

Abstract
Platelets and the lungs have an intimate relationship. Platelets are anucleate mammalian blood cells that continuously circulate through pulmonary vessels and that have major effector activities in hemostasis and inflammation. The lungs are reservoirs for megakaryocytes, the requisite precursor cell in thrombopoiesis, which is the intricate process by which platelets are generated. Platelets contribute to basal barrier integrity of the alveolar capillaries, which selectively restricts the transfer of water, proteins, and red blood cells out of the vessels. Platelets also contribute to pulmonary vascular repair. Although platelets bolster hemostatic and inflammatory defense of the healthy lung, experimental evidence and clinical evidence indicate that these blood cells are effectors of injury in a variety of pulmonary disorders and syndromes. Newly discovered biological capacities of platelets are being explored in the context of lung defense, disease, and remodeling.
Keywords: megakaryocytes, thrombopoiesis, alveolus, endothelial cells, barrier function, permeability, inflammation, hemorrhage, acute lung injury, ALI, acute respiratory distress syndrome, ARDS
INTRODUCTION
Platelets are specialized anucleate blood cells that are unique to mammals (1). As with all blood cells, platelets have an intimate relationship with the lungs. Platelets transit the pulmonary vessels and are present in alveolar capillaries together with erythrocytes and leukocytes. Megakaryocytes—the parent cells that spawn platelets—are found in the mammalian lungs. There is evidence that the lungs are sites of thrombopoiesis, the intricate process of platelet production, and that the lungs of humans and nonhuman mammals are reservoirs for platelets and release them in response to certain stimuli. There is also evidence that platelets regulate pulmonary vascular permeability and the barrier function of the alveolar capillaries and influence pulmonary vasoreactivity. Platelets likely have specialized activities in lung repair. Recent experimental models and clinical observations indicate that platelets are key in pulmonary defenses and integrity but that these cells can also be effectors of injury in lung diseases. Interest in platelet activities in pulmonary pathology has been particularly keen in studies of acute lung injury (ALI) and the acute respiratory distress syndrome (ARDS) (see sidebar on ARDS), but platelets also contribute to the pathophysiology of a variety of other lung disorders and to systemic syndromes that involve the lungs.
Several reviews catalog a wealth of observations relevant to platelets and the lungs (2–12). Here we focus on some of these issues and draw on both experimental and clinical inquiries.
PLATELET BIOLOGY, OLD AND NEW: A SYNOPSIS
Platelets are highly specialized effector cells in physiological hemostasis and are critical in pathological thrombosis. The primary-hemostatic activities of platelets, recognized for more than a century, have been recently considered in a comprehensive fashion (1) and cannot be discussed in detail here because of space limitations. Platelets are also major inflammatory cells with activities that span the immune continuum (4, 13–15). The inflammatory activities of platelets contribute to lung defense but, in addition, mediate damage in ALI/ARDS and other clinical and experimental syndromes of lung inflammation (6–8, 12). Molecular mechanisms that link the interplay of hemostatic and inflammatory activities of platelets in physiology and disease were recently reviewed (7, 12, 15).
ACUTE RESPIRATORY DISTRESS SYNDROME.
Acute respiratory distress syndrome (ARDS) is a common and lethal form of acute respiratory failure. In humans, acute lung injury (ALI)/ARDS is a spectrum based on clinical, physiological, and radiographic diagnostic criteria and indices of severity. ALI/ARDS can be modeled in mice and other experimental animals. ALI/ARDS involves injury to the alveolar capillary endothelium and epithelium, resulting in barrier disruption and increased-permeability pulmonary edema, with consequent severe hypoxemia and other pathophysiological sequelae. Inflammatory injury to the alveolar endothelium and epithelium, deposition of fibrin and other proteins in vessels and on the epithelial surface, and dysregulated repair of alveolar cells and structures are important pathogenetic mechanisms in ALI/ARDS. Experimental and clinical evidence indicates that activated platelets contribute to initiation and/or amplification of alveolar damage in ALI/ARDS and that alterations in platelet number and function influence the natural history of this syndrome.
Newly discovered and previously unrecognized facets of platelet biology have emerged from recent studies. Such discoveries include evidence for a remarkably rich transcriptome that includes pre-mRNAs, mRNAs, and microRNAs; diverse pathways for processing RNA transcripts; specialized mechanisms of translation; and the capacity to synthesize new proteins and alter the constitutive platelet proteome in response to activating signals (15–18). The platelet genome is now being extensively mined in basic and clinical investigations (17, 19, 20) and is being utilized as a tool for molecular characterization of rare platelet phenotypes and diseases of platelet dysfunction (21). Anucleate platelets and previously uncharacterized circulating precursor cells can, surprisingly, generate functional daughter progeny (22, 23). Platelets can signal and induce functional cellular responses outside of the vasculature by releasing soluble factors and microparticles in addition to directly interacting with leukocytes, erythrocytes, and endothelial cells (reviewed in References 12 and 15). Most of these newly identified activities of platelets were revealed by studies of human cells, establishing immediate translational relevance, but have also been demonstrated in mouse models. Other examples of the new biology of platelets are likely to be discovered (16).
PULMONARY MEGAKARYOCYTES AND THE LUNG AS AN ORGAN OF THROMBOPOIESIS
The megakaryocyte is the central cell in thrombopoiesis, the intricately regulated series of events by which platelets are generated. Megakaryocytes are unique, polyploid hematopoietic cells that are found only in mammals and are specialized to produce and release platelets and immediate platelet precursors into the circulation (reviewed in Reference 24). The global process of thrombopoiesis includes megakaryocyte precursor development from stem cells and cell fate determination; the remarkable events of endomitosis and the development of polyploidy; internal membrane, tubular, cytoskeletal, and granule evolution directed by key transcription factors and transcriptional programs; regulated apoptosis; and additional intricate and time-dependent events (24). Cytokines regulate megakaryocyte development and platelet formation at multiple steps. Megakaryopoiesis is driven by thrombopoietin (TPO), although TPO and other cytokines critical for megakaryocyte development are not required for the late stages of thrombopoiesis in vitro (24).
There is considerable evidence that the late steps in platelet production occur by the formation, by megakaryocytes, of elongate processes termed proplatelets (although this mechanism is not uniformly agreed upon) (Figure 1). Proplatelets form in vivo, and megakaryopoiesis and proplatelet extension can be modeled and observed in vitro (reviewed in Reference 24). The molecular events involved in the release of individual platelets from proplatelets are complex and not completely worked out (24). Shear enhances platelet release in vitro (23, 25) and appears to contribute to platelet release from proplatelet tips in marrow sinusoids in vivo on the basis of intravital microscopic studies of mice (26). Platelet generation by megakaryocytes, by proplatelets that have separated from the megakaryocyte cell body, or by novel precursor cells may occur in the bloodstream (22–24) (see below). Although physicians and basic physiologists know the bone marrow to be the traditional site of platelet production, in vitro modeling demonstrates that central events in thrombopoiesis are directed by cell-autonomous mechanisms and do not require the specific bone marrow environment (24). Many studies indicate that the lung is a site of platelet biogenesis. A central feature suggesting this is that megakaryocytes are found in vessels in the lungs of nonhuman mammals and human subjects.
Figure 1.

Thrombopoiesis. (a) Cellular intermediates in thrombopoiesis. See text and Reference 24 for further details. (b) Photomicrographs of human cells involved in thrombopoiesis. The photographs are from different experiments and at different magnifications and do not precisely illustrate relative cellular sizes. (Left to right) CD34+ hematopoietic stem cell (HSC) precursors stained for integrin subunit αIIb; CD34+HSC-derived megakaryocyte stained for aIIb and spliceosome factors; CD34+HSC-derived megakaryocyte extending proplatelets, stained for mammalian target of rapamycin; freshly isolated platelets—both elongate platelets undergoing fission and resting discoid platelets—from venous blood stained for actin. See References 22, 137, and 138 for further details. Reprinted with permission from References 22, 137, and 138. (c) Electron micrographs of human platelets undergoing fission. See Reference 22 for further details. Reprinted with permission from Reference 22.
The first description of intravascular megakaryocytes in the lung is attributed to Aschoff (discussed in References 24, 27, and 28). Fueled by Aschoff’s observations, studies of experimental mammals and in humans have consistently reported the presence of megakaryocytes in lung vessels, although the numbers of lung megakaryocytes and their significance to thrombopoiesis in health and disease have been controversial. An early and often-cited study provided observations largely from anesthetized cats but also from dogs and utilized blood sampling and histological examination of lungs from these animals. Giant cells, interpreted to be megakaryocytes, were found in lung sections from all animals studied, in variable numbers that were interpreted to be due to sampling differences and individual variation (27). The investigators further noted that the morphology of the intravascular giant cells in the lung is somewhat different from that of megakaryocytes in the marrow or spleen. The researchers also pointed out that the cellular morphology included nuclei with attached cytoplasmic processes that were identified in serial sections in addition to apparently naked intravascular nuclei. On the basis of these and other observations, Howell & Donahue (27) challenged an idea in the field that held that megakaryocytes lose their cytoplasm as a result of platelet formation in the marrow and that naked giant nuclei then somehow exit the marrow and accumulate in lung vessels. Countering this idea, they proposed that megakaryocytes with retained cytoplasm are frequently present in the pulmonary circulation and may be in a state of active platelet production (27).
Subsequent studies indicated that megakaryocytes are present in the lungs of rabbits, rats, and mice (28–31; reviewed in References 24 and 30). Camera lucida drawings in early reports of lung megakaryocytes included cells with cytoplasmic extensions consistent with proplatelets (27, 28). Histological surveys reported that 6–18 megakaryocytes cm−2 can be detected in paraffin-embedded rabbit lung sections, with an approximately three- to sixfold increase in lungs from animals perturbed to become thrombocytopenic (reviewed in Reference 30). Nevertheless, an analysis of mouse lungs resulted in the conclusion that megakaryocytes, including naked nuclei and large cytoplasmic fragments, are rare in pulmonary blood vessels of animals of the C57BL/6 background both under basal conditions and when the animals are made thrombocytopenic with platelet antiserum, a chemotherapeutic agent, or with radiation (30). This interpretation was based on immunostaining (polyclonal antibodies against the integrin β3 subunit or whole platelet membranes) or hematoxylin and eosin staining of plastic-embedded tissue, combined with light microscopy. More recently, an ultrastructural study reported that intact megakaryocytes, bare nuclei, and cytoplasmic fragments were present in the lungs of unperturbed CB6FL/J mice under basal conditions and when thrombocytosis was induced by phlebotomy or treatment with TPO (31). Megakaryocyte nuclei almost devoid of cytoplasm and with a morphological appearance suggestive of apoptosis were present in pulmonary capillaries of animals with accelerated thrombopoiesis but were more difficult to identify in control mice with basal platelet numbers. These studies (30, 31) suggest that detection of megakaryocytes in murine lungs is influenced by the physiological state of the animal and perhaps by specific microscopic detection methods (30) and other technical variables. Staining with antibodies against determinants that are highly selective or specific for megakaryocytes and platelets, such as integrin subunit αIIb (GPIIb; CD41), GPIb, or GPVI (1), was not done in these studies. A parallel point is that there are quantitative differences in sizes and numbers of megakaryocytes and platelets in mice and humans (32).
Megakaryocytes have also been routinely identified in examinations of human lungs in diagnostic or forensic necropsies, and their numbers are increased in ALI/ARDS, burns, disseminated intravascular coagulation, and thrombosis (reviewed in References 7, 24, 30, and 33). Furthermore, megakaryocytes are routinely detected in diagnostic lung biopsies (7, 33). In addition, examination of selectively sampled vascular compartments consistently demonstrates circulating megakaryocytes in peripheral and central venous blood of humans and indicates that these cells are ~10-fold more abundant in pulmonary artery blood compared with aortic blood (34–39). The subjects in some of these reports were largely patients undergoing diagnostic procedures, with fewer completely healthy individuals included if central blood sampling was done. In one study, megakaryocytes were found in right atrial or high vena caval blood in each of 23 subjects undergoing diagnostic cardiac catheterization. Approximately half of the megakaryocytes had an intact cytoplasm (35). Microscopic images of blood megakaryocytes from some of these reports included cells with cytoplasmic extensions that suggested proplatelets (35, 37, 38). Data from these studies indicate that approximately 90% of intact megakaryocytes in pulmonary artery blood are retained in the lung microvasculature and that many fewer transit the lungs and reach the arterial circulation. These estimates are based on cell counts in blood from the right and left sides of the lungs (39; reviewed in Reference 30), although in one study of patients undergoing central venous and femoral arterial catheterization, as many as 21–56% of the megakaryocytes in caval blood were thought to pass through the pulmonary vessels and to recirculate (37). The number of circulating megakaryocytes was reported to be elevated in the peripheral blood of patients with inflammatory lung conditions; this was consistent with histological examination of lungs from patients with pulmonary inflammation in autopsy studies (40). A fascinating possibility suggested by greater numbers of megakaryocytes in blood on the right side of the pulmonary vascular bed is that increased delivery of megakaryocytes to the distal extremities via arterial blood is a mechanistic contributor to hypertrophic pulmonary osteoarthropathy associated with intracardiac or intrapulmonary shunts (reviewed in Reference 31).
A common interpretation from these and related clinical and experimental observations, and one originally proposed by Aschoff, is that megakaryocytes originate in the marrow, are carried to the lungs in venous blood, are trapped in pulmonary microvessels—the first capillary bed to be encountered and the one that receives the entire circulating blood volume under normal conditions—and are lodged there because of their size (31, 37, 39, 41) (Figures 2 and 3). In a particularly informative study, megakaryocytes elutriated from pulmonary artery blood of subjects undergoing cardiac catheterization ranged in diameter from 16 to 88 μm, with a mean of 38.7 μm, and were further characterized on the basis of cellular features (e.g., fibrinogen in storage granules as expected for megakaryocytes, expression of integrin αIIbβ3) and apparent maturity (39). This size range predicts retention in the lung vessels. The pulmonary capillary bed slows the transit time of leukocytes and dynamically retains polymorphonuclear leukocytes (PMNs; also known as neutrophils), which are considerably smaller than megakaryocytes (reviewed in Reference 11; see also the next section) (Figure 2). Thus, the retention of mature megakaryocytes in pulmonary microvessels because of their diameters is expected (Figure 2). The nuclear size and complexity and, potentially, cytoskeletal rigidity of megakaryocytes (24) may also limit their deformability and contribute to retention in the pulmonary circulation (Figure 3). In an experiment in dogs, sequential perfusion of first the right lung and then the left lung resulted in the accumulation of most of the blood megakaryocytes in the right lung, suggesting trapping or filtration of megakaryocytes by pulmonary vessels (42). Differential blood sampling in humans undergoing cardiopulmonary bypass also supported the interpretation that megakaryocytes are retained in pulmonary vessels and indicated that the lungs selectively remove large megakaryocytes (43). Although histological observations demonstrate that pulmonary capillaries are common sites of megakaryocyte accumulation in the lung, the diameters of megakaryocytes in central venous blood (39) are larger than those of many pulmonary arterioles. Consistent with this finding, histological studies also show megakaryocytes in larger vessels and in capillaries. Although physical trapping of megakaryocytes in lung microvessels is likely a major mechanism of accumulation, surface adhesion molecules on megakaryocytes may also contribute to intravascular retention in the lung, particularly in conditions in which the pulmonary endothelium is activated because of inflammation, infection, or injury (7, 10, 11). In vitro experiments demonstrated molecular interactions between activated human endothelial cells and megakaryocytes and a megakaryocyte cell line and showed that these interactions result in intercellular adhesion and promote megakaryocyte maturation (44).
Figure 2.
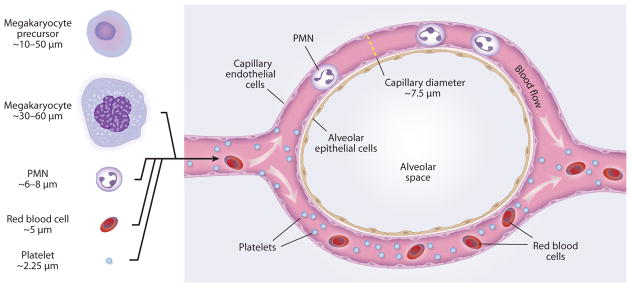
Blood cell retention in lungs. The majority of megakaryocytes that reach the lungs appear to be retained in capillaries and arterioles (see Figure 3). Polymorphonuclear leukocytes (PMN) transit through pulmonary capillaries but must deform, and their transit times are delayed compared with those of red blood cells, creating a substantial marginated pool. Most mature platelets readily pass through alveolar capillaries. Transit of released proplatelets (see Figure 1) has not been characterized. See text and Reference 11 for further details. Diameters of human megakaryocytes, megakaryocyte precursors, and platelets are from References 24, 32, and 39.[Megakaryocyte and platelet diameters are smaller in mice (32).] Human PMN and capillary diameters are from Reference 139.
Figure 3.

Lung megakaryocytes. Megakaryocytes are found in human lung microvessels and may spawn platelets and platelet precursors in this location. Megakaryocyte nuclei with little or no cytoplasm are also detected in microvessels in human lungs. The process of platelet biogenesis from proplatelet extensions and released proplatelets has, however, not been formally demonstrated in the lungs of humans or experimental animals.
Reports in experimental animals of megakaryocytes transmigrating through endothelial openings in marrow vessels, thereby apparently gaining access to the circulation, support the concept that megakaryocytes are released from the marrow and are carried to the pulmonary vessels (reviewed in References 24 and 41). Cell stage–specific responsiveness of developing versus mature megakaryocytes to stromal cell–derived factor 1 (SDF-1) may provide a mechanism for the retention of early megakaryocytes in the marrow and the later release of fully differentiated megakaryocytes that are competent for thrombopoiesis into the circulation, with subsequent transfer to the lungs via the blood (24). Nevertheless, megakaryocyte precursors may be released from the marrow into the circulating blood under some circumstances and may be trapped in the pulmonary microvascular bed because these cells are also large (10–50 μm) (24) (Figure 2). If so, molecular interactions between lung endothelial cells and megakaryocyte precursors may make unique signaling contributions that trigger or support intrapulmonary megakaryopoiesis. This issue has not been definitively investigated, however.
Together, the clinical and experimental investigations to date demonstrate that megakaryocytes reach the lungs and are retained in significant numbers in pulmonary vessels in many, and likely all, mammals. Nevertheless, the contribution of pulmonary megakaryocytes to thrombopoiesis (Figures 1 and 3) and total platelet production remains controversial. The predictions and conclusions range from insignificant (although perhaps only for the mouse) (30) to substantial (31, 35, 39, 45), and the quantitative estimates vary from a small fraction to 100% (reviewed in Reference 24). A central observation is that platelet numbers are higher in the pulmonary veins and systemic arterial vessels than in the pulmonary artery in experimental animals and in humans. Comparison of platelet counts in venous versus arterial blood, the right versus left ventricles, and the inferior vena cava versus the aorta in cats, dogs, rabbits, and rats yielded platelet numbers that were higher on the left side than on the right side of the lungs in most determinations (27, 29, 46, 47). In one of these reports, retrograde perfusion of the lungs resulted in the recovery of large numbers of megakaryocytes with degenerative nuclear changes in the effluent, suggesting that platelets are released from intrapulmonary megakaryocytes (29). In an analysis of human blood compartments, paired arm vein and radial artery determinants were done on samples from one of the investigators in an early study, with platelet counts usually higher in the radial artery sample and arterovenous differences similar to those recorded in cats (27). On the basis of measurements of platelet numbers in arterial and right ventricular blood from patients, and maneuvers that included cross-transfusion and epinephrine administration, Bierman (2) concluded that human lungs either are sites of platelet production or are reservoirs for platelets. Examination of paired samples from the right atrium or pulmonary artery and the aorta of subjects undergoing diagnostic cardiac catheterization demonstrated greater platelet numbers in the aortic blood from nine of ten patients; in addition, greater numbers of megakaryocytes were found in the central venous blood samples compared with the central arterial samples, consistent with other observations as outlined above (38). On the basis of some of these studies and additional data, a computational analysis led to the conclusion that a physical fragmentation process causes megakaryocytes in pulmonary vessels to release all platelets in the circulation (48). A calculation based on the volume of megakaryocyte cytoplasm in the human pulmonary circulation indicated that the expected daily production rate of platelets can occur in human lungs (39).
A more recent study supports the possibility that the lungs are a site of platelet production from megakaryocytes. Infusion of large and small murine fetal liver megakaryocytes with associated proplatelets, or primary murine bone marrow megakaryocytes, into transgenic mice with an endogenous platelet phenotype (expression of human integrin subunit αIIb) that allowed quantitation of platelets generated from the wild-type infused cells resulted in significant increases in circulating platelet numbers (49). The exogenous megakaryocytes were injected into recipient mice via retro-orbital vessels or the tail vein, implying that they would be carried to the central venous and pulmonary circulation, and infused megakaryocytes labeled exogenously with BrdU or identified by staining of integrin subunit αIIb were detected in lung sections of recipient ani-mals. Most of the BrdU-labeled cells accumulated in the lungs, with few found in the spleen or liver (which are sites of intravascular megakaryocyte accumulation in experimental animals and humans, as documented by previous histological studies). This finding suggests that the lungs are the dominant site of thrombopoiesis in this model but does not exclude platelet shedding in the venous circulation prior to impaction of infused megakaryocytes in pulmonary microvessels. Nevertheless, BrdU-labeled large megakaryocytes with cytoplasm were visible in the lungs for at least 30 min after infusion, implying pulmonary release of some, and perhaps all, of the newly generated platelets. Platelets released by infused megakaryocytes had the expected surface markers, had near-normal circulating half-lives, and were functional in an in vivo thrombosis model (49). These results are interesting and informative from a mechanistic standpoint and have implications for transfusion therapy because functional megakaryocytes are currently more easily generated ex vivo than are platelets (49, 50).
There is evidence that platelet genesis from proplatelets released from megakaryocytes, subsets of platelets, and/or novel immediate platelet precursors occurs in the bloodstream (reviewed in Reference 24) (Figure 1). Such evidence first came from studies of platelet-rich plasma from rat and human blood (51). More recently, we found that individual human platelets from peripheral venous blood transform into elongate, multilobed cells that are similar to detached proplatelets, that segregate organelles into the lobes, and that undergo fission to yield daughter platelets (22) (Figure 1b,c). Analysis of murine fetal liver–derived megakaryocytes with and without attached proplatelets, free proplatelets, and individual platelets also identified free discoid cells that have the capacity to morph into barbell-shaped precursors that undergo fission into platelets; these intermediate discoid cells are termed preplatelets (23, 24). Murine preplatelets are larger than mature platelets (3–10 mu;m versus 1.5 mu;m) (24, 32) and may be retained in pulmonary alveolar capillaries (see the next section). The process that yields fission-dependent platelet formation appears to be similar in mice and humans (22–24, 52). Shear potentiates the generation of daughter platelets by this mechanism in vitro (23, 52). Whether pulmonary vessels—a low-shear environment—provide particularly favorable rheological conditions for the fission process and/or contribute molecular fission factors remains unknown. Proplatelet numbers were higher in prepulmonary blood than on the systemic side of the lungs in rats, whereas platelet numbers were higher in systemic blood (53), supporting the possibility that pulmonary vessels have this role.
PLATELETS IN THE LUNG VASCULATURE: IS THERE A MARGINATED POOL?
The concept of marginated (also termed marginal) pools of specific blood cells in the lungs is most clearly established for PMNs. Marginated pools are an increased concentration of leukocytes in the pulmonary blood volume in normal, uninflamed lungs compared with their concentration in the blood volume in peripheral vessels (reviewed in Reference 11). Many studies indicate that PMNs are present in pulmonary capillaries in concentrations that are 30–100-fold greater than their concentration in arterial and venous conduit vessels in humans and experimental animals under physiological conditions (11).
The marginated pool of leukocytes in the lungs is established by leukocyte diameter and deformability, alveolar capillary diameter (2–15 mu;m, with an average of 7.5 mu;m at maximal dilation) (Figure 2), blood flow, lung region, and capillary and alveolar distending pressures (reviewed in Reference 11). PMN transit times are retarded compared with those of red blood cells (RBCs), concentrating the leukocytes in alveolar capillaries (Figure 2). In a study of radiolabeled PMNs, monocytes, lymphocytes, and platelets reinfused into the venous circulation of spontaneously breathing rabbits, each blood cell type was distributed to a lung marginated pool, but the lung pool of platelets was 13 times smaller than the circulating pool, whereas the lung PMN marginated pool was threefold larger than the circulating pool (54). The differences were attributed to the relative diameters of PMNs and platelets (11, 54). Although the platelet lung marginated pool was the smallest of the blood cells examined and was thought to be physiologically insignificant, 33% of the platelets were retained on first passage through the pulmonary circulation (54). In instrumented human subjects, epinephrine infusion increased platelet numbers in arterial blood, consistent with the possibility of a marginal pool mobilized by increased pulmonary blood flow (55, 56), and forced changes in ventilation differentially influenced the numbers of PMNs and platelets in left ventricular versus pulmonary artery blood samples (57). Whether such maneuvers influence platelet release from intrapulmonary megakaryocytes (Figure 3) or other precursors (see the previous section) is unknown. Experiments in ventilated rabbits, mice, and dogs involving the infusion of labeled platelets and analysis by intravital microscopy and other approaches (6, 58–60) also suggested the retention of a small pool of platelets in lung capillaries (6). These patterns of initial retention in the lungs may be due to platelet activation ex vivo during labeling before reinfusion, resulting in unphysiological adhesion in lung vessels. Nevertheless, morphometry of normal human lungs indicated that ~0.1% of alveolar capillaries were occupied by platelets that would not have been subjected to ex vivo manipulation, although this was a limited analysis (61). This figure compares to estimates of 1–2% to 15% of alveolar capillaries being occupied by the marginated PMN pool (11).
Although these studies suggest a small marginated pool of platelets in the lungs under basal conditions, the mechanism(s) involved is not defined. One possibility is that platelets utilize adhesive mechanisms to interact with pulmonary capillary endothelial cells in physiological margination (6). Against this possibility are the facts that platelets circulate with integrin αIIβ3 and other integrins in an unactivated, nonadhesive state (62) and that P-selectin, another major adhesion molecule, is in intracellular granules and not on the surfaces of quiescent, unactivated platelets (1, 13, 15). Furthermore, the pulmonary endothelium is a rich source of prostacyclin (PGI2) and nitric oxide (NO), which are major inhibitors of platelet activation (7). Some platelets, although small in diameter (2.25 mu;m in humans, 1.5 mu;m in mice) (32), may be retained in alveolar capillaries at the lower end of the size range, and their retention may be influenced by location in the lung vascular bed and by tidal changes in alveolar pressure (11).
Mechanical ventilation is used in some experimental models (58, 59) and is commonly used in the critical care of patients. The effect of mechanical ventilation on the retention of platelets and platelet precursors in alveolar capillaries has not been systematically examined.
A basal lung marginated pool of platelets may be significant, even though it is small. Relatively small numbers of platelets can have pivotal effects on alveolar capillary integrity (see the next section). In addition, a basal marginated pool of platelets may be the nidus for the recruitment of additional platelets in inflammation or injury and/or may mediate platelet-leukocyte interactions and signaling of alveolar capillary endothelium if this pool is activated (6–11, 59).
PLATELETS IN PULMONARY VASCULAR INTEGRITY AND BARRIER FUNCTION
Platelets are essential hemostatic effector cells in lung hemorrhage due to penetrating or blunt trauma, surgical or biopsy procedures, and a variety of nontraumatic conditions that involve alveolar or airway bleeding (7, 63). Here, critical activities of platelets include initial hemostatic plug formation and a variety of highly regulated activities that initiate and propagate primary hemostasis (1, 7, 15). Hemostatic activities of platelets may be particularly important in the bronchial circulation, which is composed of systemic vessels with systemic pressures, but are also critical in the lower-pressure pulmonary circulation. In addition to primary-hemostatic activities, platelets are important in maintaining endothelial integrity and barrier function; these mechanisms are related to, but are also mechanistically distinct from, these cells’ primary-hemostatic functions (15, 64).
Platelets and platelet-rich plasma, in contrast to platelet-free perfusate or platelet-poor plasma, contribute to microvascular barrier integrity; vascular preservation; and regulation of transvascular flux of fluid, solutes, and red blood cells in diverse experimental models. Such models include the edematous frog hind limb; isolated heart, kidney, and thyroid preparations; in situ capillary endothelium in muscle biopsies of animals made thrombocytopenic; and experimental animals studied by the infusion of radiolabeled albumin with measurement of its leak from systemic vessels (reviewed in References 3, 5, and 64). Early experiments also specifically examined the influence of platelets on endothelial integrity and barrier function in the lung microvascular bed (reviewed in References 3 and 5). For example, in chronically instrumented, spontaneously breathing sheep, thrombocytopenia induced by antiplatelet serum was associated with increased pulmonary vascular permeability to protein on the basis of lung lymph-to-plasma protein ratios. The increased permeability was ameliorated by the infusion of platelet-rich plasma (65). Staub popularized this preparation, the lung lymph fistula model, in the field, and the discoveries made with this model in his laboratory and others generated classic insights into the mechanisms of pulmonary edema due to increased alveolar capillary permeability (66) and into the physiological management of pulmonary edema in the clinical arena (67). In a study of isolated sheep lungs, perfusion with platelet-free solutions worsened lung fluid accumulation, indicating that platelets protect against pulmonary edema by an unknown mechanism (68). In isolated perfused rabbit lungs, human platelets reduced lung edema induced by oxidant challenge or by ischemia-reperfusion (69, 70). In vitro studies also demonstrated that intact platelets reduce labeled-albumin transfer across cultured pulmonary artery endothelial monolayers (65). Experiments with platelet lysates and supernatants from unactivated platelets indicated that platelets contain factors that reduce the permeability of cultured endothelial monolayers and that one or more of these substances can be released into solution (reviewed in Reference 64).
Experiments with various models involving pulmonary or systemic endothelium reveal evidence for at least five putative mechanisms by which platelets contribute to basal endothelial barrier function: the release of soluble molecules that maintain barrier integrity, most likely by signaling endothelial cells; the physical obstruction of gaps in the endothelial barrier; the maintenance of key structural features of endothelial cells that are required for barrier integrity; the stimulation or enhancement of endothelial cell growth; and the neutralization or elimination of agents that increase endothelial permeability and impair barrier integrity (reviewed in References 3, 5, and 64). The general interpretation of these studies is that one or more of these mechanisms is compromised if severe thrombocytopenia occurs and that this condition can lead to increased transendothelial flux of water and proteins and, in some cases, to RBC escape from vessels (64, 71) (Figure 4). Clinical observations in patients with thrombocytopenia secondary to leukemia or caused by nonneoplastic conditions (including immune thrombocytopenic purpura and amegakaryocytic thrombocytopenia) support the conclusions that local transfer of RBCs out of vessels is a consequence of compromised endothelial barrier function and that such transfer can occur without traumatic interruption of endothelial continuity (72, 73). In one study, ultrastructural changes in human skin and skeletal muscle capillaries in biopsies from thrombocytopenic patients were similar to those in experimental thrombocytopenia (73).
Figure 4.

Platelets stabilize pulmonary vascular endothelial barriers. (a) Under basal conditions, platelets express and/or release stabilizing factors that interact with receptors on alveolar capillary endothelial cells. Sphingosine-1-phosphate is a prototype stabilizing factor. Receptors for stabilizing factors are linked to intracellular signaling pathways that regulate cytoskeletal interactions, adherens junction assembly, and basal endothelial barrier properties. Homophilic binding of vascular endothelial cell cadherin (VE-cadherin) is central to stable alveolar capillary endothelial barrier function (reviewed in References 82 and 83). (b) Animal models show that in severe thrombocytopenia, basal alveolar capillary endothelial barrier function is disrupted, leading to increased permeability and transvascular escape of water, solutes, and red blood cells (RBC) (reviewed in Reference 64). Large arrows denote fluid exiting to the alveolar space, and small arrows denote fluid exiting to the interstitial space and to the lymph.
Recent studies in mouse models provide parallel observations on the effects of thrombocytopenia on endothelial barrier function and its contribution to local bleeding. In the setting of dermal, central nervous system, or pulmonary inflammation, severe thrombocytopenia (<2.5% of baseline platelet numbers) was associated with bleeding, whereas animals with an equivalent degree of thrombocytopenia but no inflammation did not bleed (74). Alveolar hemorrhage was dramatic in thrombocytopenic mice given inhaled lipopolysaccharide (LPS) and was substantial enough to cause anemia. Bleeding in these models was primarily from capillaries and postcapillary venules and was not explained by an inability to form platelet plugs on the basis of experiments with animals genetically deficient in key platelet adhesion molecules (64, 74). The latter result suggested that platelet adhesion is not required for platelet-dependent maintenance of endothelial barriers under these conditions and that other mechanisms are involved (74). Additional murine models also demonstrated vascular barrier disruption and local bleeding into tissues in severely thrombocytopenic animals (<5% of control platelet numbers) subjected to inflammatory challenge, and this phenotype was reversed by relatively small numbers (approximately 10% of the normal circulating platelet number) of transfused platelets (reviewed in Reference 64). Earlier observations reported that RBCs accumulate in thoracic duct lymph in experimental canine thrombocytopenia induced by radiation, also consistent with extravasation of erythrocytes across compromised endothelial barriers; platelet transfusion reduced RBC accumulation in the lymph (75).
Platelets express, store, and synthesize a variety of factors that operate in cell-cell interactions with endothelial cells and that alter endothelial cell functions (1, 15, 76). Several such factors may mediate the stabilization of basal endothelial permeability and barrier function on the basis of experiments with systemic or pulmonary endothelium. This list includes serotonin, sphingosine-1-phosphate (S1P), epinephrine, adenosine, angiopoietin-1 (Ang-1), and lysophosphatidic acid (3, 5, 64). Of these factors, serotonin and S1P have been most extensively studied. Intravenous and intraperitoneal injection of serotonin reduced RBC extravasation associated with thrombocytopenia in mice, and infusion of serotonin antagonists into animals with normal platelet numbers yielded local bleeding (petechiae) similar to that in thrombocytopenic animals. Experiments with cultured endothelial cells suggested that serotonin may maintain endothelial barrier function by altering the endothelial cytoskeleton (reviewed in Reference 5). Paradoxically, in earlier classic rat experiments, serotonin had the opposite effect and increased permeability of vessels to fluid, but not to large-molecular-weight tracers (77). Serotonin also increased permeability of cultured rat endothelial cells (78) and of in situ murine skin vessels (79). There may be differential sensitivity to serotonin’s barrier-altering activities on the basis of serotonin receptor subtype expression and, potentially, other biological features of endothelial cells in different-sized vessels and/or different vascular beds (77).
S1P has been extensively studied as a molecular stabilizer of endothelial barrier function (reviewed in Reference 80). In vitro, S1P binds to S1P1 (previously termed EDG-1), a G protein– coupled receptor expressed by endothelial cells, and induces actin cytoskeleton rearrangement and Rac-GTPase-dependent assembly of adherens junctions on endothelial cell plasma membranes (80, 81). S1P also induces localization of α-, β-, and γ-catenins and vascular endothelial cell cadherin (VE-cadherin) to sites of endothelial cell-cell contact (80). VE-cadherin is a major endothelial adherens junction protein and a key regulator of endothelial barrier function (reviewed in Reference 82) (Figure 4). Disruption of homophilic VE-cadherin bonds increases microvascular permeability in the lung and other organs, whereas stabilization of VE-cadherin bonds and consequent adherens junction integrity have the opposite effect (reviewed in Reference 83). Alveolar and systemic capillary membrane barrier stabilization may be a molecular strategy for the treatment of ALI/ARDS and sepsis (reviewed in Reference 83).
In vitro and in vivo experiments indicate that S1P stabilizes pulmonary endothelial cell-cell interactions that establish barrier integrity. Early studies demonstrated the enhancement of pulmonary artery and lung microvascular endothelial cell barrier integrity by S1P and identified key molecular mechanisms, including molecular interactions of VE-cadherin. These studies also showed that S1P can reverse the barrier dysfunction triggered by destabilizing agonists (84, 85; reviewed in Reference 86). Consistent with these observations, in vivo experiments with genetically altered mice demonstrated that plasma S1P regulates basal pulmonary vascular permeability to albumin and governs altered barrier integrity in response to inflammatory challenge (87). These and other in vivo experiments further show that S1P blunts or reverses pulmonary vascular leak and alveolar edema in models of lung inflammation and ALI (88–90; reviewed in Reference 86). Platelets have constitutive activity of the key S1P synthetic enzyme, sphingosine-1-kinase; lack the degradative enzyme S1P lyase; and therefore have high levels of S1P and release it on stimulation (80). In vitro experiments demonstrated that human platelets and platelet supernatants enhance cultured human pulmonary artery endothelial cell barrier function, measured by transmonolayer electrical resistance, and that this enhancement depends on S1P1 (85). These and other observations (80, 86) indicate that S1P is one of the platelet-released factors that can regulate pulmonary vascular permeability and stabilize alveolar capillary barrier integrity (3, 5, 7, 64). It was originally thought that platelets are the principal source of plasma S1P, but more recent studies indicate that—at least in mice—erythrocytes supply basal blood levels of S1P (80, 87). Nevertheless, S1P concentrations in serum are generally higher than in plasma, implying that activated platelets may locally release S1P in inflammation and thrombosis (80). Endothelial responses to S1P or to small-molecule agonists of S1P1 (91) may depend on temporal variables and biological context because of altered expression or desensitization of S1P1, barrier-destabilizing effects resulting from the engagement of other S1P receptors (including S1P2 and S1P3), or parallel signaling by other factors such as Ang-1 (80, 92, 93).
There is clear evidence that platelets can reduce endothelial barrier permeability and enhance barrier function in vitro and that sufficient numbers of circulating platelets may be critical for basal barrier integrity of alveolar capillaries and other capillary beds in vivo. Nevertheless, there is also substantial evidence that platelets can contribute to increased alveolar capillary permeability in lung inflammation and ALI (6–8, 12). Platelets and platelet supernatants stabilize or promote assembly of endothelial barriers under some conditions (see above), but platelets and platelet lysates induced edema and inflammation or were required for a maximal increase in vascular permeability in other experiments (94, 95). Mouse and other in vivo animal models demonstrated that platelets increase alveolar capillary permeability (6–8, 12). Human and murine platelets synthesize interleukin 1β (reviewed in Reference 12), a cytokine with major endothelial barrier–destabilizing activities.
The mechanisms that account for barrier-enhancing activities on the one hand and barrier-destabilizing activities on the other hand are not clear (7) but in many cases may involve critical interactions of platelets with inflammatory effector cells (e.g., PMNs, monocytes), triggering barrier-disrupting activities and/or the release of endothelium-destabilizing factors (12, 15, 64) (Figure 5). Differential expression of endothelial receptors for platelet factors that alter barrier function, differential activity (e.g., desensitization) of these receptors, and linkage of these receptors to alternative functional pathways in response to different ligand concentrations may be additional key mechanisms that alternatively influence barrier stabilization versus destabilization. S1P signaling is a prime example (80, 90, 92), and these possibilities may also apply to serotonin (see above). In addition, the signaling context that governs endothelial responses to platelet factors or to other cellular or plasma factors may change during time-dependent stages of disease (80, 83, 92). Differential release of platelet-signaling factors from unique granular compartments (96) may lead to endothelial barrier destabilization under some conditions and to endothelial barrier stabilization under others (64). As another potential mechanism, the platelet transcriptome and proteome can change in disease (reviewed in References 12 and 15). As examples, multiple differentially expressed transcripts were found in platelets from subjects with sickle cell disease, in contrast to platelets from controls (97), and platelets from patients with sepsis express mature tissue factor mRNA and tissue factor activity, whereas those from controls do not (98). Thus, megakaryocytes and/or platelets appear to be reprogrammed in certain disease conditions. In such contexts, the profile of platelet-signaling factors that can influence endothelial barrier function and permeability may switch from a predominantly barrier-stabilizing portfolio (Figure 4) to a predominantly destabilizing one (Figure 5) in experimental or clinical lung inflammation and injury (7), although this possibility has not been investigated.
Figure 5.

Platelets contribute to alveolar capillary membrane injury and increased-permeability lung edema in ALI/ARDS. On the basis of clinical observations and studies in experimental models, activated platelets mediate ALI by mechanisms that include aggregation, the release of mediators, signaling of endothelial cells, and interactions with polymorphonuclear leukocytes (PMN) and monocytes, triggering or amplifying inflammatory damage (reviewed in References 6–8, 12, and 103).
PLATELETS IN PULMONARY VASCULAR REACTIVITY, REPAIR, AND REMODELING
A unique animal model, the fawn-hooded rat, has platelet storage and functional alterations associated with the spontaneous development of severe pulmonary hypertension (99), suggesting links between platelet activation and pulmonary vascular function. Platelets store or synthesize factors that have vasoactive properties (reviewed in References 3, 5, and 100). Serotonin, thromboxane A2 (TxA2), and platelet-derived growth factor (PDGF) have pulmonary vasoconstrictor activities when released by activated platelets, and plasma serotonin levels are elevated in some patients with pulmonary hypertensive disorders (100, 101). In a recently described mouse model of severe pulmonary hypertension, serotonin levels were substantially increased in platelet-rich plasma from affected animals compared with plasma from controls (102). Acute pulmonary hypertension, a common physiological alteration in early ARDS, is induced by sepsis and other underlying conditions (3, 5, 103) and exacerbates the leak of fluid and protein across injured alveolar capillaries with impaired barrier function (see the last section) (66, 67, 83). Platelets and platelet products, including serotonin and TxA2, have been implicated in pulmonary vascular pressor responses to LPS infusion and in other experimental models of ALI/ARDS, but there are differences in outcomes that may depend on species and other variables (3, 5, 104, 105). As in the systemic circulation, pulmonary vasoreactive responses to factors released by platelets in physiological or pathological models depend on whether the endothelium is intact and capable of synthesizing vasodilators, including PGI2 and NO, or, in contrast, is denuded or functionally compromised (106).
Platelets are central effector cells in wound healing (107) and have only recently identified activities in vascular development, angiogenesis, and vascular repair and remodeling (reviewed in References 7, 15, and 64). In mouse models, platelets recruit circulating vascular progenitor cells to thrombi, sites of vascular damage, or ischemic vessels, using mechanisms that depend on P-selectin, integrins, and SDF-1α (CXCL12) (108–112). Platelets also induce functional changes in vascular progenitor and precursor cells (108, 110), as they do in target leukocytes and other cell types (113, 114; reviewed in Reference 15). Nevertheless, these biological potentials have not been examined in the context of pulmonary vascular repair or physiological remodeling. If platelets or megakaryocytes (see the first section of this review) can favorably affect lung repair, they could be used therapeutically and could potentially be engineered to deliver reparative factors, or signals for reparative precursor cells, in lung injury states (7, 115).
Platelets may contribute to dysregulated and pathological vascular repair or remodeling mechanisms, including obstructive remodeling in chronic pulmonary hypertensive disorders (100, 116, 117). Platelet mitogens, including serotonin and PDGF, may cause pulmonary vascular smooth-muscle proliferation in chronic pulmonary hypertension, although each can originate from other cells besides platelets (100, 102, 116, 118). Platelet-dependent mechanisms, including inflammatory signaling between platelets and monocytes (12, 15, 113, 114), may contribute to chronic thromboembolic pulmonary hypertension (119). Recent studies utilizing mouse models identified serotonin from platelets as a mediator that links vascular damage to extravascular tissue fibrosis (120), and there is evidence for serotonin signaling in experimental pulmonary fibrosis (121). Therefore, platelets may be effector cells in pulmonary fibrotic diseases and in chronic pulmonary vascular syndromes.
PLATELETS IN LUNG DISEASE: A SYNOPSIS
The involvement of platelets in human lung diseases appears diverse and protean (3–8, 10, 12, 100, 103, 107). Alterations in platelet numbers and function occur in primary lung infections, such as influenza (122), and in systemic infectious syndromes that involve the lungs and pleurae, including bacterial sepsis, malaria, and dengue (reviewed in References 12, 15, and 103). As mentioned above, there is considerable interest in dissecting the mechanisms of localization, cellular interactions, and effector activities of platelets in experimental and clinical ALI/ARDS (6–8, 12, 61, 83, 103). As a specific example, platelet infusion is a cause of transfusion-related ALI (TRALI) (12). Innovative experimental modeling in mice (123, 124) provides key insights into the interactions of platelets and myeloid leukocytes in TRALI that are broadly relevant to inflammatory alveolar injury (6–8, 12). In experimental TRALI, platelets were reported to induce the formation of neutrophil extracellular traps (125), a mechanism that may also be important in ALI induced by sepsis and other triggers (83). If we turn from the alveoli to the airways, a substantial body of literature supports the idea that platelets are effector cells in asthma and cystic fibrosis (4, 6, 10, 126–130). Pulmonary hypertensive disorders (100) are briefly discussed in the last section. Pathological lymphangiogenesis in the lung is induced by inflammation (reviewed in Reference 131) and may be regulated by platelets on the basis of recent experimental observations (reviewed in References 15 and 64). Animal models (132, 133) and ongoing translational investigation (119) promise to uncover pivotal platelet-dependent mechanisms central to pulmonary embolism and its acute and chronic sequelae. Platelets are determinants of lung metastasis in experimental and clinical neoplasia (134, 135). These are only some of the lung syndromes in which platelets have putative or documented activities. Platelets may also contribute to iatrogenic complications, including ventilator-induced lung injury (136), an important clinical problem (83). Many of the disease-promoting and/or disease-enhancing activities of platelets in lung disorders appear to result from dysregulated cellular and molecular mechanisms that operate in physiological hemostasis, barrier maintenance, and inflammation (7, 12, 14, 15, 64).
SUMMARY POINTS.
Platelets are key effector cells in hemostasis, inflammation, and tissue repair and continuously circulate through the lungs.
The lungs may be a key site of thrombopoiesis, the complex process of platelet production, and megakaryocytes—which are specialized precursor cells—are found in the lungs.
There may be a small marginated pool of platelets that is transiently retained in lung capillaries and that is dynamically altered on the basis of physiological and pathological conditions.
Experimental and clinical observations show that platelets contribute to basal barrier function of the alveolar capillary endothelium and to disruption of barrier integrity in lung inflammation and injury.
Platelets likely have specialized activities in repair and remodeling of pulmonary, alveolar, and bronchial vessels and, potentially, in remodeling of extravascular lung structures, although this latter issue has not been critically explored.
Platelets have protean activities in experimental lung injury and in clinical lung disease.
FUTURE ISSUES.
It is unknown whether there are molecular signals from the lungs that trigger megakaryocyte release from the marrow under basal conditions and in disease or whether the lung endothelium generates biochemical factors that locally promote or modulate thrombopoiesis.
“Locking” megakaryocytes in the marrow so that they cannot transit to the pulmonary vessels may be a mechanism of thrombocytopenia, and injured or inflamed lungs may mechanistically contribute to impaired thrombopoiesis in other ways.
Conversely, the lungs may actively contribute to the thrombocytosis that is sometimes associated with inflammation or neoplasia.
It is unknown whether megakaryocytes in the pulmonary microcirculation have other functions besides the putative release of platelets, proplatelets, and/or preplatelets; such additional functions may involve specific activities in inflammation or repair. It is also unknown whether intercellular signaling between megakaryocytes and leukocytes or between megakaryocytes and red blood cells in the pulmonary vascular milieu results in functional alterations.
The mechanisms that influence platelet-dependent vascular barrier stabilization versus platelet-dependent alveolar capillary barrier impairment need to be worked out.
It is unknown whether platelet transfusions have a beneficial effect in human subjects with disrupted alveolar capillary barrier function and increased-permeability pulmonary edema or, in contrast, whether platelet transfusion is actually a hazard in these conditions.
Interactions of platelets with stem or precursor cell populations in lung repair are additional topics for future investigation.
Changes in the transcriptome, proteome, and metabolome of platelets in lung diseases need to be examined, and reprogramming of megakaryocytes and platelets in lung syndromes merits exploration.
Acknowledgments
The authors thank Alexandra Greer for skillful preparation of the manuscript and Diana Lim for artistic expertise and creative contributions in drafting the figures. We thank our colleagues, collaborators, research team, and the junior members of our group for critical discussions and scientific interactions. Work described in articles cited here was supported by the National Institutes of Health (R37HL044525, R01HL092746, R01HL066277, R01HL100121, and R01HL091754).
Glossary
- Platelets
anucleate circulating blood cells that mediate primary hemostasis, inflammation, and tissue repair
- Megakaryocytes
highly specialized cells that release platelets and platelet precursors into the circulation
- Thrombopoiesis
the complex process of platelet production
- Barrier function
the property of alveolar capillary endothelium and other vessels that selectively restricts the transfer of blood constituents out of the vessel
- Primary hemostasis
the arrest of bleeding from injured vessels; accomplished by plug formation by activated platelets and ancillary hemostatic measures
- Immune continuum
the spectrum of innate and adaptive immune responses and activities that mediate acute and chronic inflammation
- Endothelial cells
the lining cells of alveolar capillaries and other vessels in the lungs and other organs
- Proplatelets
pseudopod-like extensions from megakaryocytes; are thought to be critical precursors of platelets
- Polymorphonuclear leukocytes (PMNs)
circulating myeloid effector cells that are critical in innate immune responses to microbial invasion and tissue injury
- Marginated pool
a subset of circulating blood cells that is transiently retained in the capillary bed of the lungs or other organs
- Pulmonary edema
the accumulation of water and, in increased-permeability pulmonary edema, other blood constituents in lung interstitium and alveoli
Footnotes
DISCLOSURE STATEMENT
The authors are not aware of any affiliations, memberships, funding, or financial holdings that might be perceived as affecting the objectivity of this review.
Contributor Information
Andrew S. Weyrich, Email: andy.weyrich@u2m2.utah.edu.
Guy A. Zimmerman, Email: guy.zimmerman@u2m2.utah.edu.
LITERATURE CITED
- 1.Michelson AD, editor. Platelets. 3. San Diego: Elsevier/Academic; 2013. In press. [Google Scholar]
- 2.Bierman HR. The hematologic role of the lung in man. Am J Surg. 1955;89:130–40. doi: 10.1016/0002-9610(55)90515-8. [DOI] [PubMed] [Google Scholar]
- 3.Heffner JE, Repine JE. Platelets. In: Crystal RG, editor. The Lung: Scientific Foundations. New York: Raven; 1991. pp. 617–30. [Google Scholar]
- 4.Herd CM, Page CP. Pulmonary immune cells in health and disease: platelets. Eur Respir J. 1994;7:1145–60. [PubMed] [Google Scholar]
- 5.Heffner JE, Repine JE. Platelets. In: Crystal RG, editor. The Lung: Scientific Foundations. 2 New York: Raven; 1997. pp. 947–59. [Google Scholar]
- 6.Tabuchi A, Kuebler WM. Endothelium-platelet interactions in inflammatory lung disease. Vascul Pharmacol. 2008;49:141–50. doi: 10.1016/j.vph.2008.06.004. [DOI] [PubMed] [Google Scholar]
- 7.Bozza FA, Shah AM, Weyrich AS, Zimmerman GA. Amicus or adversary: platelets in lung biology, acute injury, and inflammation. Am J Respir Cell Mol Biol. 2009;40:123–34. doi: 10.1165/rcmb.2008-0241TR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zarbock A, Ley K. The role of platelets in acute lung injury (ALI) Front Biosci. 2009;14:150–58. doi: 10.2741/3236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Voelkel NF, Rounds S, editors. The Pulmonary Endothelium: Function in Health and Disease. New York: Wiley & Sons; [Google Scholar]
- 10.Nicolls MR, Tamosiuniene T, Babu AN, Voelkel NF. Interactions of pulmonary endothelial cells with immune cells and platelets: implications for disease pathogenesis. 2009:417–36. See Ref. 9. [Google Scholar]
- 11.Rodrigues RS, Zimmerman GA. Pulmonary endothelial interactions with leukocytes and platelets. 2009:146–66. See Ref. 9. [Google Scholar]
- 12.Vieira-de-Abreu A, Rondina MT, Weyrich AS, Zimmerman GA. Inflammation. 2013 See Ref. 1 In press. [Google Scholar]
- 13.Weyrich AS, Zimmerman GA. Platelets: signaling cells in the immune continuum. Trends Immunol. 2004;25:489–95. doi: 10.1016/j.it.2004.07.003. [DOI] [PubMed] [Google Scholar]
- 14.Semple JW, Italiano JE, Jr, Freedman J. Platelets and the immune continuum. Nat Rev Immunol. 2011;11:264–74. doi: 10.1038/nri2956. [DOI] [PubMed] [Google Scholar]
- 15.Vieira-de-Abreu A, Campbell RA, Weyrich AS, Zimmerman GA. Platelets: versatile effector cells in hemostasis, inflammation, and the immune continuum. Semin Immunopathol. 2012;34:5–30. doi: 10.1007/s00281-011-0286-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zimmerman GA, Weyrich AS. Signal-dependent protein synthesis by activated platelets: new pathways to altered phenotype and function. Arterioscler Thromb Vasc Biol. 2008;28:S17–24. doi: 10.1161/ATVBAHA.107.160218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Provost P. Platelet microRNA. 2013 See Ref. 1. In press. [Google Scholar]
- 18.Weyrich AS. Platelet proteomics. 2013 See Ref. 1. In press. [Google Scholar]
- 19.Ouwehand W, Bray PF. Platelet genomics. 2013 See Ref. 1. In press. [Google Scholar]
- 20.Rowley JW, Oler AJ, Tolley ND, Hunter BN, Low EN, et al. Genome-wide RNA-seq analysis of human and mouse platelet transcriptomes. Blood. 2011;118:e101–11. doi: 10.1182/blood-2011-03-339705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kahr WH, Hinckley J, Li L, Schwertz H, Christensen H, et al. Mutations in NBEAL2, encoding a BEACH protein, cause gray platelet syndrome. Nat Genet. 2011;43:738–40. doi: 10.1038/ng.884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schwertz H, Koster S, Kahr WH, Michetti N, Kraemer BF, et al. Anucleate platelets generate progeny. Blood. 2010;115:3801–9. doi: 10.1182/blood-2009-08-239558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Thon JN, Montalvo A, Patel-Hett S, Devine MT, Richardson JL, et al. Cytoskeletal mechanics of proplatelet maturation and platelet release. J Cell Biol. 2010;191:861–74. doi: 10.1083/jcb.201006102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Italiano JE, Hartwig JH. Megakaryocyte development and platelet formation. 2013 doi: 10.1083/jcb.201304054. See Ref. 1. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dunois-Larde C, Capron C, Fichelson S, Bauer T, Cramer-Borde E, Baruch D. Exposure of human megakaryocytes to high shear rates accelerates platelet production. Blood. 2009;114:1875–83. doi: 10.1182/blood-2009-03-209205. [DOI] [PubMed] [Google Scholar]
- 26.Junt T, Schulze H, Chen Z, Massberg S, Goerge T, et al. Dynamic visualization of thrombopoiesis within bone marrow. Science. 2007;317:1767–70. doi: 10.1126/science.1146304. [DOI] [PubMed] [Google Scholar]
- 27.Howell WH, Donahue DD. The production of blood platelets in the lungs. J Exp Med. 1937;65:177–203. doi: 10.1084/jem.65.2.177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jordan HE. The origin and significance of the megakaryocytes of the lung. Anat Rec. 1940;77:99–101. [Google Scholar]
- 29.Pedersen NT. The pulmonary vessels as a filter for circulating megakaryocytes in rats. Scand J Haematol. 1974;13:225–31. doi: 10.1111/j.1600-0609.1974.tb00263.x. [DOI] [PubMed] [Google Scholar]
- 30.Davis RE, Stenberg PE, Levin J, Beckstead JH. Localization of megakaryocytes in normal mice and following administration of platelet antiserum, 5-fluorouracil, or radiostrontium: evidence for the site of platelet production. Exp Hematol. 1997;25:638–48. [PubMed] [Google Scholar]
- 31.Zucker-Franklin D, Philipp CS. Platelet production in the pulmonary capillary bed: new ultra-structural evidence for an old concept. Am J Pathol. 2000;157:69–74. doi: 10.1016/S0002-9440(10)64518-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Thon JN, Italiano JE. Platelet formation. Semin Hematol. 2010;47:220–26. doi: 10.1053/j.seminhematol.2010.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mandal RV, Mark EJ, Kradin RL. Megakaryocytes and platelet homeostasis in diffuse alveolar damage. Exp Mol Pathol. 2007;83:327–31. doi: 10.1016/j.yexmp.2007.08.005. [DOI] [PubMed] [Google Scholar]
- 34.Scheinin TM, Koivuniemi AP. Megakaryocytes in the pulmonary circulation. Blood. 1963;22:82–87. [PubMed] [Google Scholar]
- 35.Kaufman RM, Airo R, Pollack S, Crosby WH. Circulating megakaryocytes and platelet release in the lung. Blood. 1965;26:720–31. [PubMed] [Google Scholar]
- 36.Melamed MR, Cliffton EE, Mercer C, Koss LG. The megakaryocyte blood count. Am J Med Sci. 1966;252:301–9. doi: 10.1097/00000441-196609000-00009. [DOI] [PubMed] [Google Scholar]
- 37.Pedersen NT. Occurrence of megakaryocytes in various vessels and their retention in the pulmonary capillaries in man. Scand J Haematol. 1978;21:369–75. doi: 10.1111/j.1600-0609.1978.tb00381.x. [DOI] [PubMed] [Google Scholar]
- 38.Kallinikos-Maniatis A. Megakaryocytes and platelets in central venous and arterial blood. Acta Haematol. 1969;42:330–35. doi: 10.1159/000208817. [DOI] [PubMed] [Google Scholar]
- 39.Levine RF, Eldor A, Shoff PK, Kirwin S, Tenza D, Cramer EM. Circulating megakaryocytes: delivery of large numbers of intact, mature megakaryocytes to the lungs. Eur J Haematol. 1993;51:233–46. doi: 10.1111/j.1600-0609.1993.tb00637.x. [DOI] [PubMed] [Google Scholar]
- 40.Hansen M, Pedersen NT. Circulating megakaryocytes in patients with pulmonary inflammation and in patients subjected to cholecystectomy. Scand J Haematol. 1979;23:211–16. doi: 10.1111/j.1600-0609.1979.tb02693.x. [DOI] [PubMed] [Google Scholar]
- 41.Trowbridge EA. Pulmonary platelet production: a physical analogue of mitosis? Blood Cells. 1988;13:451–65. [PubMed] [Google Scholar]
- 42.Kaufman RM, Airo R, Pollack S, Crosby WH, Doberneck R. Origin of pulmonary megakaryocytes. Blood. 1965;25:767–75. [PubMed] [Google Scholar]
- 43.Wilde NT, Burgess R, Keenan DJ, Lucas GS. The effect of cardiopulmonary bypass on circulating megakaryocytes. Br J Haematol. 1997;98:322–27. doi: 10.1046/j.1365-2141.1997.2373055.x. [DOI] [PubMed] [Google Scholar]
- 44.Avraham H, Cowley S, Chi SY, Jiang S, Groopman JE. Characterization of adhesive interactions between human endothelial cells and megakaryocytes. J Clin Investig. 1993;91:2378–84. doi: 10.1172/JCI116470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.George JN. Platelets. Lancet. 2000;355:1531–39. doi: 10.1016/S0140-6736(00)02175-9. [DOI] [PubMed] [Google Scholar]
- 46.Sharnoff JG, Scardino V. Platelet-count differences in blood of the rabbit right and left heart ventricles. Nature. 1960;187:334–35. doi: 10.1038/187334a0. [DOI] [PubMed] [Google Scholar]
- 47.Warheit DB, Barnhart MI. Ultrastructure of circulating and platelet-forming megakaryocytes: a combined correlative SEM-TEM and SEM histochemical study. Ann N Y Acad Sci. 1981;370:30–41. doi: 10.1111/j.1749-6632.1981.tb29718.x. [DOI] [PubMed] [Google Scholar]
- 48.Trowbridge EA, Martin JF, Slater DN. Evidence for a theory of physical fragmentation of megakaryocytes, implying that all platelets are produced in the pulmonary circulation. Thromb Res. 1982;28:461–75. doi: 10.1016/0049-3848(82)90163-3. [DOI] [PubMed] [Google Scholar]
- 49.Fuentes R, Wang Y, Hirsch J, Wang C, Rauova L, et al. Infusion of mature megakaryocytes into mice yields functional platelets. J Clin Investig. 2010;120:3917–22. doi: 10.1172/JCI43326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Leavitt AD. Are there more tricks in the bag for treating thrombocytopenia? J Clin Investig. 2010;120:3807–10. doi: 10.1172/JCI45179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Behnke O, Forer A. From megakaryocytes to platelets: Platelet morphogenesis takes place in the bloodstream. Eur J Haematol Suppl. 1998;61:3–23. doi: 10.1111/j.1600-0609.1998.tb01052.x. [DOI] [PubMed] [Google Scholar]
- 52.Schwertz H, Weyrich AS. Platelet precursors display bipolar behavior. J Cell Biol. 2010;191:699–700. doi: 10.1083/jcb.201010082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Handagama PJ, Feldman BF, Jain NC, Farver TB, Kono CS. Circulating proplatelets: isolation and quantitation in healthy rats and in rats with induced acute blood loss. Am J Vet Res. 1987;48:962–65. [PubMed] [Google Scholar]
- 54.Doerschuk CM, Downey GP, Doherty DE, English D, Gie RP, et al. Leukocyte and platelet margination within microvasculature of rabbit lungs. J Appl Physiol. 1990;68:1956–61. doi: 10.1152/jappl.1990.68.5.1956. [DOI] [PubMed] [Google Scholar]
- 55.Bierman HR, Kelly KH, King FW, Petrakis NL. The pulmonary circulation as a source of leucocytes and platelets in man. Science. 1951;114:276–77. doi: 10.1126/science.114.2959.276. [DOI] [PubMed] [Google Scholar]
- 56.Bierman HR, Kelly KH, Cordes FL, Byron RL, Jr, Polhemus JA, Rappoport S. The release of leukocytes and platelets from the pulmonary circulation by epinephrine. Blood. 1952;7:683–92. [PubMed] [Google Scholar]
- 57.Markos J, Hooper RO, Kavanagh-Gray D, Wiggs BR, Hogg JC. Effect of raised alveolar pressure on leukocyte retention in the human lung. J Appl Physiol. 1990;69:214–21. doi: 10.1152/jappl.1990.69.1.214. [DOI] [PubMed] [Google Scholar]
- 58.Tabuchi A, Mertens M, Kuppe H, Pries AR, Kuebler WM. Intravital microscopy of the murine pulmonary microcirculation. J Appl Physiol. 2008;104:338–46. doi: 10.1152/japplphysiol.00348.2007. [DOI] [PubMed] [Google Scholar]
- 59.Eichhorn ME, Ney L, Massberg S, Goetz AE. Platelet kinetics in the pulmonary microcirculation in vivo assessed by intravital microscopy. J Vasc Res. 2002;39:330–39. doi: 10.1159/000065545. [DOI] [PubMed] [Google Scholar]
- 60.Kien M, Hechtman H, Shepro D. Platelet-microvasculature interaction during one passage through the dog lung. Microvasc Res. 1971;3:209–10. doi: 10.1016/0026-2862(71)90025-2. [DOI] [PubMed] [Google Scholar]
- 61.Bachofen M, Weibel ER. Structural alterations of lung parenchyma in the adult respiratory distress syndrome. Clin Chest Med. 1982;3:35–56. [PubMed] [Google Scholar]
- 62.Shattil SJ, Kim C, Ginsberg MH. The final steps of integrin activation: the end game. Nat Rev Mol Cell Biol. 2010;11:288–300. doi: 10.1038/nrm2871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Jean-Baptiste E. Clinical assessment and management of massive hemoptysis. Crit Care Med. 2000;28:1642–47. doi: 10.1097/00003246-200005000-00066. [DOI] [PubMed] [Google Scholar]
- 64.Ho-Tin-Noe B, Demers M, Wagner DD. How platelets safeguard vascular integrity. J Thromb Haemost. 2011;9(Suppl 1):56–65. doi: 10.1111/j.1538-7836.2011.04317.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lo SK, Burhop KE, Kaplan JE, Malik AB. Role of platelets in maintenance of pulmonary vascular permeability to protein. Am J Physiol Heart Circ Physiol. 1988;254:H763–71. doi: 10.1152/ajpheart.1988.254.4.H763. [DOI] [PubMed] [Google Scholar]
- 66.Staub NC. Pulmonary edema: physiologic approaches to management. Chest. 1978;74:559–64. doi: 10.1378/chest.74.5.559. [DOI] [PubMed] [Google Scholar]
- 67.Ware LB, Matthay MA. Clinical practice. Acute pulmonary edema. N Engl J Med. 2005;353:2788–96. doi: 10.1056/NEJMcp052699. [DOI] [PubMed] [Google Scholar]
- 68.Pearse DB, Brower RG, Adkinson NF, Jr, Sylvester JT. Spontaneous injury in isolated sheep lungs: role of perfusate leukocytes and platelets. J Appl Physiol. 1989;66:1287–96. doi: 10.1152/jappl.1989.66.3.1287. [DOI] [PubMed] [Google Scholar]
- 69.Heffner JE, Cook JA, Halushka PV. Human platelets modulate edema formation in isolated rabbit lungs. J Clin Investig. 1989;84:757–64. doi: 10.1172/JCI114233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zamora CA, Baron D, Heffner JE. Washed human platelets prevent ischemia-reperfusion edema in isolated rabbit lungs. J Appl Physiol. 1991;70:1075–84. doi: 10.1152/jappl.1991.70.3.1075. [DOI] [PubMed] [Google Scholar]
- 71.Nachman RL, Rafii S. Platelets, petechiae, and preservation of the vascular wall. N Engl J Med. 2008;359:1261–70. doi: 10.1056/NEJMra0800887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Roy AJ, Djerassi I. Effects of platelet transfusions: plug formation and maintenance of vascular integrity. Proc Soc Exp Biol Med. 1972;139:137–42. doi: 10.3181/00379727-139-36096. [DOI] [PubMed] [Google Scholar]
- 73.Kitchens CS, Pendergast JF. Human thrombocytopenia is associated with structural abnormalities of the endothelium that are ameliorated by glucocorticosteroid administration. Blood. 1986;67:203–6. [PubMed] [Google Scholar]
- 74.Goerge T, Ho-Tin-Noe B, Carbo C, Benarafa C, Remold-O’Donnell E, et al. Inflammation induces hemorrhage in thrombocytopenia. Blood. 2008;111:4958–64. doi: 10.1182/blood-2007-11-123620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Jackson DP, Sorensen DK, Cronkite EP, Bond VP, Fliedner TM. Effectiveness of transfusions of fresh and lyophilized platelets in controlling bleeding due to thrombocytopenia. J Clin Investig. 1959;38:1689–97. doi: 10.1172/JCI103947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Maguire PB, Belton O, O’Donoghue N, Austini S. Platelet-endothelial interactions. In: Aird WC, editor. Endothelial Biomedicine. Cambridge, UK: Cambridge Univ. Press; 2007. pp. 597–601. [Google Scholar]
- 77.Majno G, Palade GE. Studies on inflammation. 1 The effect of histamine and serotonin on vascular permeability: an electron microscopic study. J Biophys Biochem Cytol. 1961;11:571–605. doi: 10.1083/jcb.11.3.571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Lee HZ, Lin WC, Yeh FT, Wu CH. 2-Phenyl-4-quinolone prevents serotonin-induced increases in endothelial permeability to albumin. Eur J Pharmacol. 1998;354:205–13. doi: 10.1016/s0014-2999(98)00452-x. [DOI] [PubMed] [Google Scholar]
- 79.Zhao D, Qin L, Bourbon PM, James L, Dvorak HF, Zeng H. Orphan nuclear transcription factor TR3/Nur77 regulates microvessel permeability by targeting endothelial nitric oxide synthase and destabilizing endothelial junctions. Proc Natl Acad Sci USA. 2011;108:12066–71. doi: 10.1073/pnas.1018438108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Obinata H, Hla T. Sphingosine 1-phosphate in coagulation and inflammation. Semin Immunopathol. 2012;34:73–91. doi: 10.1007/s00281-011-0287-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Lee MJ, Thangada S, Claffey KP, Ancellin N, Liu CH, et al. Vascular endothelial cell adherens junction assembly and morphogenesis induced by sphingosine-1-phosphate. Cell. 1999;99:301–12. doi: 10.1016/s0092-8674(00)81661-x. [DOI] [PubMed] [Google Scholar]
- 82.Vestweber D, Winderlich M, Cagna G, Nottebaum AF. Cell adhesion dynamics at endothelial junctions: VE-cadherin as a major player. Trends Cell Biol. 2009;19:8–15. doi: 10.1016/j.tcb.2008.10.001. [DOI] [PubMed] [Google Scholar]
- 83.Matthay MA, Ware LB, Zimmerman GA. The acute respiratory distress syndrome. J Clin Investig. 2012;122:2731–40. doi: 10.1172/JCI60331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Garcia JG, Liu F, Verin AD, Birukova A, Dechert MA, et al. Sphingosine 1-phosphate promotes endothelial cell barrier integrity by Edg-dependent cytoskeletal rearrangement. J Clin Investig. 2001;108:689–701. doi: 10.1172/JCI12450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Schaphorst KL, Chiang E, Jacobs KN, Zaiman A, Natarajan V, et al. Role of sphingosine-1 phosphate in the enhancement of endothelial barrier integrity by platelet-released products. Am J Physiol Lung Cell Mol Physiol. 2003;285:L258–67. doi: 10.1152/ajplung.00311.2002. [DOI] [PubMed] [Google Scholar]
- 86.Wolfson RK, Lang G, Jacobson J, Garcia JGN. Therapeutic strategies to limit lung endothelial cell permeability. 2009:337–54. See Ref. 9. [Google Scholar]
- 87.Camerer E, Regard JB, Cornelissen I, Srinivasan Y, Duong DN, et al. Sphingosine-1-phosphate in the plasma compartment regulates basal and inflammation-induced vascular leak in mice. J Clin Investig. 2009;119:1871–79. doi: 10.1172/JCI38575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Peng X, Hassoun PM, Sammani S, McVerry BJ, Burne MJ, et al. Protective effects of sphingosine 1-phosphate in murine endotoxin-induced inflammatory lung injury. Am J Respir Crit Care Med. 2004;169:1245–51. doi: 10.1164/rccm.200309-1258OC. [DOI] [PubMed] [Google Scholar]
- 89.Tauseef M, Kini V, Knezevic N, Brannan M, Ramchandaran R, et al. Activation of sphingosine kinase-1 reverses the increase in lung vascular permeability through sphingosine-1-phosphate receptor signaling in endothelial cells. Circ Res. 2008;103:1164–72. doi: 10.1161/01.RES.0000338501.84810.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Sammani S, Moreno-Vinasco L, Mirzapoiazova T, Singleton PA, Chiang ET, et al. Differential effects of sphingosine 1-phosphate receptors on airway and vascular barrier function in the murine lung. Am J Respir Cell Mol Biol. 2010;43:394–402. doi: 10.1165/rcmb.2009-0223OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Teijaro JR, Walsh KB, Cahalan S, Fremgen DM, Roberts E, et al. Endothelial cells are central orchestrators of cytokine amplification during influenza virus infection. Cell. 2011;146:980–91. doi: 10.1016/j.cell.2011.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Shea BS, Brooks SF, Fontaine BA, Chun J, Luster AD, Tager AM. Prolonged exposure to sphingo-sine 1-phosphate receptor-1 agonists exacerbates vascular leak, fibrosis, and mortality after lung injury. Am J Respir Cell Mol Biol. 2010;43:662–73. doi: 10.1165/rcmb.2009-0345OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Li X, Stankovic M, Bonder CS, Hahn CN, Parsons M, et al. Basal and angiopoietin-1-mediated endothelial permeability is regulated by sphingosine kinase-1. Blood. 2008;111:3489–97. doi: 10.1182/blood-2007-05-092148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Braunstein PW, Cuenoud HF, Joris I, Majno G. Platelets, fibroblasts, and inflammation: tissue reactions to platelets injected subcutaneously. Am J Pathol. 1980;99:53–66. [PMC free article] [PubMed] [Google Scholar]
- 95.Kim MH, Curry FR, Simon SI. Dynamics of neutrophil extravasation and vascular permeability are uncoupled during aseptic cutaneous wounding. Am J Physiol Cell Physiol. 2009;296:C848–56. doi: 10.1152/ajpcell.00520.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Italiano JE, Jr, Richardson JL, Patel-Hett S, Battinelli E, Zaslavsky A, et al. Angiogenesis is regulated by a novel mechanism: Pro- and antiangiogenic proteins are organized into separate platelet αgranules and differentially released. Blood. 2008;111:1227–33. doi: 10.1182/blood-2007-09-113837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Raghavachari N, Xu X, Harris A, Villagra J, Logun C, et al. Amplified expression profiling of platelet transcriptome reveals changes in arginine metabolic pathways in patients with sickle cell disease. Circulation. 2007;115:1551–62. doi: 10.1161/CIRCULATIONAHA.106.658641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Rondina MT, Schwertz H, Harris ES, Kraemer BF, Campbell RA, et al. The septic milieu triggers expression of spliced tissue factor mRNA in human platelets. J Thromb Haemost. 2011;9:748–58. doi: 10.1111/j.1538-7836.2011.04208.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Sato K, Webb S, Tucker A, Rabinovitch M, O’Brien RF, et al. Factors influencing the idiopathic development of pulmonary hypertension in the fawn hooded rat. Am Rev Respir Dis. 1992;145:793–97. doi: 10.1164/ajrccm/145.4_Pt_1.793. [DOI] [PubMed] [Google Scholar]
- 100.Peacock AJ, Egermayer P, Town GI. The role of platelets in pulmonary hypertension. In: Weir EK, Reeve HL, Reeves JT, editors. Interactions of Blood and the Pulmonary Circulation. New York: Futura; 2002. pp. 187–201. [Google Scholar]
- 101.McGoon MD, Vanhoutte PM. Aggregating platelets contract isolated canine pulmonary arteries by releasing 5-hydroxytryptamine. J Clin Investig. 1984;74:828–33. doi: 10.1172/JCI111499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Ciuclan L, Bonneau O, Hussey M, Duggan N, Holmes AM, et al. A novel murine model of severe pulmonary arterial hypertension. Am J Respir Crit Care Med. 2011;184:1171–82. doi: 10.1164/rccm.201103-0412OC. [DOI] [PubMed] [Google Scholar]
- 103.Harris ES, Rondina MT, Schwertz H, Weyrich AS, Zimmerman GA. Pathogenesis of sepsis and sepsis-induced acute lung injury. In: Choi AMK, editor. Acute Respiratory Distress Syndrome. 2 New York: Informa Healthcare; 2010. pp. 369–419. [Google Scholar]
- 104.Heffner JE, Shoemaker SA, Canham EM, Patel M, McMurtry IF, et al. Acetyl glyceryl ether phosphorylcholine-stimulated human platelets cause pulmonary hypertension and edema in isolated rabbit lungs. Role of thromboxane A2. J Clin Investig. 1983;71:351–57. doi: 10.1172/JCI110776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Snapper JR, Hinson JM, Jr, Hutchison AA, Lefferts PL, Ogletree ML, Brigham KL. Effects of platelet depletion on the unanesthetized sheep’s pulmonary response to endotoxemia. J Clin Investig. 1984;74:1782–91. doi: 10.1172/JCI111597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Vanhoutte PM, Houston DS. Platelets, endothelium, and vasospasm. Circulation. 1985;72:728–34. doi: 10.1161/01.cir.72.4.728. [DOI] [PubMed] [Google Scholar]
- 107.Nurden AT. Platelets, inflammation and tissue regeneration. Thromb Haemost. 2011;105(Suppl 1):13–33. doi: 10.1160/THS10-11-0720. [DOI] [PubMed] [Google Scholar]
- 108.de Boer HC, Verseyden C, Ulfman LH, Zwaginga JJ, Bot I, et al. Fibrin and activated platelets cooperatively guide stem cells to a vascular injury and promote differentiation towards an endothelial cell phenotype. Arterioscler Thromb Vasc Biol. 2006;26:1653–59. doi: 10.1161/01.ATV.0000222982.55731.f1. [DOI] [PubMed] [Google Scholar]
- 109.Massberg S, Konrad I, Schurzinger K, Lorenz M, Schneider S, et al. Platelets secrete stromal cell-derived factor 1α and recruit bone marrow-derived progenitor cells to arterial thrombi in vivo. J Exp Med. 2006;203:1221–33. doi: 10.1084/jem.20051772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Stellos K, Langer H, Daub K, Schoenberger T, Gauss A, et al. Platelet-derived stromal cell-derived factor-1 regulates adhesion and promotes differentiation of human CD34+ cells to endothelial progenitor cells. Circulation. 2008;117:206–15. doi: 10.1161/CIRCULATIONAHA.107.714691. [DOI] [PubMed] [Google Scholar]
- 111.Stellos K, Gnerlich S, Kraemer B, Lindemann S, Gawaz M. Platelet interaction with progenitor cells: vascular regeneration or inquiry? Pharmacol Rep. 2008;60:101–8. [PubMed] [Google Scholar]
- 112.Jin DK, Shido K, Kopp HG, Petit I, Shmelkov SV, et al. Cytokine-mediated deployment of SDF-1 induces revascularization through recruitment of CXCR4+ hemangiocytes. Nat Med. 2006;12:557–67. doi: 10.1038/nm1400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Weyrich AS, Elstad MR, McEver RP, McIntyre TM, Moore KL, et al. Activated platelets signal chemokine synthesis by human monocytes. J Clin Investig. 1996;97:1525–34. doi: 10.1172/JCI118575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Dixon DA, Tolley ND, Bemis-Standoli K, Martinez ML, Weyrich AS, et al. Expression of COX-2 in platelet-monocyte interactions occurs via combinatorial regulation involving adhesion and cytokine signaling. J Clin Investig. 2006;116:2727–38. doi: 10.1172/JCI27209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Shi Q, Wilcox DA, Fahs SA, Weiler H, Wells CW, et al. Factor VIII ectopically targeted to platelets is therapeutic in hemophilia A with high-titer inhibitory antibodies. J Clin Investig. 2006;116:1974–82. doi: 10.1172/JCI28416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Barst RJ. PDGF signaling in pulmonary arterial hypertension. J Clin Investig. 2005;115:2691–94. doi: 10.1172/JCI26593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Tuder RM, Erzurum SC. Pulmonary endothelium and pulmonary hypertension. 2009:449–60. See Ref. 9. [Google Scholar]
- 118.Eddahibi S, Guignabert C, Barlier-Mur AM, Dewachter L, Fadel E, et al. Cross talk between endothelial and smooth muscle cells in pulmonary hypertension: critical role for serotonin-induced smooth muscle hyperplasia. Circulation. 2006;113:1857–64. doi: 10.1161/CIRCULATIONAHA.105.591321. [DOI] [PubMed] [Google Scholar]
- 119.Lang IM. Pulmonary endothelium in thromboembolism. 2009:471–83. See Ref. 9. [Google Scholar]
- 120.Dees C, Akhmetshina A, Zerr P, Reich N, Palumbo K, et al. Platelet-derived serotonin links vascular disease and tissue fibrosis. J Exp Med. 2011;208:961–72. doi: 10.1084/jem.20101629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Fabre A, Marchal-Somme J, Marchand-Adam S, Quesnel C, Borie R, et al. Modulation of bleomycin-induced lung fibrosis by serotonin receptor antagonists in mice. Eur Respir J. 2008;32:426–36. doi: 10.1183/09031936.00126907. [DOI] [PubMed] [Google Scholar]
- 122.Rondina MT, Brewster B, Grissom CK, Zimmerman GA, Kastendieck DH, et al. In vivo platelet activation in critically ill patients with primary 2009 influenza A(H1N1) Chest. 2012;141:1490–95. doi: 10.1378/chest.11-2860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Looney MR, Su X, Van Ziffle JA, Lowell CA, Matthay MA. Neutrophils and their Fc receptors are essential in a mouse model of transfusion-related acute lung injury. J Clin Investig. 2006;116:1615–23. doi: 10.1172/JCI27238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Looney MR, Nguyen JX, Hu Y, Van Ziffle JA, Lowell CA, Matthay MA. Platelet depletion and aspirin treatment protect mice in a two-event model of transfusion-related acute lung injury. J Clin Investig. 2009;119:3450–61. doi: 10.1172/JCI38432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Caudrillier A, Kessenbrock K, Gilliss BM, Nguyen JX, Marques MB, et al. Platelets induce neutrophil extracellular traps in transfusion-related acute lung injury. J Clin Investig. 2012;122:2661–71. doi: 10.1172/JCI61303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Davi G, Iapichino L, Balsamo V, Ganci A, Giammarresi C, et al. Thromboxane biosynthesis and pulmonary function in cystic fibrosis. Adv Prostaglandin Thromboxane Leukot Res. 1995;23:369–71. [PubMed] [Google Scholar]
- 127.Pitchford SC, Yano H, Lever R, Riffo-Vasquez Y, Ciferri S, et al. Platelets are essential for leukocyte recruitment in allergic inflammation. J Allergy Clin Immunol. 2003;112:109–18. doi: 10.1067/mai.2003.1514. [DOI] [PubMed] [Google Scholar]
- 128.O’Sullivan BP, Michelson AD. The inflammatory role of platelets in cystic fibrosis. Am J Respir Crit Care Med. 2006;173:483–90. doi: 10.1164/rccm.200508-1243PP. [DOI] [PubMed] [Google Scholar]
- 129.Pitchford SC, Momi S, Baglioni S, Casali L, Giannini S, et al. Allergen induces the migration of platelets to lung tissue in allergic asthma. Am J Respir Crit Care Med. 2008;177:604–12. doi: 10.1164/rccm.200702-214OC. [DOI] [PubMed] [Google Scholar]
- 130.Johansson MW, Han ST, Gunderson KA, Busse WW, Jarjour NN, Mosher DF. Platelet activation, P-selectin, and eosinophil β1-integrin activation in asthma. Am J Respir Crit Care Med. 2012;185:498–507. doi: 10.1164/rccm.201109-1712OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Tammela T, Alitalo K. Lymphangiogenesis: molecular mechanisms and future promise. Cell. 2010;140:460–76. doi: 10.1016/j.cell.2010.01.045. [DOI] [PubMed] [Google Scholar]
- 132.Murciano JC, Harshaw D, Neschis DG, Koniaris L, Bdeir K, et al. Platelets inhibit the lysis of pulmonary microemboli. Am J Physiol Lung Cell Mol Physiol. 2002;282:L529–39. doi: 10.1152/ajplung.00112.2001. [DOI] [PubMed] [Google Scholar]
- 133.von Bruhl ML, Stark K, Steinhart A, Chandraratne S, Konrad I, et al. Monocytes, neutrophils, and platelets cooperate to initiate and propagate venous thrombosis in mice in vivo. J Exp Med. 2012;209:819–35. doi: 10.1084/jem.20112322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Gay LJ, Felding-Habermann B. Contribution of platelets to tumour metastasis. Nat Rev Cancer. 2011;11:123–34. doi: 10.1038/nrc3004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Lonsdorf AS, Kramer BF, Fahrleitner M, Schonberger T, Gnerlich S, et al. Engagement of ανbβ3 (GPIIb/IIIa) with a β3 integrin mediates interaction of melanoma cells with platelets: a connection to hematogenous metastasis. J Biol Chem. 2012;287:2168–78. doi: 10.1074/jbc.M111.269811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Yiming MT, Lederer DJ, Sun L, Huertas A, Issekutz AC, Bhattacharya S. Platelets enhance endothelial adhesiveness in high tidal volume ventilation. Am J Respir Cell Mol Biol. 2008;39:569–75. doi: 10.1165/rcmb.2007-0332OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Denis MM, Tolley ND, Bunting M, Schwertz H, Jiang H, et al. Escaping the nuclear confines: signal-dependent pre-mRNA splicing in anucleate platelets. Cell. 2005;122:379–91. doi: 10.1016/j.cell.2005.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Weyrich AS, Denis MM, Schwertz H, Tolley ND, Foulks J, et al. mTOR-dependent synthesis of Bcl-3 controls the retraction of fibrin clots by activated human platelets. Blood. 2007;109:1975–83. doi: 10.1182/blood-2006-08-042192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Hogg JC, Doerschuk CM. Leukocyte traffic in the lung. Annu Rev Physiol. 1995;57:97–114. doi: 10.1146/annurev.ph.57.030195.000525. [DOI] [PubMed] [Google Scholar]