptations of these tissues for this parasite. Localizing the tissues where expression occurs has facilitated the process to elucidate functions of these unknown proteins. The resolution gained at the parasite stage and tissue levels can explain, in part, observations on the nematode gene space, which continued to expand with new gene information from addition of each new nematode species (Martin et al. 2012); novel adaptations for tissue and/or stage specific functions are likely to contribute a high percentage of the gene sequences underlying this observation. Nevertheless, we cannot exclude that apparent species specific adaptations represented in this discussion actually reflect lineage specific adaptations (at some level). The rapidly accumulating gene sequence data for nematodes should help resolve this issue.
Predicted functions in A. suum tissues
Our analysis resolved a large number of predicted proteins that characterized each of the tissues investigated, while gender specific proteins constituted a major subcategory for each of the tissues. When considering all predicted proteins that were specific to each A. suum tissue, including gender specific proteins, a wide range of functions were discerned. In many cases, the predicted proteins and functions were expected based on current knowledge. For instance, predicted proteases, possibly involved in nutrient digestion, were commonly identified in both male and female intestine. This finding is consistent with previous results (Yin et al. 2008). The microarray data agreed with and extended previous results on A. suum gene expression. IntFam-241 is a collection of intestinal gene families that are apparently conserved across multiple, diverse nematode species (Yin et al. 2008). Of the 241 intestinal gene families, 61% (148) were represented on the microarray and confirmed to be expressed in male and female intestine, with over half demonstrating over-expression (64%, 95/148) in the intestine compared to other tissues (Figure S1). This comparison shows how this research expanded information for a subset of previously described genes and proteins, in this case IntFam-241.
Many predicted proteins from reproductive tissues were consistent with other research results (Costa et al. 2008; Evans et al. 2008; Li et al. 2011), and as will be discussed below. Perhaps one of the more unexpected findings in a broad sense was the underrepresentation of transcripts for contractile proteins, such as spectrins, from the pharynx which is predominately comprised of abundant radial muscle cells (Brownlee et al. 1995), also has the associated nerve ring. We postulate that the pharyngeal contractile proteins are stable and therefore steady state mRNA levels stay relatively low once enough proteins have accumulated during the process of initial organogenesis. Although the significance of this result is not easily reconciled without additional information, such as half-lives of contractile proteins in the pharynx, the result reminds us that transcript data provides but one dimension of information for functional investigations of this kind. In any case, we now have a substantial data set on proteins and functions that contribute to specific characteristics of the A. suum tissues investigated.
In addition to predicted proteins expressed by individual tissues, this analysis resolved a number of cases in which expression of transcripts for different protein family members (paralogs) were specific to individual tissues. Protein families with low average pairwise expression distance suggested conserved expression patterns and accordingly conserved functions. In contrast, high average pairwise expression distance within the protein family members suggested that diverse biological functions are served by different family members. This information will be valuable for functional annotation of the genome.
Gender specific functions
Gender specific gene expression has not been previously investigated for parasitic nematodes in context of such a wide range of tissues. Perhaps not surprisingly, gender specific genes were identified for each of the tissues investigated. Less predictable was the relatively large number of gender-specific elements that were detected in the male and female intestine. It appears that the intestine has a much larger role in support of gender specific functions than the other non-reproductive (somatic) tissues investigated. It is well known that yolk proteins are synthesized in the intestine and transported to the ovary in the hermaphroditic C. elegans (Kimble and Sharrock 1983), which likely involves a number of support systems that have not been fully elucidated. In addition, it is unclear how many other intestinal products are transported to the ovary, and the adult female A. suum system may provide a means to investigate these issues. It also remains unclear if products flow only one or both ways and if exchange occurs between the intestine and other tissues. Results from comparisons made possible with male A. suum indicate that relationships between the male intestine and other tissues may be as complicated, which has not been previously appreciated.
Predicted GO categories exclusively over-expressed in female intestine were related to one-carbon metabolic processes, mannose biosynthetic process, and lipid transport. In contrast, examples of predicted proteins exclusively over-expressed in male intestine were related to aspartic- and metallo-endopeptidase and isocitrate metabolic process. Thus, specific functions were identified that may differ between intestinal tissues of the sexes. From whole worm studies, female worm associated lipid transport functions were also reported in Trichuris globulosa (Sarwal et al. 1989).
The reproductive tissues of A. suum exhibited the highest number of gender-related expression patterns. Each reproductive tissue over-expressed a distinct set of genes, relatively or exclusively, in a gender specific manner. By comparison to other tissues, more gender related genes were over-expressed in the reproductive tissues of A. suum, similar to what was observed in B. malayi (Jiang et al. 2008). Furthermore, functional categorization represented by sequences over-expressed in A. suum reproductive tissues shared many similarities with results from T. vitrinus (Nisbet and Gasser 2004) and B. malayi (Michalski and Weil 1999). Therefore, it appears that the results from A. suum address basic reproductive processes that are conserved among phylogenetically disparate nematode species.
The uterus had the largest number of tissue specific over-expressed elements. The predicted A. suum proteins in this set of sequences from which the oligonucleotide elements were derived had a relatively low number of examples of homology to known C. elegans proteins. This disparity may reflect differences between reproductive strategies involving self-fertilization in C. elegans and cross-fertilization in A. suum. Also, uterine eggs in A. suum are single celled and do not divide until released into the environment, whereas uterine eggs are multicellular in C. elegans. The only enriched GO term achieving statistical significance in the uterus was regulation of ARF GTPase activity, instead of any terms related to egg development. The enrichment analysis may reflect limits of the GO database, and deeper understanding will require more individualized assessment of genes identified in the analysis. ARF GTPases are involved in membrane trafficking and organelle structuring, and the functions of individual ARF proteins are related to the effector molecules with which they interact (Donaldson et al. 1995).
The testes possess a large number of exclusively over-expressed elements that are enriched in diverse biological processes including steroid process, amino acid phosphorylation and dephosphorylation. These observations are consistent with what has been found in whole male Trichostrongylus vitrinus in which transcripts for protein kinases/phosphatases were relatively overexpressed (Nisbet and Gasser 2004), thus apparently playing important roles in sperm development. Another male-specific transcript identified in T. vitrinus encoded a major sperm protein (MSP, (Nisbet and Gasser 2004) (Reinke et al. 2000)). MSPs are nematode-specific cytoskeletal proteins involved in sperm movement (Roberts and Stewart 2000). Our study showed that A. suum major sperm protein (MSP) was exclusively enriched in the testes, consistent with its function as a sperm mobility-related protein as found in other nematode species.
Probes exclusively over-expressed in the ovary were enriched for both chitin metabolic process and homophilic cell adhesion process. These two processes have been reported in oocytes of other nematodes and are crucial for egg development (Anderson et al. 1999; Harris et al. 2000).
GO term analysis also indicated an enrichment for glycolysis in three reproductive tissues - ovary, testes, and seminal vesicle. Nevertheless, the nature of these enrichments could be very different among the three tissues. In the testes, gluconeogenesis and tricarboxylic acid cycle were also enriched, which is curious given the anaerobic environment in which A. suum lives, but consistent with findings on tricarboxylic acid enzymes (Komuniecki et al. 1993). If carried through fertilization this enrichment could reflect preadaptation for the zygote when released into the environment. Nevertheless while gluconeogenesis has been documented in developing eggs (Passey and Fairbairn 1957), we cannot exclude that this process also occurs in sperm. In the ovary, glycolysis was the only enriched term related to energy, suggesting that glycolysis is used as the main energy source, which is consistent with the anaerobic environment in which these worms exist. In the seminal vesicle, glycolysis enrichment was accompanied by responses to oxidative stress, indicating reliance on glycolysis as an energy source for sperm movement, also.
A. suum tissue and gender-related genes and proteins were ordered into predicted, higher level networks of protein interactions. Although it is likely that the expression databases generated here are incomplete, the density of genes identified led to predictions of apparent protein interactive networks that distinguished individual tissues, and those that are shared across all or subsets of tissues, including gender related networks. Proteins that represent centers of convergence for (networks and subnetworks) were identified for all tissues, either in general or tissue-or geneder-specific contexts. One example is the GEI-4 protein common to twenty functional modules identified for all A. suum tissues investigated. In C. elegans, the GEI-4 protein interacts with structural proteins, adaptor proteins and hormone receptors, and has been implicated in embryogenesis, fertility, and vulval morphogenesis by regulating intermediate filament dynamics (Tsuboi et al. 2002; Kamath et al. 2003). Although GEI-4 shows a more general presence across tissues, it may also participate in tissue specific-functions as shown in Table 6, based on differential expression of proteins with which it interacts. An example of a network center that might be involved in gender specific functions is provided by PQN-54, which emerged as a predicted core protein for eleven functional modules in female A. suum. The function of PQN-54 is relatively vague, although its expression in the C. elegans pharynx is recognized (Li et al. 2004b) and it has been implicated in the unfolded protein and innate immune responses of C. elegans related to bacterial infections (Haskins et al. 2008). Functions of this kind may be relevant to the health of parasitic nematodes that naturally contain endogenous bacteria in their gut or other tissues. Centers of these networks that may integrate multiple cellular pathways present attractive subjects of research aimed at disrupting cellular functions in pathogens. The results presented here provide an entry into further considering this application with A. suum and related parasites.
In summary, our study provides useful information about genes that are commonly expressed among the adult A. suum tissues investigated and those genes that differ in expression relative tissue and/or gender. Predicted functions for encoded proteins provided expectations of functions that are uniquely carried out by different tissues and in relation to gender. Gene ontology and functional module analyses placed findings in context of cellular processes and networks of protein interactions. The constellation of comparisons among tissues, between genders and across species produced many new perspectives on molecular functions of importance to the biology of A. suum and likely other parasitic and free-living nematodes. We also anticipate that results presented here will add valuable information for annotation of the A. suum genome.
Supplementary Material
Primer sequences for qRT-PCR
Coefficients of microarray elements and the functional annotation
Top scored active modules identified from (A) adult male A. suum tissues only or (B) adult female A. suum tissues only. Rows represent the identified active modulse and columns represent the individual tissues (Hea, head; Int, intestine; Pha, pharynx; Ova, ovary; SemVes, seminal vesicle; Ute, uterus; Tes, testis). Each black filled area indicates the existence of an active module in a specific tissue. Modules are sorted according to the descending z scores
Examples of the active modules common to the adult male A. suum tissues only that are centered at gene gei-4. Node indicates the protein members in the active module. Edge indicates the interaction between protein members. Conserved protein members between A. suum and C. elegans are highlighted by grey color. Interaction files for modules A, B and C are available at www.nematode.net (nematode.net/Ascaris_suum.html)
Examples of the active modules common to the adult female A. suum tissues only that are centered at gene pgn-4. Node indicates the protein members in the active module. Edge indicates the interaction between protein members. Conserved protein members between A. suum and C. elegans are highlighted by grey color. Interaction files for modules A, B and C are available at www.nematode.net (nematode.net/Ascaris_suum.html)
Heatmap showing expression data for the 214 nucleotide sequence contigs that collectively formed 95 different A. suum intestinal gene families (Yin et al. 2008) found to be differentially expressed among the ten tissues investigated. Rows represent genes and columns represent different tissues. The gradation from green (highest similarity) to red (lowest similarity) depicts distance in relation to individual contigs and overall expression profiles among tissues. FemPha, female pharynx; MalPha, male pharynx; FemHea, female head; MalHea, male head; FemInt, female intestine; MalInt, male intestine; SemVes, seminal vesicle; Tes, testis; Ova, ovary; Ute, uterus.
qPCR validation of selected genes that showed the highest level of expression differences in microarray assays.
Interpro annotations of top 20 tissues specific expressed genes
IDs of family members expressed significantly differently
GO associations of A. suum elements represented on the microarray
Example genes that directly interact with gei-4 that are not included in Table 5 and display differential expression across adult A. suum tissues
Acknowledgments
The analysis presented in this study were supported by NIH/NAIAD and NIH/NIGMS grants to M.M. We thank the Washington University microarray core Dr. Seth Crosby and Michael Heinz for their service and technical assistance and Dr. Richard Komuniecki for providing the A. suum tissues.
References
- Anderson R, Schaible K, Heasman J, Wylie C. Expression of the homophilic adhesion molecule, Ep-CAM, in the mammalian germ line. J Reprod Fertil. 1999;116:379–384. doi: 10.1530/jrf.0.1160379. [DOI] [PubMed] [Google Scholar]
- Aranda B, Achuthan P, Alam-Faruque Y, Armean I, Bridge A, Derow C, Feuermann M, Ghanbarian AT, Kerrien S, Khadake J, Kerssemakers J, Leroy C, Menden M, Michaut M, Montecchi-Palazzi L, Neuhauser SN, Orchard S, Perreau V, Roechert B, van Eijk K, Hermjakob H. The IntAct molecular interaction database in 2010. Nucl Acids Res. 2010;38:D525–531. doi: 10.1093/nar/gkp878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arbeitman MN, Furlong EE, Imam F, Johnson E, Null BH, Baker BS, Krasnow MA, Scott MP, Davis RW, White KP. Gene expression during the life cycle of Drosophila melanogaster. Science. 2002;297:2270–2275. doi: 10.1126/science.1072152. [DOI] [PubMed] [Google Scholar]
- Bader GD, Betel D, Hogue CWV. BIND: the Biomolecular Interaction Network Database. Nucl Acids Res. 2003;31:248–250. doi: 10.1093/nar/gkg056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bao Z, Eddy SR. Automated de novo identification of repeat sequence families in sequenced genomes. Genome Res. 2002;12:1269–1276. doi: 10.1101/gr.88502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bethony J, Brooker S, Albonico M, Geiger SM, Loukas A, Diemert D, Hotez PJ. Soil-transmitted helminth infections: ascariasis, trichuriasis, and hookworm. The Lancet. 2006;367:1521–1532. doi: 10.1016/S0140-6736(06)68653-4. [DOI] [PubMed] [Google Scholar]
- Brindley PJ, Mitreva M, Ghedin E, Lustigman S. Helminth genomics: The implications for human health. PLoS Negl Trop Dis. 2009;3:e538. doi: 10.1371/journal.pntd.0000538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brownlee DJ, Holden-Dye L, Walker RJ, Fairweather I. The pharynx of the nematode Ascaris suum: structure and function. Acta Biol Hung. 1995;46:195–204. [PubMed] [Google Scholar]
- Cantacessi C, Zou FC, Hall RS, Zhong W, Jex AR, Campbell BE, Ranganathan S, Sternberg PW, Zhu XQ, Gasser RB. Bioinformatic analysis of abundant, gender-enriched transcripts of adult Ascaris suum (Nematoda) using a semi-automated workflow platform. Molecular and Cellular Probes. 2009;23:205–217. doi: 10.1016/j.mcp.2009.03.003. [DOI] [PubMed] [Google Scholar]
- Capron A, Riveau G, Capron M, Trottein F. Schistosomes: the road from host-parasite interactions to vaccines in clinical trials. Trends in Parasitology. 2005;21:143–149. doi: 10.1016/j.pt.2005.01.003. [DOI] [PubMed] [Google Scholar]
- Choi YJ, Ghedin E, Berriman M, McQuillan J, Holroyd N, Mayhew GF, Christensen BM, Michalski ML. A deep sequencing approach to comparatively analyze the transcriptome of lifecycle stages of the filarial worm, Brugia malayi. PLoS Negl Trop Dis. 2011;5:e1409. doi: 10.1371/journal.pntd.0001409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa AF, Gomes-Ruiz AC, Rabelo EM. Identification of gender-regulated genes in Ancylostoma braziliense by real-time RT-PCR. Vet Parasitol. 2008;153:277–284. doi: 10.1016/j.vetpar.2008.02.005. [DOI] [PubMed] [Google Scholar]
- Crompton DW. Ascaris and ascariasis. Adv Parasitol. 2001;48:285–375. doi: 10.1016/s0065-308x(01)48008-0. [DOI] [PubMed] [Google Scholar]
- Ding L, Sabo A, Berkowicz N, Meyer RR, Shotland Y, Johnson MR, Pepin KH, Wilson RK, Spieth J. EAnnot: a genome annotation tool using experimental evidence. Genome Res. 2004;14:2503–2509. doi: 10.1101/gr.3152604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dold C, Holland CV. Ascaris and ascariasis. Microbes Infect. 2011;13:632–637. doi: 10.1016/j.micinf.2010.09.012. [DOI] [PubMed] [Google Scholar]
- Donaldson JG, Radhakrishna H, Peters PJ. The ARF GTPases: Defining roles in membrane traffic and organelle structure. Cold Spring Harbor Symposia on Quantitative Biology. 1995;60:229–234. doi: 10.1101/sqb.1995.060.01.026. [DOI] [PubMed] [Google Scholar]
- Evans H, Mello LV, Fang Y, Wit E, Thompson FJ, Viney ME, Paterson S. Microarray analysis of gender- and parasite-specific gene transcription in Strongyloides ratti. Int J Parasitol. 2008;38:1329–1341. doi: 10.1016/j.ijpara.2008.02.004. [DOI] [PubMed] [Google Scholar]
- Fincham JE, Markus MB, Adams VJ. Could control of soil-transmitted helminthic infection influence the HIV/AIDS pandemic. Acta Tropica. 2003;86:315–333. doi: 10.1016/s0001-706x(03)00063-9. [DOI] [PubMed] [Google Scholar]
- Finn RD, Mistry J, Schuster-Bockler B, Griffiths-Jones S, Hollich V, Lassmann T, Moxon S, Marshall M, Khanna A, Durbin R, Eddy SR, Sonnhammer ELL, Bateman A. Pfam: clans, web tools and services. Nucleic Acids Research. 2006;34:D247–D251. doi: 10.1093/nar/gkj149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gentleman RC, Carey VJ, Bates DM, Bolstad B, Dettling M, Dudoit S, Ellis B, Gautier L, Ge YC, Gentry J, Hornik K, Hothorn T, Huber W, Iacus S, Irizarry R, Leisch F, Li C, Maechler M, Rossini AJ, Sawitzki G, Smith C, Smyth G, Tierney L, Yang JYH, Zhang JH. Bioconductor: open software development for computational biology and bioinformatics. Genome Biology. 2004;5 doi: 10.1186/gb-2004-5-10-r80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghedin E, Hailemariam T, DePasse JV, Zhang X, Oksov Y, Unnasch TR, Lustigman S. Brugia malayi gene expression in response to the targeting of the Wolbachia endosymbiont by tetracycline treatment. PLoS Negl Trop Dis. 2009;3:e525. doi: 10.1371/journal.pntd.0000525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagan P, Appleton CC, Coles GC, Kusel JR, Tchuem-TchuentÈ L-A. Schistosomiasis control: keep taking the tablets. Trends in Parasitology. 2004;20:92–97. doi: 10.1016/j.pt.2003.11.010. [DOI] [PubMed] [Google Scholar]
- Harris MT, Lai K, Arnold K, Martinez HF, Specht CA, Fuhrman JA. Chitin synthase in the filarial parasite, Brugia malayi. Molecular and Biochemical Parasitology. 2000;111:351–362. doi: 10.1016/s0166-6851(00)00328-5. [DOI] [PubMed] [Google Scholar]
- Haskins KA, Russell JF, Gaddis N, Dressman HK, Aballay A. Unfolded protein response genes regulated by CED-1 are required for Caenorhabditis elegans innate immunity. Dev Cell. 2008;15:87–97. doi: 10.1016/j.devcel.2008.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hotez PJ. Neglected diseases and poverty in “The Other America”: the greatest health disparity in the United States? PLoS Negl Trop Dis. 2007;1:e149. doi: 10.1371/journal.pntd.0000149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang CQ, Gasser RB, Cantacessi C, Nisbet AJ, Zhong W, Sternberg PW, Loukas A, Mulvenna J, Lin RQ, Chen N, Zhu XQ. Genomic-bioinformatic analysis of transcripts enriched in the third-stage larva of the parasitic nematode Ascaris suum. PLoS Negl Trop Dis. 2008;2:e246. doi: 10.1371/journal.pntd.0000246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang X, Wang J, Aluru S, Yang SP, Hillier L. PCAP: a whole-genome assembly program. Genome Res. 2003;13:2164–2170. doi: 10.1101/gr.1390403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ideker T, Ozier O, Schwikowski B, Siegel AF. Discovering regulatory and signalling circuits in molecular interaction networks. Bioinformatics. 2002;18:S233–240. doi: 10.1093/bioinformatics/18.suppl_1.s233. [DOI] [PubMed] [Google Scholar]
- Jasmer DP, Yao C, Rehman A, Johnson S. Multiple lethal effects induced by a benzimidazole anthelmintic in the anterior intestine of the nematode Haemonchus contortus. Mol Biochem Parasitol. 2000;105:81–90. doi: 10.1016/s0166-6851(99)00169-3. [DOI] [PubMed] [Google Scholar]
- Jex AR, Liu S, Li B, Young ND, Hall RS, Li Y, Yang L, Zeng N, Xu X, Xiong Z, Chen F, Wu X, Zhang G, Fang X, Kang Y, Anderson GA, Harris TW, Campbell BE, Vlaminck J, Wang T, Cantacessi C, Schwarz EM, Ranganathan S, Geldhof P, Nejsum P, Sternberg PW, Yang H, Wang J, Gasser RB. Ascaris suum draft genome. Nature. 2011;479:529–533. doi: 10.1038/nature10553. [DOI] [PubMed] [Google Scholar]
- Jiang D, Li B-W, Fischer PU, Weil GJ. Localization of gender-regulated gene expression in the filarial nematode Brugia malayi. International Journal for Parasitology. 2008;38:503–512. doi: 10.1016/j.ijpara.2007.09.010. [DOI] [PubMed] [Google Scholar]
- Kamath RS, Fraser AG, Dong Y, Poulin G, Durbin R, Gotta M, Kanapin A, Le Bot N, Moreno S, Sohrmann M, Welchman DP, Zipperlen P, Ahringer J. Systematic functional analysis of the Caenorhabditis elegans genome using RNAi. Nature. 2003;421:231–237. doi: 10.1038/nature01278. [DOI] [PubMed] [Google Scholar]
- Kimber MJ, McKinney S, McMaster S, Day TA, Fleming CC, Maule AG. flp gene disruption in a parasitic nematode reveals motor dysfunction and unusual neuronal sensitivity to RNA interference. FASEB J. 2007;21:1233–1243. doi: 10.1096/fj.06-7343com. [DOI] [PubMed] [Google Scholar]
- Kimble J, Sharrock WJ. Tissue-specific synthesis of yolk proteins in Caenorhabditis elegans. Dev Biol. 1983;96:189–196. doi: 10.1016/0012-1606(83)90322-6. [DOI] [PubMed] [Google Scholar]
- Knox DP, Redmond DL, Newlands GF, Skuce PJ, Pettit D, Smith WD. The nature and prospects for gut membrane proteins as vaccine candidates for Haemonchus contortus and other ruminant trichostrongyloids. Int J Parasitol. 2003;33:1129–1137. doi: 10.1016/s0020-7519(03)00167-x. [DOI] [PubMed] [Google Scholar]
- Komuniecki PR, Johnson J, Kamhawi M, Komuniecki R. Mitochondrial heterogeneity in the parasitic nematode, Ascaris suum. Exp Parasitol. 1993;76:424–437. doi: 10.1006/expr.1993.1051. [DOI] [PubMed] [Google Scholar]
- Leles D, Gardner SL, Reinhard K, Iniguez A, Araujo A. Are Ascaris lumbricoides and Ascaris suum a single species? Parasit Vectors. 2012;5:42. doi: 10.1186/1756-3305-5-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li BW, Rush AC, Jiang DJ, Mitreva M, Abubucker S, Weil GJ. Gender-associated genes in filarial nematodes are important for reproduction and potential intervention targets. PLoS Negl Trop Dis. 2011;5:e947. doi: 10.1371/journal.pntd.0000947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li BW, Rush AC, Tan J, Weil GJ. Quantitative analysis of gender-regulated transcripts in the filarial nematode Brugia malayi by real-time RT-PCR. Mol Biochem Parasitol. 2004a;137:329–337. doi: 10.1016/j.molbiopara.2004.07.002. [DOI] [PubMed] [Google Scholar]
- Li BW, Wang Z, Rush AC, Mitriva M, Weil GJ. Transcription profiling reveals stage- and functiondependent expression patterns in the filarial nematode brugia malayi. BMC Genomics. 2012;13:184. doi: 10.1186/1471-2164-13-184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, Armstrong CM, Bertin N, Ge H, Milstein S, Boxem M, Vidalain PO, Han JD, Chesneau A, Hao T, Goldberg DS, Li N, Martinez M, Rual JF, Lamesch P, Xu L, Tewari M, Wong SL, Zhang LV, Berriz GF, Jacotot L, Vaglio P, Reboul J, Hirozane-Kishikawa T, Li Q, Gabel HW, Elewa A, Baumgartner B, Rose DJ, Yu H, Bosak S, Sequerra R, Fraser A, Mango SE, Saxton WM, Strome S, Van Den Heuvel S, Piano F, Vandenhaute J, Sardet C, Gerstein M, Doucette-Stamm L, Gunsalus KC, Harper JW, Cusick ME, Roth FP, Hill DE, Vidal M. A map of the interactome network of the metazoan C. elegans. Science. 2004b;303:540–543. doi: 10.1126/science.1091403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma X, Zhu Y, Li C, Shang Y, Meng F, Chen S, Miao L. Comparative transcriptome sequencing of germline and somatic tissues of the Ascaris suum gonad. BMC Genomics. 2011;12:481. doi: 10.1186/1471-2164-12-481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin J, Abubucker S, Heizer E, Taylor CM, Mitreva M. Nematode. net update 2011: addition of data sets and tools featuring next-generation sequencing data. Nucleic Acids Res. 2012;40:D720–728. doi: 10.1093/nar/gkr1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michalski ML, Weil GJ. Gender-specific gene expression in Brugia malayi. Molecular and Biochemical Parasitology. 1999;104:247–257. doi: 10.1016/s0166-6851(99)00149-8. [DOI] [PubMed] [Google Scholar]
- Mitreva M, Jasmer DP, Appleton J, Martin J, Dante M, Wylie T, Clifton SW, Waterston RH, McCarter JP. Gene discovery in the adenophorean nematode Trichinella spiralis: an analysis of transcription from three life cycle stages. Molecular and Biochemical Parasitology. 2004;137:277–291. doi: 10.1016/j.molbiopara.2004.05.015. [DOI] [PubMed] [Google Scholar]
- Mitreva M, Zarlenga DS, McCarter JP, Jasmer DP. Parasitic nematodes--From genomes to control. Veterinary Parasitology. 2007;148:31–42. doi: 10.1016/j.vetpar.2007.05.008. [DOI] [PubMed] [Google Scholar]
- Morimoto M, Zarlenga D, Beard H, Alkharouf N, Matthews BF, Urban JF. Ascaris suum: cDNA microarray analysis of 4th stage larvae (L4) during self-cure from the intestine. Experimental Parasitology. 2003;104:113–121. doi: 10.1016/s0014-4894(03)00139-5. [DOI] [PubMed] [Google Scholar]
- Neveu C, Jaubert S, Abad P, Castagnone-Sereno P. A set of genes differentially expressed between avirulent and virulent Meloidogyne incognita near-isogenic lines encode secreted proteins. Mol Plant Microbe Interact. 2003;16:1077–1084. doi: 10.1094/MPMI.2003.16.12.1077. [DOI] [PubMed] [Google Scholar]
- Nisbet AJ, Gasser RB. Profiling of gender-specific gene expression for Trichostrongylus vitrinus (Nematoda: Strongylida) by microarray analysis of expressed sequence tag libraries constructed by suppressive-subtractive hybridisation. International Journal for Parasitology. 2004;34:633–643. doi: 10.1016/j.ijpara.2003.12.007. [DOI] [PubMed] [Google Scholar]
- Passey RF, Fairbairn D. The conversion of fat to carbohydrate during embryonation of ascaris eggs. Can J Biochem Physiol. 1957;35:511–525. [PubMed] [Google Scholar]
- Prufer K, Muetzel B, Do HH, Weiss G, Khaitovich P, Rahm E, Paabo S, Lachmann M, Enard W. FUNC: a package for detecting significant associations between gene sets and ontological annotations. BMC Bioinformatics. 2007;8 doi: 10.1186/1471-2105-8-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajeevan MS, Ranamukhaarachchi DG, Vernon SD, Unger ER. Use of real-time quantitative PCR to validate the results of cDNA array and differential display PCR technologies. Methods. 2001;25:443–451. doi: 10.1006/meth.2001.1266. [DOI] [PubMed] [Google Scholar]
- Reinke V, Smith HE, Nance J, Wang J, Van Doren C, Begley R, Jones SJM, Davis EB, Scherer S, Ward S, Kim SK. A Global Profile of Germline Gene Expression in C. elegans. Molecular Cell. 2000;6:605–616. doi: 10.1016/s1097-2765(00)00059-9. [DOI] [PubMed] [Google Scholar]
- Roberts TM, Stewart M. Acting like actin: The dynamics of the nematode major sperm protein (MSP) cytoskeleton indicate a push-pull mechanism for amoeboid cell motility. The Journal of Cell Biology. 2000;149:7–12. doi: 10.1083/jcb.149.1.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarwal R, Sanyal SN, Khera S. Lipid metabolism in Trichuris globulosa (Nematoda) Journal of Helminthology. 1989;63:287–297. doi: 10.1017/s0022149x00009160. [DOI] [PubMed] [Google Scholar]
- Scheibel T, Lindquist SL. The role of conformational flexibility in prion propagation and maintenance for Sup35p. Nat Struct Biol. 2001;8:958–962. doi: 10.1038/nsb1101-958. [DOI] [PubMed] [Google Scholar]
- Serio TR, Lindquist SL. [PSI+], SUP35, and chaperones. Adv Protein Chem. 2001;57:335–366. doi: 10.1016/s0065-3233(01)57027-8. [DOI] [PubMed] [Google Scholar]
- Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Research. 2003;13:2498–2504. doi: 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shingles J, Lilley CJ, Atkinson HJ, Urwin PE. Meloidogyne incognita: Molecular and biochemical characterisation of a cathepsin L cysteine proteinase and the effect on parasitism following RNAi. Experimental Parasitology. 2007;115:114–120. doi: 10.1016/j.exppara.2006.07.008. [DOI] [PubMed] [Google Scholar]
- Smyth GK. Linear models and empirical Bayes methods for assessing differential expression in microarray experiments. Statistical Applications in Genetics and Molecular Biology. 2004;3 doi: 10.2202/1544-6115.1027. [DOI] [PubMed] [Google Scholar]
- Spirin V, Mirny LA. Protein complexes and functional modules in molecular networks. Proc Natl Acad Sci U S A. 2003;100:12123–12128. doi: 10.1073/pnas.2032324100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas PD, Campbell MJ, Kejariwal A, Mi H, Karlak B, Daverman R, Diemer K, Muruganujan A, Narechania A. PANTHER: A library of protein families and subfamilies indexed by function. Genome Research. 2003;13:2129–2141. doi: 10.1101/gr.772403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuboi D, Qadota H, Kasuya K, Amano M, Kaibuchi K. Isolation of the interacting molecules with GEX-3 by a novel functional screening. Biochem Biophys Res Commun. 2002;292:697–701. doi: 10.1006/bbrc.2002.6717. [DOI] [PubMed] [Google Scholar]
- Walhout AJ, Sordella R, Lu X, Hartley JL, Temple GF, Brasch MA, Thierry-Mieg N, Vidal M. Protein interaction mapping in C. elegans using proteins involved in vulval development. Science. 2000;287:116–122. doi: 10.1126/science.287.5450.116. [DOI] [PubMed] [Google Scholar]
- Wang J, Czech B, Crunk A, Wallace A, Mitreva M, Hannon GJ, Davis RE. Deep small RNA sequencing from the nematode Ascaris reveals conservation, functional diversification, and novel developmental profiles. Genome Res. 2011;21:1462–1477. doi: 10.1101/gr.121426.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Abubucker S, Martin J, Wilson RK, Hawdon J, Mitreva M. Characterizing Ancylostoma caninum transcriptome and exploring nematode parasitic adaptation. BMC Genomics. 2010;11:307. doi: 10.1186/1471-2164-11-307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin Y, Martin J, Abubucker S, Scott AL, McCarter JP, Wilson RK, Jasmer DP, Mitreva M. Intestinal Transcriptomes of Nematodes: Comparison of the Parasites Ascaris suum and Haemonchus contortus with the Free-living Caenorhabditis elegans. PLoS Neglected Tropical Diseases. 2008;2 doi: 10.1371/journal.pntd.0000269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zdobnov EM, Apweiler R. InterProScan - an integration platform for the signature-recognition methods in InterPro. Bioinformatics. 2001;17:847–848. doi: 10.1093/bioinformatics/17.9.847. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Primer sequences for qRT-PCR
Coefficients of microarray elements and the functional annotation
Top scored active modules identified from (A) adult male A. suum tissues only or (B) adult female A. suum tissues only. Rows represent the identified active modulse and columns represent the individual tissues (Hea, head; Int, intestine; Pha, pharynx; Ova, ovary; SemVes, seminal vesicle; Ute, uterus; Tes, testis). Each black filled area indicates the existence of an active module in a specific tissue. Modules are sorted according to the descending z scores
Examples of the active modules common to the adult male A. suum tissues only that are centered at gene gei-4. Node indicates the protein members in the active module. Edge indicates the interaction between protein members. Conserved protein members between A. suum and C. elegans are highlighted by grey color. Interaction files for modules A, B and C are available at www.nematode.net (nematode.net/Ascaris_suum.html)
Examples of the active modules common to the adult female A. suum tissues only that are centered at gene pgn-4. Node indicates the protein members in the active module. Edge indicates the interaction between protein members. Conserved protein members between A. suum and C. elegans are highlighted by grey color. Interaction files for modules A, B and C are available at www.nematode.net (nematode.net/Ascaris_suum.html)
Heatmap showing expression data for the 214 nucleotide sequence contigs that collectively formed 95 different A. suum intestinal gene families (Yin et al. 2008) found to be differentially expressed among the ten tissues investigated. Rows represent genes and columns represent different tissues. The gradation from green (highest similarity) to red (lowest similarity) depicts distance in relation to individual contigs and overall expression profiles among tissues. FemPha, female pharynx; MalPha, male pharynx; FemHea, female head; MalHea, male head; FemInt, female intestine; MalInt, male intestine; SemVes, seminal vesicle; Tes, testis; Ova, ovary; Ute, uterus.
qPCR validation of selected genes that showed the highest level of expression differences in microarray assays.
Interpro annotations of top 20 tissues specific expressed genes
IDs of family members expressed significantly differently
GO associations of A. suum elements represented on the microarray
Example genes that directly interact with gei-4 that are not included in Table 5 and display differential expression across adult A. suum tissues