

Abstract
This minireview is dedicated to the memory of Henryk Eisenberg and honors his major contributions to many areas of biophysics and to the analysis of macromolecular states and interactions in particular. This work reviews the ATP and ADP states of a ubiquitous protein, actins, and considers the present evidence for and against unique, nucleotide-dependent conformations of this protein. The effects of ATP and ADP on specific structural elements of actins, its loops and clefts, as revealed by mutational, crosslinking, spectroscopic, and EPR methods are discussed. It is concluded that the existing evidence points to dynamic equilibria of these structural elements among various conformational states in both ATP- and ADP-actins, with the nucleotides impacting the equilibria distributions.
Keywords: actin, nucleotide states, structure
INTRODUCTION
Actin is a highly conserved, abundant, and functionally versatile protein expressed in virtually all eukaryotic cells. Eukaryotic organisms produce at least one isoform of actin and all known actins are highly conserved. Thus, although S. cerevisiae yeast actin and human actins are separated by billion years of evolution, they have 87% identical amino acids (325 out of 374 a.a.), with 94% overall sequence conservation (353 out of 374 a.a.). Mammalian and avian non-muscle β-isoforms of actin differ by a single conservative substitution (R336K), whereas their muscle α-skeletal isoforms are identical. Functional versatility of actin is defined by its amazing structural plasticity, tuned and maintained by more than a 150 interacting partners, as well as by its ability to support myosin-based motility and generate force and motion (motility) on its own.
Actin was first isolated from skeletal muscle seven decades ago, in early 1940s. Straub and Szent-Györgyi have discovered then that actin is an essential protein for muscle contractility and can exist in both fibrous (F-) and globular (G-) states.1–3 It was not until early seventies when actin was also found in nonmuscle cells and organisms—first in Acanthamoeba4 and then in various mammalian nonmuscle cells.5 At that time it was realized that in contrast to muscle cells, the motility of nonmuscle cells relies primarily on actin and less on myosin.6 Because actin polymerizes or aggregates under crystallization conditions, the first X-ray structure of G-actin was solved only in 1990,7 in a complex with an actin binding protein DNase I which blocks the polymerization, leading to the first atomic model of actin filaments.8
Structure of G-Actin
Actin protomer has a globular shape, flattened in one dimension, and consists of two major domains—large or inner and small or outer domains, when referred to their size or orientation in the filament. The small and the large domains of actin are further subdivided into subdomains 1, 2 (SD1,2) and 3, 4 (SD3,4), respectively (Figure 1). A nucleotide (ATP or ADP) with a tightly bound divalent cation (Mg2+ under physiological conditions) is positioned deep inside the cleft between SD 2 and SD4 and is clamped between two phosphate binding loops—P1 and P2 loops (residues 11–16 and 154–161, respectively). The nucleotide binding cleft (NBC) is separated by a hinge region (including residues 137–146 and 334–337) from another, less deep cleft—often referred to as a hydrophobic cleft or patch—enriched in hydrophobic amino acid residues (Figure 1). This patch is a primary binding site for many actin-binding proteins (ABPs) and small actin-specific macrolide toxins.9 It should not be confused with the hydrophobic plug or loop (H-plug; a.a. residues 264–271) of actin, which projects from the back of actin at the border between SD3 and 4 and establishes important interstrand contacts with SD2 and SD1 of other subunits in F-actin.10–15 Among actin subdomains, SD2 is the smallest and by far the most flexible one. The outer part of SD2—the DNase I binding loop (D-loop; residues 40–51)—is disordered in most X-ray structures of actin published to date, but has been also observed in an α-helix, β-turn, and various loop conformations. Both the N- and C-termini of actin are located in SD1, albeit on opposite faces of the molecule. Similar to the D-loop, these termini are flexible and are often disordered in X-ray structures. The other three structurally variable regions of actin are the H-plug (defined above), WH2-binding loop (W-loop; residues 165–172), and the V-stretch (residues 227–237).16 Notably, all these flexible regions of actin, with the exception of the V-stretch, form interfaces between actin protomers in the filament.
FIGURE 1.
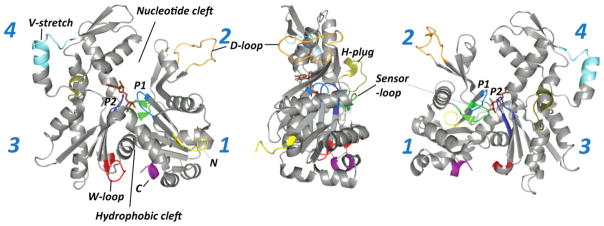
G-actin structure. Structure of actin monomer is shown in three projections. The flexible segments of actin are colored as follows: the phosphate binding loops P1 (a.a. 11–16) and P2 (a.a. 154–161) are in light and dark blue, respectively; the sensor loop (a.a. 70–78) is in green; the H-plug (a.a. 264–271) is in olive; the D-loop (a.a. 40–51) is in orange; the W-loop (a.a. 165–172) is in red; the V-stretch (a.a. 227–237) is in cyan; the N- and C-terminal regions are colored in yellow and purple, respectively.
Tightly Bound Divalent Cation
Several molecules of mono- and divalent cations bind to actin at low affinity sites (Kd = 0.15 mM and above)17,18 and play role in filament stabilization, while one molecule of a divalent cation binds with high afiiniity to the NBC in complex with the bound nucleotide.17,19,20 The tightly bound divalent cation is coordinated with the γ- and β-phosphate groups of ATP, but only with the β-phosphate of ADP.18 The physiologically relevant high affinity divalent cation is Mg2+; but most preparations yield the less polymerizable CaATP-actin21 and, therefore, both species have been studied extensively. High affinity Ca2+ and Mg2+ bind to ATP-actin with low nanomolar and low micromolar affinities respectively,17 and the divalent cation dissociation defines an overall rate of nucleotide release from actin.22 Complexes of actin with Ba2+, Mn2+, Cr2+, Cu2+, and Li2+ cations affect actin structure and dynamics in various ways, and therefore have been used to solve specific experimental challenges.23–26
Role of Nucleotide Hydrolysis in Filament Polarity and Treadmilling
The interaction of actin with nucleotides was independently discovered in 1950 by two groups, who reported that ATP is an essential part of actin and it is hydrolyzed to ADP and Pi upon actin polymerization.27,28 Although the ATPase activity of G-actin is very low, it is activated upon polymerization by a factor of 40,000 and MgATP-F-actin hydrolyzes ATP with a rate constant of 0.3 s−1.23 The inorganic phosphate release is about two orders of magnitude slower than the hydrolysis. Consequently, the newly assembled part of the filament contains mostly ADP-Pi protomers with a cap of ATP subunits at the very tip, while older filaments have mainly ADP-protomers. The ADP-Pi actin is structurally similar to ATP actin29 and both are more structurally stable that ADP-F-actin.30 The release of phosphate triggers intra- and intermolecular conformational rearrangements in the filament that result in less stable and more flexible (shorter persistence length) ADP-actin filaments.31 The stability of filaments can be described by actin’s critical concentration for polymerization (Cc), which is defined as the concentration of free monomers in equilibrium with the filaments, i.e., less stable filaments have a higher Cc. As a result of ATP hydrolysis, actin filaments are functionally polar and the ADP-enriched end (minus or pointed end) of the filament disassembles more readily (Cc~0.7 μM). This supplies G-actin subunits, which add then to the opposite filament end (plus or barbed end; Cc~0.1 μM) after ADP in their cleft is exchanged to ATP. Therefore, in steady state and in the presence of ATP in solution, shortening of the pointed end is balanced by elongation of the barbed end of the filament (Figure 2). This directed, ATP hydrolysis-driven turnover of actin protomers is called treadmilling (recently reviewed in Ref. 32). In eukaryotic cells, actin treadmilling is a basic underlying force for cell migration, invasion, as well as endo- and exocytosis. Treadmilling occurs continuously even in resting cells33 and constitutes a major energy drain, causing up to 50% of total ATP consumption in cells of various tissues (e.g. platelets and neuronal cells).34,35
FIGURE 2.
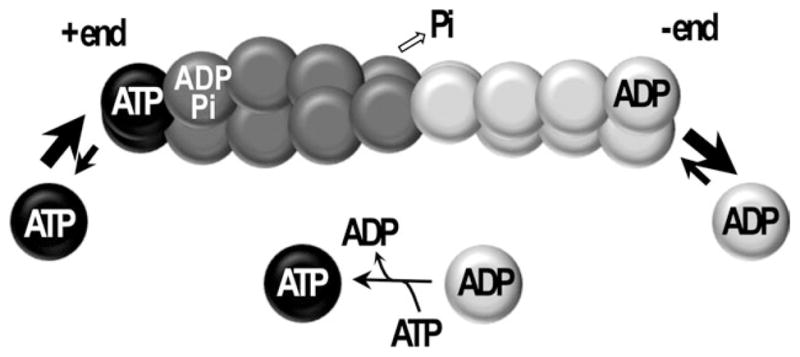
Schematic representation of actin treadmilling. Under steady state conditions in the presence of ATP, actin protomers containing ADP dissociate predominantly from the minus end of the filament, exchange the nucleotide in the cleft from ADP to ATP, and then ATP-containing protomers associate predominantly at the plus end of the filament. The dissociation from the minus end is potentiated by cofilin; ADP to ATP exchange is promoted in solution by profilin. Both proteins sense nucleotide-dependent conformations of actin. Overall, ATP hydrolysis by filamentous actin fuels the translocation of protomers from one end of the filament to the other.
ATP hydrolysis is not required for polymerization per se, as actin containing a nonhydrolyzable nucleotide analog AMP-PNP polymerizes with similar kinetics to ATP-actin.36,37 Moreover, nucleotide-free actin polymerizes rapidly38 and forms filaments with normal appearance,39 providing that the integrity of actin monomers—compromised by the absence of a nucleotide—is stabilized by a high concentration of sucrose.38–40 Among the nucleotide-free, ADP-Pi, and ADP F-actins the latter is the least stable and has the highest critical concentration for polymerization, suggesting that ADP bound to the cleft favors a conformation (or a subset of distinct conformations) that yields less stable filaments.38
In the cell, treadmilling of actin is regulated and accelerated by orders of magnitude by various actin binding proteins (ABPs) that preferentially recognize one of the nucleotide-related conformational states of actin.32 Intriguingly, it was proposed that ATP hydrolysis and Pi release do not introduce significant gradient of filament instability under crowded conditions of cellular environment, and that actin filament treadmilling in the cell is defined primarily by regulatory ABPs that selectively recognize and intensify specific nucleotide-dependent conformational transitions.41 Therefore, a primary role of ATP hydrolysis in F-actin may be to create age-dependent conformational changes that can be recognized by ABPs and are required to maintain a proper balance between freshly polymerized and old populations of actin filaments in a regulated equilibrium with G-actin.
Nucleotide-Dependent Conformational States of Actin
Open and Closed Conformation of the Nucleotide Binding Cleft in G-Actin
Since the discovery of treadmilling and nucleotide-specific recognition of actin by various ABPs, attempts were made to reconcile these functional traits with structural conformations of actin. Some examples of ABPs preference for nucleotide specific states of actin include the much better binding of cofilin and profilin to ADP- and ATP-G-actins, respectively. Cofilin’s inhibition of nucleotide exchange on G-actin is consistent with its proposed shifting of the actin’s cleft to a closed state. On the other hand, the acceleration of nucleotide exchange by profilin is in line with its documented opening of the nucleotide cleft on G-actin. More detailed discussion of ABPs effect on actin’s conformation, dynamics and its cleft states is outside the scope of this review.
Structural similarity between actin and proteins of the actin superfamily, such as hexokinase, hsc70,42 as well as Arp2, Arp3,43 and ParM,44 each of which adopts an open conformation in Apo-state, allowed to formulate an analogy-based hypothesis of global closed-to-open transitions of the cleft region upon Pi release following ATP hydrolysis. According to this hypothesis, the nucleotide binding cleft of actin closes when the two major domains are bridged by ATP, but assumes a more open conformation, with wider separation of the domains, due to an attenuated domain bridging upon hydrolysis and a subsequent release of γ-phosphate.
This hypothesis has been indirectly supported by the finding that the nucleotide binding cleft of Arp3 protein, an actin-related protein of the Arp2/3 nucleation complex, adopted more open conformations in the ADP- than in the ATP-state.43 Nevertheless, an overwhelming majority of ~80 X-ray structures of actin submitted to the Protein Data Bank, including those solved in the presence of ADP,45,46 are found in a tightly closed state. Thus, two structures of ADP-actin, crystallized in the absence of ABP and small molecule toxins, have the cleft in a closed state, with distances between nucleotide surrounding loops similar or even slightly closer than those found in ATP-actin.46,47
Two structures, in which actin adopted a wide-open or partially open conformation of the cleft, were in its complexes with profilin.48–50 Profilin is an abundant cytoplasmic protein that binds to the barbed end of actin monomer, between subdomains 1 and 3, favoring the nucleotide-free actin over ATP-and ADP-actin.38 The wide-open state of the actin-profilin complex is characterized by a 2.9 Å increased separation of the tips (residues G15 and D157) of two hairpin loops located across the NBC and clamping the nucleotide (Figure 3). These loops are named by different authors either as Pi binding loops 1 and 2 (P1 and P2 loops),18,51 or as S- and G-loops,46,50,52 respectively. In the actin-profilin structure with a wide-open cleft, the major domains of actin rotate with respect to each other by nearly 10°, resulting in the opening of the NBC with a ~25% increase in solvent accessibility of the nucleotide.48 In the moderately open actin–profilin structure, the main domains are separated by 3.3°.49 Profilin accelerates nucleotide exchange of MgATP-actin and MgADP-actin by ~30- and 150-folds, respectively,53 by favoring and stabilizing the nucleotide-free conformation, which is likely to be the “true” open conformation of actin. Yet, the rate of ATP binding to this actin (106 M−1 s−1) is slower than expected for a diffusion limited reaction, suggesting that even nucleotide-free actin spends at least part of the time in a closed conformation inaccessible to nucleotide binding.23,54 This observation agrees with the concept that proteins exist in a dynamic equilibrium among various conformational states, with the occupancy of each particular conformation inversely proportional to its free energy. Transition from the open to closed conformation of the NBC in complex of actin with profilin can occur with a very small free energy change,48 suggesting that both conformations can be nearly equally populated also in the presence of profilin.
FIGURE 3.
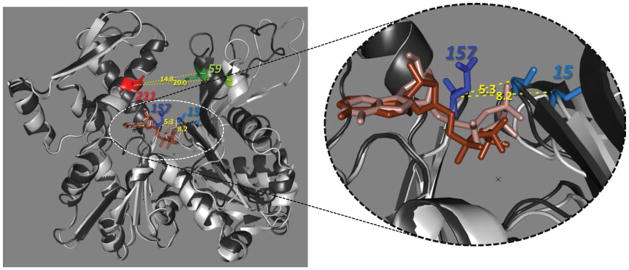
Open and closed cleft conformations of actin. X-ray structures of actin–profilin complexes superimposed in closed (black; PDB:2btf) and open (white; PDB:1hlu) states. The SD3-4 parts of each structure were coaligned to reveal the scissors-like rotation of SD2. Distances between the tips of the phosphate clamping loops P1 an P2 (Cα-atoms of G15 and D157) in both states differ by 2.9 Å (indicated in yellow). Residues Q59 and D211, colored in red and green, respectively, were mutated to cysteines in order to assess conformational transitions in the cleft. Distances between Cα-atoms of these residues are shown in yellow numbers.
Recent progress in molecular and computational modeling has resulted in the publication of several molecular dynamics (MD) simulation studies that reconstructed actin behavior in various nucleotide states. Majority of these works agreed that actin remains in a closed cleft conformation upon transition from the ATP- to ADP-bound state.51,52,55,56 It was also concluded that a removal of profilin from actin in the open conformation results in the cleft closure, irrespective of the nature of the bound nucleotide.55,57
In contrast to that, several lines of evidence from solution biochemistry appear to support the cleft opening upon transition from ATP- to ADP-actin. Thus, higher accessibility of the NBC region to proteolysis,58 higher accessibility of the bound nucleotides to collisional quenchers,59 higher nucleotide release rate,22 lower affinity to DNase I,60 and lower thermal stability30 in the ADP-state were interpreted in favor of this hypothesis. However, later work proposed that none of the methods provided definitive evidence of the NBC opening in ADP-state, suggesting that these approaches may monitor local conformational changes, rather than reporting on the open-closed transitions.61 Thus, while binding of gelsolin segment-1 to actin, or labeling C374 of actin with rhodamine maleimide (TMR-actin), inhibited nucleotide exchange in both ATP and ADP states—in agreement with the proposed cleft closure in both cases47,62—these factors neither affected the accessibility of bound nucleotides to collisional quenching, nor influenced NBC susceptibility to trypsinolysis both in ADP- and ATP-actin.61 These examples raised concerns that even in the case of collective changes observed upon ATP/ADP switch in G-actin, their interpretation in terms of nucleotide cleft closing and opening may not be fully justified.
Mutational Testing of the Open and Closed Cleft States in Actin
In an attempt to overcome the ambiguity of indirect biochemical approaches, we designed S. cerevisiae yeast actin mutants with pairs of cysteine residues located at either side of NBC. Among various combinations explored, the Q59C/D211C mutant with the C374S background (hereafter C59C211 actin) had the mildest influence on yeast cell viability and proliferation (Figures 4A and 4B). C59C211 actin polymerized spontaneously upon addition of 3 mM MgCl2, albeit with a slower rate than wild type yeast actin (data not shown). Rates of nucleotide release from the cleft of the double cysteine mutant were within experimental error of those for WT actin in three nucleotide/divalent cation states—CaATP, MgATP, and MgADP (Figure 4C), suggesting that interactions of nucleotides with the nucleotide binding cleft and the overall conformation of the cleft in mutant actin were not significantly affected by the mutations. Actin with the S60C/D211C substitutions also yielded viable yeast cells and supported their growth and proliferation, but had lower stability in the ADP-state and therefore was not used further.
FIGURE 4.

Crosslinking approach to mapping the nucleotide cleft conformation. Growth of yeast cells with cysteine mutants in the nucleotide binding loop of actin on agar plates (A) and in liquid YPD medium (B). Both single (C59 and C211) and double (C59/C211) cysteine actin mutants supported yeast growth under various conditions, at rates comparable to those of the WT and S374 actin expressing strains. Yeast cells expressing C203 mutant actin, known to have impaired polymerization, were used as a control. C: C59/C211 mutations do not affect nucleotide release rates as compared with WT actin. D: Representative gels of C59/C211 actin crosslinked with MTS reagents in different nucleotide states. The crosslinking was initiated by addition of equimolar amount of MTS reagents to C59C/211 actin in different nucleotide states and was blocked after one minute by adding N-ethylmaleimide. The intramolecularly crosslinked actin (denoted by X) has higher mobility on SDS-page than uncrosslinked actin (denoted by A). E: Percentage of C59/C211 actin crosslinked by MTS reagents. Bars represent standard errors of three independent experiments.
We mapped the distance between cysteines across the NBC in various nucleotide and polymerization states of actin using sulfhydryl-reactive methanethiosulfonate-based cross-linking reagents of various lengths (MTS1-17). The cross-linking rates depended on the length of a crosslinking reagent and a nucleotide state of actin, suggesting that the crosslinking approach is valid and sensitive to the nucleotide dependent conformational transitions in actin. It is important to recognize that shorter span reagents are more rigid and therefore more accurate molecular rulers than the longer but flexible reagents. We found that the long-span crosslinking reagents MTS6 (9.6 Å) and MTS17 (broad range span from 6 to 22 Å) linked cysteine residues across the cleft with similar efficiency, regardless of the nucleotide state of actin (Figures 4D and 4E). In contrast to that, actin crosslinking by intermediate span reagents MTS3 (6.4 Å) and MTS4 (7.8 Å) ranged from higher to lower crosslinking efficiency in the following order: F-actin-phalloidin, MgATP-G-actin, CaATP-G-actin, and MgADP-G-actin. The shortest span crosslinkers MTS1 (5.4 Å) and MTS2 (6.1 Å) did not differentiate between the ATP- and ADP-G-actin, but crosslinked F-actin-phalloidin complex more efficiently (Figures 4D and 4E). These data agree with the cleft being more open in the ADP-state of G-actin, or, in thermodynamic terms, with the open conformation being more frequently occupied in ADP- than in ATP-actin. The data prompts also the speculation that upon actin polymerization and stabilization by phalloidin the nucleotide cleft of the C59C211 mutant actin adopts closed conformation. However, a shorter distance between C59 and C211 does not necessarily impose a closed cleft conformation at the phosphate clamp region in F-actin (Figure 5; discussed below).
FIGURE 5.
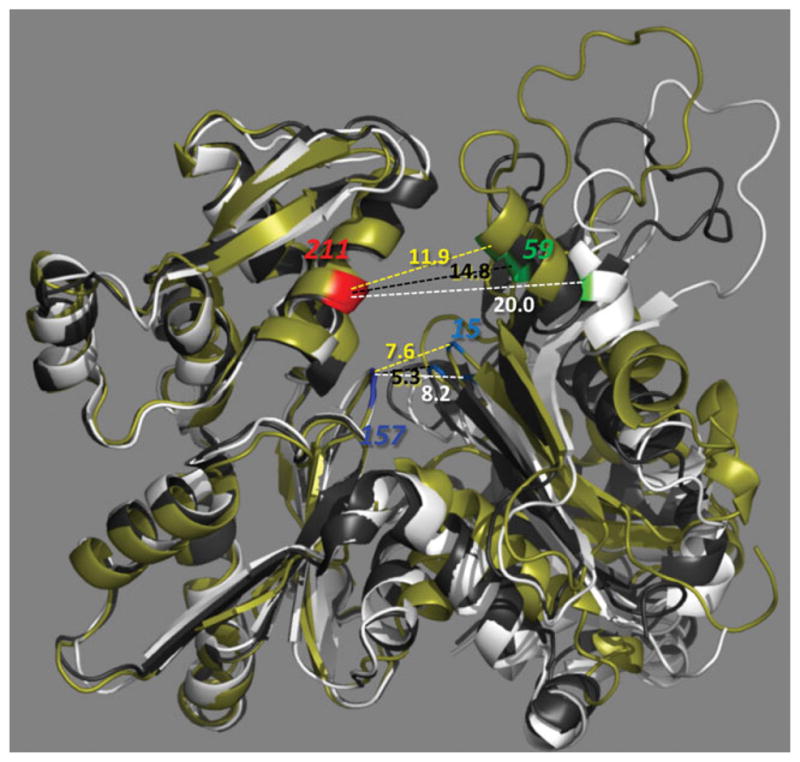
Nucleotide cleft conformation in G- and F-actin. A protomer from a recent high resolution cryo EM reconstructions of F-actin10 (colored in olive) is superimposed on X-ray structures of G-actin in the open and closed states (PDB:1hlu and 2btf; colored in white and black, respectively). Distances between Cα-atoms of G15-D157 and Q59-D211 are indicated in colors of their parent molecules. In all known X-ray structures of G-actin, the closed state of the phosphate clamp (defined as a distance between G15 and D157) correlates with a closed conformtaion at the mouth of the cleft (typically measured between Q59 and E207; Q59-D211 distance is used here to compare with the results of crosslinking experiments in Figure 4). However in F-actin, open phosphate clamp conformation (7.6 Å) may come with a shorter cleft mouth distance (11.9 Å) than the one in the closed state of G-actin (14.8 Å).
In addition to the crosslinking approach, we also attempted electron paramagnetic resonance (EPR) measurements of the distance between paramagnetic nitroxide probes attached to cysteines at positions 59 and 211. Unexpectedly, no coupling was observed between these probes even though the incorporation of two nitroxide probes per actin molecule was confirmed by mass spectrometry. We speculate that paramagnetic coupling between these probes was absent either because the distance between them exceeded the sensitivity range of a conventional steady state EPR approach (8–22 Å), or due to a broad distribution of distances across the cleft.
Open and Closed States of F-Actin
Under physiological salt conditions actin monomers polymerize spontaneously into filaments that can be described as either a single-strand left-handed helix or a double-strand right-handed helix with ~36 nm per pitch or a half-pitch, respectively. Because of the intrinsic plasticity of actin, the pitch span can vary by several nanometers in undecorated actin,63,64 and can be altered by ABPs (e.g., cofilin and drebrin65,66) by up to ~13 nm in cofilin-decorated filaments.64,67 Although it had been known for a long time that actin protomers undergo conformational transitions upon polymerization, the nature of these changes was clarified only recently, when 4–10 Å resolution structures of F-actin resolved via X-ray diffraction of oriented actin crystalline sol12 and electron cryomicroscopy10,11 became available. The G- to F-actin transition involves relative reorientation of all four subdomains,10 but the net effect can be described as a relative rotation of the two major domains by 20° in a propeller-like (or swing-door) manner,12 as opposed to a scissors-like motion upon cleft opening/closing. Consequently, a twisted conformation of G-actin becomes flattened, with the major domains aligned in one plane.12
In various modes of polymorphic F-actin, the nucleotide binding cleft can adopt either closed or open conformations.11 However, the concept of cleft opening might need to be redefined for F-actin. Analysis of data published by Fujii et al.10 suggests that opening of the cleft at the base region (in the phosphate clamp; between residues G15-D157) can be uncoupled from that at the mouth (residues Q59-D211) in F-actin. Figure 5 illustrates that the cleft in F-actin can be clearly open in the phosphate clamp region (7.6 Å vs. 8.2 Å and 5.3 Å in the open and closed states of the profilin-actin complex, respectively), but appears to be in the closed state at the mouth between the Cα-atoms of residues Q59 and D211 (11.9 Å vs. 20.0 Å and 14.8 Å in the open and closed states of G-actin, respectively).
Local Nucleotide-Dependent Conformations of Actin
Immediate Surroundings of the Nucleotide–P1 and Sensor Loops
Among several dozen X-ray structures of G-actin deposited in the Protein Data Bank, four are particularly useful for understanding the effects of ATP hydrolysis on actin structure, as they represent actin crystallized both in the ADP and ATP states and without interacting partners (ABP or a small molecule preventing polymerization). The latter is important as the absence of interaction partners minimizes the risk of interference from external factors. In two of these structures (solved in the ADP-state and the ATP-like AMP-PNP-state), actin polymerization was prevented by labeling the C374 residue with tetramethyrodamine maleimide45,47 (TMR), which intercalates in the hydrophobic patch between SD1 and SD3. In two others (solved in ADP and ATP states) the transition to polymer was blocked by site-directed muta-genesis of two residues that play an important role in a major longitudinal contact between SD3 and SD4 in F-actin (A204E and P243K; AP-actin46). Both ADP state structures of the uncomplexed actins had the cleft tightly closed, prompting the authors to speculate that ADP does not cause the opening of the cleft.45,46
TMR- and AP-actin structures converged on the β-hairpin loop containing S14 (P1-loop) as a direct sensor of ATP hydrolysis. S14 bonds to β-phosphate of ADP, but shifts away to avoid steric clashes with the γ-phosphate of ATP, so its hydroxyl chain is rotated by 130°.46 This shift translates to a conformational transition in the H73 containing loop (a.a. 70–78), named accordingly the sensor loop47 (Figure 1). H73 is methylated in actins of most eukaryotes, but not in yeast actin,68,69 and this posttranslational modification plays a critical role in modulation of Pi release after ATP hydrolysis.18 Thus, it was speculated that the absence of this modification is accountable for the undelayed release of Pi upon polymerization of yeast actin.18,70
Nucleotide dependent conformational rearrangements in the vicinity of the sensor loop were proposed to be translated into structural changes in distant regions of the actin molecule. These, in turn, play critical roles in the modulation of F-actin conformational states and their recognition by ABPs. Among the distant nucleotide-sensitive regions that have been identified are the DNase-binidng loop (D-loop), WH2-binding loop (W-loop), and the N- and C-terminal ends of actin (Figure 1).
DNase-I Binding Loop
Converging on the involvement of the S14-hairpin loop and the sensor loop in sensing conformational changes linked to the hydrolysis of ATP, the analysis of the TMR- and AP-actin led to different conclusions on potential allosteric effects of these loops on peripheral regions of actin.45,46 Thus, conformational perturbations caused by the removal of γ-phosphate dissipated before reaching the D-loop in ADP-AP-actin, but appeared to induce the formation of a short α-helix (residues 40–48) in ADP-TMR-actin45 (Figure 6). It was suggested that the α-helical conformation of D-loop was induced by either crystal contacts or TMR-probe intercalation between SD1 and SD3.46,71 However, the structured D-loop correlates well with ADP-actin being less susceptible to in-solution cleavage with subtilisin between residues 47 and 48 in the D-loop58,72 and this protection cannot be explained by either crystal contacts or labeling actin with TMR.61
FIGURE 6.
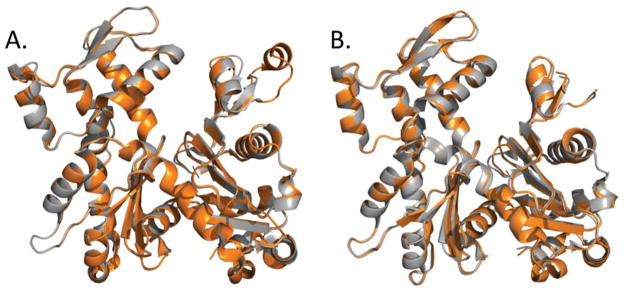
X-ray structures of uncomplexed actin in the ATP- and ADP-states. A: The D-loop of TMR-actin adopts α-helical conformation in the ADP- (orange; PDB:1j6z), but not the ATP-state (gray; PDB: 1nwk), suggesting a possible stabilization of the helix by ADP. B: The D-loop of a polymerization incompetent AP actin mutant (A204E/P243K) is disordered both in ATP- and ADP-bound states (PDB:3el2 and 2hf3; colored in gray and orange, respectively).
Subsequent molecular dynamics simulation studies either supported the loop-to-helix transition52 or found the helix conformation to be highly unstable in both ATP and ADP states.51,55 These two opposite views can be potentially reconciled by a prediction that the helical D-loop of ADP-actin is stabilized mainly by contacts with other protomers in the filament structure.9 This interpretation was supported by coarse grained analysis,73 molecular dynamics,74 and metadynamics56 simulations of F-actin trimers, but was also challenged by MD simulations performed by other authors.16
Recent high-resolution cryo-EM reconstruction of unmodified F-actin in combination with a single particle approach (IHRSR—Iterative Helical Reals Space Reconstruction) revealed six modes of actin filament. In two of these modes the electron density was most consistent with the D-loop adopting a helical conformation; in one of the modes it was consistent with a loop conformation; whereas in two other cases either the D-loop or even the entire SD2 were completely disordered.11 A connection between these various conformations and the nucleotide state of actin was not established, but it has been proposed that a disordered SD2 correlated with the more open conformation of the cleft.11
Recently, we undertook a comprehensive replacement of all residues in the D-loop of S. cerevisiae yeast actin with cysteines and tested the D-loop conformation in various states using fluorescent probe and EPR analysis.75 Among all single cysteine replacements that included residues 40–50, only the M44C actin was lethal for yeast, in agreement with the predicted key role of this residue in the loop-to-helix conformational transitions.56 Neither in G- nor in F-actin, independent of the nucleotide state, were we able to detect periodic changes in the properties of the loop (i.e., spin label mobility, cysteine reactivity, and cysteine conjugated acrylodan emission parameters) that would be predicted for α-helical conformation. In all cases we found an equivalent environment and solvent exposure of D-loop residues, attesting to dynamic structural disorder in the D-loop.75
Our EPR data suggested strongly that most D-loop residues in F-actin coexist in multiple conformations, with various degrees of freedom, separated by low energy barriers.75 This observation is in line with structural polymorphism in F-actin that stems to a large extent from a heterogeneity of the D-loop region.11,76,77 Our experiments con-firmed also that all residues tested in the D-loop (residues 40–50 with the exception of M44) are involved in extensive contacts within F-actin, in agreement with several recent high resolution cryo-EM reconstruction studies.11,18
It is possible that the observed absence of helical periodicity in our experiments is isoform-specific and results from an overall higher conformational plasticity of yeast actin as compared with α-skeletal actin. This property of yeast actin is revealed particularly in a decreased protection of the D-loop in the ADP-state of G-actin from proteolytic cleavage with subtilisin (our unpublished observation). We cannot also exclude completely the possibility that D-loop mutations and/or labeling with fluorescent and EPR probes could destabilize to some extent the conformational stability of this loop.
Recently, the role of D-loop in the activation of ATP hydrolysis upon actin polymerization was proposed based on a high resolution cryo-EM reconstruction of ADP-Pi filament, mutagenesis, and X-ray crystallography experiments.18 According to this hypothesis, a newly incorporated protomer of actin causes flattening of the penultimate protomer and enables interactions of its D-loop with the Pro-rich loop (residues 108–112) of the subunit above it. As a result of this interaction, the Pro-rich loop moves downward, eliminates stacking interactions between P109 and H161, and allows catalytic nucleophilic water molecule to attack the bond between β- and γ- phosphates of ATP, causing ATP hydrolysis.18 The Pro-rich loop together with the sensor loop represents the back door region that controls Pi release78 and a subsequent upward move of the sensor loop enables Pi release by fully opening the back door.18
WH2-Domain Binding Loop (W-Loop)
Based on the analysis of multiple X-ray structures of G-actin, the W-loop (residues 165–172) was identified as one of the six regions on actin surface with a high degree of conformational freedom.16 Regions of high flexibility (intrinsically disordered regions) are common features of protein–protein interaction sites. Accordingly, the W-loop is a prime interaction site for the WH2 domain proteins and also participates in the binding of profilin,48 cofilin and twinfilin,79 vitamin D binding protein,80 and MAL,81 most of which are known to have different affinities for ATP- and ADP-actin. Moreover, the W-loop is part of both longitudinal and lateral interprotomer interfaces in several recent filament models.10,11 A recent MD simulation predicted that the W-loop region adopts a loop conformation in ATP-actin, but forms a β-turn both in ADP-Pi and ADP states due to the formation of backbone hydrogen bonds between Y166 and Y169.52 Similarly, in MD simulations of various F-acitn models W-loop forms either a β-hairpin or a bend.16 The predicted nucleotide-dependent conformational changes in the W-loop were confirmed experimentally by probing this region with point mutagenesis and labeling with fluorescent probes.82 It has been established that the exchange of ATP to ADP caused conformational transitions in the W-loop that can be reversed by addition of inorganic phosphate or ATP.82
Nucleotide-dependent changes in W-loop conformation at the periphery of SD3 originate apparently from structural perturbations in the vicinity of the senor loop. Specifically, residues located at the beginning (G158 and V159) and the end (R177, D179) of a β-hairpin containing the W-loop reorient to accommodate nucleotide-dependent rearrangements of H73 in the sensor loop.18,46,52 ADP-ribosylation of R177 on actin by Salmonella enterica toxin SpvB altered the conformational state of the loop.82 It appears then that this residue, is not only involved in regulation of a back-door mechanism of Pi release in F-actin,78 but also represents a link in the signal transition chain between the nucleotide cleft and the W-loop.
Conformational perturbations in the W-loop caused by point mutagenesis and/or probe attachment can be reciprocally transmitted to the cleft region,82 implicating the W-loop as one of the key elements in the regulation of nucleotide exchange by several ABPs (e.g., profilin, cofilin, gelsolin segment-1). Analysis of recently refined X-ray structures of acto-profilin complexes, both in wide-open and closed state,50 allowed the authors to propose a mechanism of cleft opening by profilin. It was speculated that a 1.0 Å shift in the position of the W-loop, initiated by profilin binding to actin, is propagated in a rigid manner all the way to the cleft and causes 1.0 and 2.9 Å movements of the P1- and P2-loops and cleft opening.50 This long-distance influence originates from strong ionic interaction between R88 on profilin and E167 on actin that displaces T148 and transmits the conformational signal to the cleft.50
The C- and N-Terminal Regions
Both the N and C-termini of actin are among its regions with the highest degree of conformational freedom. The C-terminus is allosterically coupled to the D-loop and the NBC. The rate of tryptic cleavage of the C-terminus, between residues 372 and 373, is affected by both the high affinity cation (Ca2+ vs. Mg2+) and a nucleotide (ATP vs. ADP) bound to NBC.58 Structural basis for this sensitivity was elucidated by detecting a 5 Å shift in the position of the C-terminus in MD simulated transition from the ADP to ATP state.52 Interestingly, the C-terminus of Arp3 is 5 a.a. longer than that of actin and this extension was proposed to act as an “internal profilin” that interacts with the hydrophobic patch region between SD1 and SD3 and promotes open conformation of the nucleotide cleft.51 The structural connection between NBC and C-terminus facilitates apparently the well-established allosteric relationships between the C-terminus and the D-loop.83,84 In polymorphic F-actin, these two regions form one of the alternative contacts between SD1 and SD2 of longitudinally adjacent promoters,11 suggesting that linked conformational transitions in these regions might play a critical role in the nucleotide state- or ABP-moderated cooperativity in F-actin.11,85–87
The N-terminal region of actin is the least conserved part of the molecule that plays an important role in electrostatic interactions between actin and myosin.88–90 All vertebrates have two nonmuscle actin isoforms—β- and γ-cytoplasmic actins. The only difference between them is confined to the N-terminal region: β-actin begins with tri-aspartic acid tripeptide and a valine at position 10 whereas γ-actin has a tri-glutamic N-terminal tripeptide and an isoleucine at position 10. Allosteric interconnection between the N-terminus and the nucleotide binding site of G-actin is not firmly established, but it has been recently found that γ-actin containing Ca2+, but not Mg2+ as the high affinity cation bound in the NBC, exchanges nucleotide ~1.5-folds slower, releases Pi upon polymerization at a two times slower rate, and polymerizes with a significant delay as compared with the β-isoform.91 It was speculated that the difference between the isoforms may be important at the sites with high transient concentrations of calcium, such as the γ-actin-rich cochlear hair cell stereocilium.91 In F-actin, three different conformations of the N-terminus were observed, which correlated with particular conformations of the D-loop.11 However, it remains to be established whether these correlations are regulated via intramolecular or interprotomer allosteric interactions. Some of these questions may be pursued with C1 yeast actin mutant, which appeared to have unchanged polymerization and myosin binding properties.92
CONCLUSIONS
Intriguingly, all of the above considered structural elements of actin, its clefts and loops that sense and/or are impacted by the nucleotide state of this protein, are also sites of docking or attachment of many actin binding proteins. This raises the possibility that for different ABPs their recognition of ATP and ADP states of actin may be due to different combinations of these nucleotide sensing elements.
Acknowledgments
Contract grant sponsor: USPHS
Contract grant number: R01 GM077190
The authors would like to thank Dr. Christian Altenbach for his generous help in conducting and analyzing the EPR experiments.
Footnotes
Heini was an ideal mentor; inspiring, supportive, offering ideas, opening new vistas on how to solve challenging problems, and yet encouraging full independence and self-reliance of his students and postdoctoral fellows. He created an environment of intellectual ambition and curiosity and true excitement about learning how biomolecules function. The informal and supportive environment included also his family—his wife Nutzi and children Shai and Danny—as well as his students and postdocs families. Heini’s support and friendship were a gift and asset that has accompanied me (E.R.) in all the years after I left his group. His legacy is in the outstanding publications and the scientists whose careers he helped to launch.
Dedication: We dedicate this review to the memory of Henryk (Heini) Eisenberg, a truly outstanding scientist, with exceptional ability to develop and apply rigorous thermodynamic and physical approaches to the analysis of biological macromolecules and their interactions. One of us (E.R.) was fortunate to have been introduced as his graduate student to the fascinating world of self-assembly of biomolecules, specifically glutamic dehydrogenase. Heini’s own interest in the dynamic equilibria states of this enzyme and their regulation by nucleotides was stimulated during his sabbatical at NIH and the collaboration with Gordon M. Tomkins. With his creative mastery of thermodynamics and light scattering theories and the ability to model and predict the link between the measured parameters and the arrangement of the enzyme’s subunits, Heini was ahead of his time in mapping protein substructures in the absence of crystallography data.
References
- 1.Straub FB. Studies Inst Med Chem Univ Szeged. 1942;2:3–15. [Google Scholar]
- 2.Straub FB. Studies Inst Med Chem Univ Szeged. 1943;3:23–37. [Google Scholar]
- 3.Szent-György A. Acta Physiol Scand. 1945;9:25. [Google Scholar]
- 4.Pollard TD, Shelton E, Weihing RR, Korn ED. J Mol Biol. 1970;50:91–97. doi: 10.1016/0022-2836(70)90106-3. [DOI] [PubMed] [Google Scholar]
- 5.Lazarides E, Weber K. Proc Natl Acad Sci USA. 1974;71:2268–2272. doi: 10.1073/pnas.71.6.2268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tilney LG. Soc Gen Physiologists Series. 1975;30:339–388. [PubMed] [Google Scholar]
- 7.Kabsch W, Mannherz HG, Suck D, Pai EF, Holmes KC. Nature. 1990;347:37–44. doi: 10.1038/347037a0. [DOI] [PubMed] [Google Scholar]
- 8.Holmes KC, Popp D, Gebhard W, Kabsch W. Nature. 1990;347:44–49. doi: 10.1038/347044a0. [DOI] [PubMed] [Google Scholar]
- 9.Dominguez R. Trends Biochem Sci. 2004;29:572–578. doi: 10.1016/j.tibs.2004.09.004. [DOI] [PubMed] [Google Scholar]
- 10.Fujii T, Iwane AH, Yanagida T, Namba K. Nature. 2010;467:724–728. doi: 10.1038/nature09372. [DOI] [PubMed] [Google Scholar]
- 11.Galkin VE, Orlova A, Schroder GF, Egelman EH. Nat Struct Mol Biol. 2010;17:1318–1323. doi: 10.1038/nsmb.1930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Oda T, Iwasa M, Aihara T, Maeda Y, Narita A. Nature. 2009;457:441–445. doi: 10.1038/nature07685. [DOI] [PubMed] [Google Scholar]
- 13.Shvetsov A, Galkin VE, Orlova A, Phillips M, Bergeron SE, Rubenstein PA, Egelman EH, Reisler E. J Mol Biol. 2008;375:793–801. doi: 10.1016/j.jmb.2007.10.076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Scoville D, Stamm JD, Toledo-Warshaviak D, Altenbach C, Phillips M, Shvetsov A, Rubenstein PA, Hubbell WL, Reisler E. Biochemistry. 2006;45:13576–13584. doi: 10.1021/bi061229f. [DOI] [PubMed] [Google Scholar]
- 15.Shvetsov A, Musib R, Phillips M, Rubenstein PA, Reisler E. Biochemistry. 2002;41:10787–10793. doi: 10.1021/bi020205f. [DOI] [PubMed] [Google Scholar]
- 16.Splettstoesser T, Holmes KC, Noe F, Smith JC. Proteins. 2011;79:2033–2043. doi: 10.1002/prot.23017. [DOI] [PubMed] [Google Scholar]
- 17.Carlier MF, Pantaloni D, Korn ED. J Biol Chem. 1986;261:10778–10784. [PubMed] [Google Scholar]
- 18.Murakami K, Yasunaga T, Noguchi TQ, Gomibuchi Y, Ngo KX, Uyeda TQ, Wakabayashi T. Cell. 2010;143:275–287. doi: 10.1016/j.cell.2010.09.034. [DOI] [PubMed] [Google Scholar]
- 19.Gershman LC, Selden LA, Kinosian HJ, Estes JE. Adv Exp Med Biol. 1994;358:35–49. doi: 10.1007/978-1-4615-2578-3_4. [DOI] [PubMed] [Google Scholar]
- 20.Estes JE, Selden LA, Kinosian HJ, Gershman LC. J Muscle Res Cell Motility. 1992;13:272–284. doi: 10.1007/BF01766455. [DOI] [PubMed] [Google Scholar]
- 21.Pardee JD, Spudich JA. Methods Enzymol. 1982;85(Part B):164–181. doi: 10.1016/0076-6879(82)85020-9. [DOI] [PubMed] [Google Scholar]
- 22.Kinosian HJ, Selden LA, Estes JE, Gershman LC. J Biol Chem. 1993;268:8683–8691. [PubMed] [Google Scholar]
- 23.Blanchoin L, Pollard TD. Biochemistry. 2002;41:597–602. doi: 10.1021/bi011214b. [DOI] [PubMed] [Google Scholar]
- 24.Romero S, Didry D, Larquet E, Boisset N, Pantaloni D, Carlier MF. J Biol Chem. 2007;282:8435–8445. doi: 10.1074/jbc.M609886200. [DOI] [PubMed] [Google Scholar]
- 25.Kaur H, Kumar S, Singh K, Bharadwaj LM. Int J Biol Macromol. 2011;48:793–797. doi: 10.1016/j.ijbiomac.2011.03.004. [DOI] [PubMed] [Google Scholar]
- 26.DalleDonne I, Milzani A, Colombo R. Arch Biochem Biophys. 1998;351:141–148. doi: 10.1006/abbi.1997.0545. [DOI] [PubMed] [Google Scholar]
- 27.Straub FB, Feuer G. Kiserletes Orvostudomany. 1950;2:141–151. [PubMed] [Google Scholar]
- 28.Laki K, Bowen WJ, Clark A. J Gen Physiol. 1950;33:437–443. doi: 10.1085/jgp.33.5.437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Belmont LD, Orlova A, Drubin DG, Egelman EH. Proc Natl Acad Sci USA. 1999;96:29–34. doi: 10.1073/pnas.96.1.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Levitsky DI, Pivovarova AV, Mikhailova VV, Nikolaeva OP. FEBS J. 2008;275:4280–4295. doi: 10.1111/j.1742-4658.2008.06569.x. [DOI] [PubMed] [Google Scholar]
- 31.Isambert H, Venier P, Maggs AC, Fattoum A, Kassab R, Pantaloni D, Carlier MF. J Biol Chem. 1995;270:11437–11444. doi: 10.1074/jbc.270.19.11437. [DOI] [PubMed] [Google Scholar]
- 32.Bugyi B, Carlier MF. Ann Rev Biophys. 2010;39:449–470. doi: 10.1146/annurev-biophys-051309-103849. [DOI] [PubMed] [Google Scholar]
- 33.Belmont LD, Drubin DG. J Cell Biol. 1998;142:1289–1299. doi: 10.1083/jcb.142.5.1289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Daniel JL, Molish IR, Robkin L, Holmsen H. Eur J Biochem/FEBS. 1986;156:677–684. doi: 10.1111/j.1432-1033.1986.tb09631.x. [DOI] [PubMed] [Google Scholar]
- 35.Bernstein BW, Bamburg JR. J Neurosci. 2003;23:1–6. doi: 10.1523/JNEUROSCI.23-01-00002.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cooke R. Biochemistry. 1975;14:3250–3256. doi: 10.1021/bi00685a035. [DOI] [PubMed] [Google Scholar]
- 37.Cooke R. J Supramol Struct. 1975;3:146–153. doi: 10.1002/jss.400030207. [DOI] [PubMed] [Google Scholar]
- 38.Kinosian HJ, Selden LA, Gershman LC, Estes JE. Biochemistry. 2004;43:6253–6260. doi: 10.1021/bi036117s. [DOI] [PubMed] [Google Scholar]
- 39.De La Cruz EM, Mandinova A, Steinmetz MO, Stoffler D, Aebi U, Pollard TD. J Mol Biol. 2000;295:517–526. doi: 10.1006/jmbi.1999.3390. [DOI] [PubMed] [Google Scholar]
- 40.Kasai M, Nakano E, Oosawa F. Biochimica et biophysica acta. 1965;94:494–503. doi: 10.1016/0926-6585(65)90058-0. [DOI] [PubMed] [Google Scholar]
- 41.Frederick KB, Sept D, De La Cruz EM. J Mol Biol. 2008;378:540–550. doi: 10.1016/j.jmb.2008.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bork P, Sander C, Valencia A. Proc Natl Acad Sci USA. 1992;89:7290–7294. doi: 10.1073/pnas.89.16.7290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nolen BJ, Littlefield RS, Pollard TD. Proc Natl Acad Sci USA. 2004;101:15627–15632. doi: 10.1073/pnas.0407149101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.van den Ent F, Moller-Jensen J, Amos LA, Gerdes K, Lowe J. EMBO J. 2002;21:6935–6943. doi: 10.1093/emboj/cdf672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Otterbein LR, Graceffa P, Dominguez R. Science. 2001;293:708–711. doi: 10.1126/science.1059700. [DOI] [PubMed] [Google Scholar]
- 46.Rould MA, Wan Q, Joel PB, Lowey S, Trybus KM. J Biol Chem. 2006;281:31909–31919. doi: 10.1074/jbc.M601973200. [DOI] [PubMed] [Google Scholar]
- 47.Graceffa P, Dominguez R. J Biol Chem. 2003;278:34172–34180. doi: 10.1074/jbc.M303689200. [DOI] [PubMed] [Google Scholar]
- 48.Chik JK, Lindberg U, Schutt CE. J Mol Biol. 1996;263:607–623. doi: 10.1006/jmbi.1996.0602. [DOI] [PubMed] [Google Scholar]
- 49.Baek K, Liu X, Ferron F, Shu S, Korn ED, Dominguez R. Proc Natl Acad Sci USA. 2008;105:11748–11753. doi: 10.1073/pnas.0805852105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Porta JC, Borgstahl GE. J Mol Biol. 2012;418:103–116. doi: 10.1016/j.jmb.2012.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dalhaimer P, Pollard TD, Nolen BJ. J Mol Biol. 2008;376:166–183. doi: 10.1016/j.jmb.2007.11.068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zheng X, Diraviyam K, Sept D. Biophys J. 2007;93:1277–1283. doi: 10.1529/biophysj.107.109215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Selden LA, Kinosian HJ, Estes JE, Gershman LC. Biochemistry. 1999;38:2769–2778. doi: 10.1021/bi981543c. [DOI] [PubMed] [Google Scholar]
- 54.Kinosian HJ, Selden LA, Gershman LC, Estes JE. Biochemistry. 2000;39:13176–13188. doi: 10.1021/bi001520+. [DOI] [PubMed] [Google Scholar]
- 55.Splettstoesser T, Noe F, Oda T, Smith JC. Proteins. 2009;76:353–364. doi: 10.1002/prot.22350. [DOI] [PubMed] [Google Scholar]
- 56.Pfaendtner J, Branduardi D, Parrinello M, Pollard TD, Voth GA. Proc Natl Acad Sci USA. 2009;106:12723–12728. doi: 10.1073/pnas.0902092106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Minehardt TJ, Kollman PA, Cooke R, Pate E. Biophys J. 2006;90:2445–2449. doi: 10.1529/biophysj.105.072900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Strzelecka-Golaszewska H, Moraczewska J, Khaitlina SY, Mossakowska M. Eur J Biochem/FEBS. 1993;211:731–742. doi: 10.1111/j.1432-1033.1993.tb17603.x. [DOI] [PubMed] [Google Scholar]
- 59.Root DD, Reisler E. Protein Sci. 1992;1:1014–1022. doi: 10.1002/pro.5560010807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Schuler H, Schutt CE, Lindberg U, Karlsson R. FEBS Lett. 2000;476:155–159. doi: 10.1016/s0014-5793(00)01717-8. [DOI] [PubMed] [Google Scholar]
- 61.Kudryashov DS, Reisler E. Biophys J. 2003;85:2466–2475. doi: 10.1016/s0006-3495(03)74669-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bryan J. J Cell Biol. 1988;106:1553–1562. doi: 10.1083/jcb.106.5.1553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Galkin VE, VanLoock MS, Orlova A, Egelman EH. Current Biol CB. 2002;12:570–575. doi: 10.1016/s0960-9822(02)00742-x. [DOI] [PubMed] [Google Scholar]
- 64.Galkin VE, Orlova A, Lukoyanova N, Wriggers W, Egelman EH. J Cell Biol. 2001;153:75–86. doi: 10.1083/jcb.153.1.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Sharma S, Grintsevich EE, Phillips ML, Reisler E, Gimzewski JK. Nano Lett. 2011;11:825–827. doi: 10.1021/nl104159v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Grintsevich EE, Galkin VE, Orlova A, Ytterberg AJ, Mikati MM, Kudryashov DS, Loo JA, Egelman EH, Reisler E. J Mol Biol. 2010;398:542–554. doi: 10.1016/j.jmb.2010.03.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.McGough A, Pope B, Chiu W, Weeds A. J Cell Biol. 1997;138:771–781. doi: 10.1083/jcb.138.4.771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kalhor HR, Niewmierzycka A, Faull KF, Yao X, Grade S, Clarke S, Rubenstein PA. Arch Biochem Biophys. 1999;370:105–111. doi: 10.1006/abbi.1999.1370. [DOI] [PubMed] [Google Scholar]
- 69.Yao X, Grade S, Wriggers W, Rubenstein PA. J Biol Chem. 1999;274:37443–37449. doi: 10.1074/jbc.274.52.37443. [DOI] [PubMed] [Google Scholar]
- 70.Nyman T, Schuler H, Korenbaum E, Schutt CE, Karlsson R, Lindberg U. J Mol Biol. 2002;317:577–589. doi: 10.1006/jmbi.2002.5436. [DOI] [PubMed] [Google Scholar]
- 71.Sablin EP, Dawson JF, VanLoock MS, Spudich JA, Egelman EH, Fletterick RJ. Proc Natl Acad Sci USA. 2002;99:10945–10947. doi: 10.1073/pnas.152329899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Schwyter D, Phillips M, Reisler E. Biochemistry. 1989;28:5889–5895. doi: 10.1021/bi00440a027. [DOI] [PubMed] [Google Scholar]
- 73.Chu JW, Voth GA. Proc Natl Acad Sci USA. 2005;102:13111–13116. doi: 10.1073/pnas.0503732102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Pfaendtner J, Lyman E, Pollard TD, Voth GA. J Mol Biol. 2010;396:252–263. doi: 10.1016/j.jmb.2009.11.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Oztug Durer ZA, Kudryashov DS, Sawaya MR, Altenbach C, Hubbell W, Reisler E. Biophys J. 2012;103:930–939. doi: 10.1016/j.bpj.2012.07.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Oda T, Maeda Y. Structure. 2010;18:761–767. doi: 10.1016/j.str.2010.05.009. [DOI] [PubMed] [Google Scholar]
- 77.Saunders MG, Voth GA. Structure. 2012;20:641–653. doi: 10.1016/j.str.2012.02.008. [DOI] [PubMed] [Google Scholar]
- 78.Wriggers W, Schulten K. Proteins. 1999;35:262–273. [PubMed] [Google Scholar]
- 79.Paavilainen VO, Oksanen E, Goldman A, Lappalainen P. J Cell Biol. 2008;182:51–59. doi: 10.1083/jcb.200803100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Otterbein LR, Cosio C, Graceffa P, Dominguez R. Proc Natl Acad Sci USA. 2002;99:8003–8008. doi: 10.1073/pnas.122126299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Mouilleron S, Guettler S, Langer CA, Treisman R, McDonald NQ. EMBO J. 2008;27:3198–3208. doi: 10.1038/emboj.2008.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Kudryashov DS, Grintsevich EE, Rubenstein PA, Reisler E. J Biol Chem. 2010;285:25591–25601. doi: 10.1074/jbc.M110.123869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Strzelecka-Golaszewska H, Mossakowska M, Wozniak A, Moraczewska J, Nakayama H. Biochem J. 1995;307(Part 2):527–534. doi: 10.1042/bj3070527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Mossakowska M, Moraczewska J, Khaitlina S, Strzelecka-Golaszewska H. Biochem J. 1993;289(Part 3):897–902. doi: 10.1042/bj2890897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Bugyi B, Papp G, Hild G, Lorinczy D, Nevalainen EM, Lappalainen P, Somogyi B, Nyitrai M. J Biol Chem. 2006;281:10727–10736. doi: 10.1074/jbc.M510252200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Orlova A, Prochniewicz E, Egelman EH. J Mol Biol. 1995;245:598–607. doi: 10.1006/jmbi.1994.0049. [DOI] [PubMed] [Google Scholar]
- 87.Papp G, Bugyi B, Ujfalusi Z, Barko S, Hild G, Somogyi B, Nyitrai M. Biophys J. 2006;91:2564–2572. doi: 10.1529/biophysj.106.087775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Miller CJ, Wong WW, Bobkova E, Rubenstein PA, Reisler E. Biochemistry. 1996;35:16557–16565. doi: 10.1021/bi962388+. [DOI] [PubMed] [Google Scholar]
- 89.Bertrand R, Chaussepied P, Audemard E, Kassab R. Eur J Biochem/FEBS. 1989;181:747–754. doi: 10.1111/j.1432-1033.1989.tb14787.x. [DOI] [PubMed] [Google Scholar]
- 90.Abe A, Saeki K, Yasunaga T, Wakabayashi T. Biochem Biophys Res Commun. 2000;268:14–19. doi: 10.1006/bbrc.1999.2069. [DOI] [PubMed] [Google Scholar]
- 91.Bergeron SE, Zhu M, Thiem SM, Friderici KH, Rubenstein PA. J Biol Chem. 2010;285:16087–16095. doi: 10.1074/jbc.M110.110130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Hansen JE, Marner J, Pavlov D, Rubenstein PA, Reisler E. Biochemistry. 2000;39:1792–1799. doi: 10.1021/bi991873c. [DOI] [PubMed] [Google Scholar]