

Abstract
Background/Aims
The senescence marker protein-30 (SMP30) is a 34 kDa protein originally identified in rat liver that shows decreased levels with age. Several functional studies using SMP30 knockout (Smp30Y/−) mice established that SMP30 functions as an antioxidant and protects against apoptosis. To address the potential role of SMP30 in nonalcoholic fatty liver disease (NAFLD) pathogenesis, we established Smp30Y/− mice on a Leprdb/db background (Leprdb/dbSmp30Y/− mice).
Research Design/Principal Findings
Male Leprdb/dbSmp30Y/− mice were fed a standard diet (340 kcal/100 g, fat 5.6%) for 16 weeks whereupon the lipid/lipoprotein profiles, hepatic expression of genes related to lipid metabolism and endoplasmic reticulum stress markers were analyzed by HPLC, quantitative RT-PCR and western blotting, respectively. Changes in the liver at a histological level were also investigated. The amount of SMP30 mRNA and protein in livers was decreased in Leprdb/dbSmp30Y/+ mice compared with Leprdb/+Smp30Y/+ mice. Compared with Leprdb/dbSmp30Y/+ mice, 24 week old Leprdb/dbSmp30Y/− mice showed: i) increased small dense LDL-cho and decreased HDL-cho levels; ii) fatty liver accompanied by numerous inflammatory cells and increased oxidative stress; iii) decreased mRNA expression of genes involved in fatty acid oxidation (PPARα) and lipoprotein uptake (LDLR and VLDLR) but increased CD36 levels; and iv) increased endoplasmic reticulum stress.
Conclusion
Our data strongly suggest that SMP30 is closely associated with NAFLD pathogenesis, and might be a possible therapeutic target for NAFLD.
Introduction
Metabolic syndrome has been described as the association of insulin resistance, hypertension, hyperlipidemia and obesity. Its prevalence has increased dramatically, mainly in developed countries. The hepatic manifestations of metabolic syndrome include nonalcoholic fatty liver disease (NAFLD) and its progressive variant, nonalcoholic steatohepatitis (NASH) [1], [2]. Several animal models have been proposed for NAFLD and NASH research [3]. Since leptin plays a major role in food intake and energy expenditure, total leptin deficiency or leptin resistance can lead to massive obesity, type 2 diabetes, dyslipidemia and fatty liver. Therefore, many investigations pertaining to NAFLD/NASH have been carried out in genetic leptin-deficient ob/ob mice or leptin-resistant db/db mice that were fed a high fat diet (HFD) or the methionine/choline deficiency diet [3]–[5]. However, these models differ significantly from the human NAFLD/NASH phenotype in a number of pathogenically important ways.
The senescence marker protein-30 (SMP30) is a 34 kDa protein that was originally identified in rat liver and its levels decrease with age [6]. We previously reported that SMP30 participates in Ca2+ efflux by activating the calmodulin-dependent Ca2+-pump that confers resistance to cell injury caused by high intracellular Ca2+ concentrations [7]. We identified SMP30 as a gluconolactonase (GNL) that is involved in L-ascorbic acid biosynthesis in mammals, and have established SMP30-knockout (KO) mice [8]. The livers of SMP30-KO mice are highly susceptible to tumor necrosis factor-α (TNF-a) and Fas-mediated apoptosis, indicating that SMP30 has an anti-apoptotic effect [9]. SMP30-KO mice showed mitochondrial damage and abnormal accumulation of triglycerides, cholesterol, and phospholipids in the liver [10]. In addition, we reported that decreased SMP30 levels contribute to lowered glucose tolerance [11]. These results are in agreement with several functional studies, which also established that SMP30 functions as an antioxidant and anti-apoptotic protein [12]–[15].
To address the potential role of SMP30 in NAFLD/NASH pathogenesis, we generated SMP30-KO mice on a Leprdb/db background (Leprdb/dbSmp30Y/−) and investigated the lipid/lipoprotein profiles, hepatic expression of genes relevant to lipid metabolism and histological changes in the livers of Leprdb/dbSmp30Y/− mice fed a standard diet. Here we show that despite being fed a standard diet, Leprdb/dbSmp30Y/− mice have altered lipoprotein components and severe fatty liver accompanied by increased inflammation and oxidative stress induced by mitochondrial and endoplasmic reticulum dysfunction.
Materials and Methods
Animal crossing and genotyping, and experimental protocol
We used type 2 diabetic obese Leprdb/db mice with a C57BLKS/J background. Male Leprdb/+ mice were obtained from Charles River Laboratories Japan, Inc. (Kanagawa, Japan). SMP30-knockout (KO) mice with a C57BL/6 background were established and maintained as described previously [8], [9]. Heterozygous SMP30-KO male mice do not exist, because the Smp30 gene is located on the X chromosome. SMP30-KO mice cannot synthesize vitamin C in vivo, because in mammals SMP30 is the penultimate enzyme in the vitamin C biosynthetic pathway [8]. To maintain vitamin C levels in tissues that were similar to that of wild type mice, and to eliminate any possible confounding influences of vitamin C deficiency, these mice were given free access to water supplemented with 1.5 g/L vitamin C and 10 µM ethylenediaminetetraacetic acid (EDTA) to avoid the effects of vitamin C deficiency [16]. As schematically illustrated in Figure 1A, male Leprdb/+ mice were first crossed with female Smp30−/− mice to produce male Leprdb/+Smp30Y/− mice and female Leprdb/+Smp30+/− mice. The SMP30 mutant mice genotypes were determined as described previously [9]. Next, male Leprdb/+Smp30Y/− and female Leprdb/+Smp30+/− mice were interbred to produce homozygous Leprdb/dbSmp30Y/+ and Leprdb/dbSmp30Y/− mice and heterozygote control Leprdb/+Smp30Y/− and Leprdb/+Smp30Y/+ mice. The mutant Leprdb gene was identified by restriction enzyme digestion of PCR products. In brief, Leprdb gene PCR products were amplified by PCR using genomic DNA and forward (5′-AGAACGGACACTCTTTGAAGTCTC-3′) and reverse (5′-CATTCAAACCATAGTTTAGGTTTGTGT-3′) primers. PCR products were then digested by AfaI (Takara Bio Inc., Shiga, Japan) and analyzed by agarose gel electrophoresis. The mutant Leprdb gene showed two bands of 108 bp and 27 bp while the wild type allele showed one 135 bp band.
Figure 1. Establishment of Leprdb/dbSmp30Y/− mice.

(A) Generation of Leprdb/dbSmp30Y/− mice. F1 hybrid mice (in green boxes) were crossed to produce the experimental Leprdb/+Smp30Y/+, Leprdb/+Smp30Y/−, Leprdb/dbSmp30Y/+ and Leprdb/dbSmp30Y/− mice (in red boxes). (B) Appearance of Leprdb/dbSmp30Y/− mice at 24 weeks of age. (C) Western blot analysis of SMP30 protein levels in livers from each experimental group at 24 weeks of age. (D) Vitamin C concentration in livers from each experimental group of mice at 24 weeks of age. Values are given as means ± SEM of five animals.
We prepared four groups of five eight week old male mice, with each group having four genotypes: Leprdb/+Smp30Y/+, Leprdb/+Smp30Y/−, Leprdb/dbSmp30Y/+, and Leprdb/dbSmp30Y/−. All mice were fed a vitamin C free-standard diet (CL-2; 340 kcal/100 g, fat 5.6%, CLEA Japan, Tokyo, Japan) for 16 weeks. Leprdb/+Smp30Y/− mice and Leprdb/dbSmp30Y/− mice had free access to 1.5 g/L vitamin C water containing 10 µM EDTA, whereas Leprdb/+Smp30Y/+ and Leprdb/dbSmp30Y/+ mice had 10 µM EDTA water. Mice were maintained on a 12 h light/dark cycle in a controlled environment. All experimental procedures using laboratory animals were approved by the Animal Care and Use Committee of the Tokyo Metropolitan Institute of Gerontology (Permit Number: 12016).
Blood and liver tissue collection
All mice were fasted for 16 h and anesthetized at the age of 24 weeks. Blood was obtained from the inferior vena cava, anticoagulated with EDTA, and subsequently centrifuged at 880×g for 15 min at 4°C. Mice were systemically perfused with ice-cold phosphate buffered saline through the hepatic portal vein to wash out remaining blood cells and then the livers were removed. The whole body subcutaneous fat was collected and the weight measured. The livers were immersed in RNAlater® (Life Technologies Corp., Carlsbad, CA, USA) for RNA extraction and fixed with 10% neutral buffered formalin for histological analysis or frozen in liquid nitrogen for biochemical analysis. All samples were stored at −80°C until use.
Biochemical analysis of blood
Blood glucose levels were measured using a glucometer (Glutest Every; Sanwa Kagaku Kenkyusho Co., Ltd., Aichi, Japan). Plasma insulin levels were measured using an enzyme-linked immunoassay system (Ultra sensitive mouse insulin ELISA kit; Morinaga Institute of Biological Science Inc., Kanagawa, Japan). Plasma total cholesterol (T-cho), triglyceride (TG), phospholipid (PL), aspartate aminotransferase (AST), and alanine aminotransferase (ALT) levels were measured by enzymatic assay kits (Wako Pure Chemicals Industries, Osaka, Japan).
Western blotting analysis
Livers were homogenized in ice-cold homogenization buffer (10 mM Tris-HCl (pH 8.0), 1 mM EDTA, 1 mM phenyl methanesulfonyl fluoride, and a protease inhibitor cocktail (cOmplete, EDTA-free; Roche Diagnostics GmbH, Mannheim, Germany)) for 30 seconds using a high speed homogenizer (POLYTRON® PT-MR 2100; Kinematica AG, Switzerland). The homogenate was then centrifuged at 21,000×g for 10 min at 4°C. The supernatants were boiled for 5 min with a lysis buffer containing 0.125 M Tris–HCl (pH 6.8), 4% SDS, 20% glycerol, 10% 2-mercaptoethanol, and 0.2% bromophenol blue at a ratio of 1∶1. Total protein equivalents for each sample were separated on a 14% polyacrylamide gel and transferred to a polyvinylidene difluoride membrane. The membrane was incubated with the primary antibody, followed by incubation with a horseradish peroxidase-linked goat anti-rabbit IgG (Bio-Rad Laboratories, Tokyo, Japan). The primary antibodies used were: anti-rat SMP30 rabbit polyclonal antibody (Cosmo Bio Co., Ltd., Tokyo, Japan), anti-phosphorylated eukaryotic initiation factor-2α (p-eIF2α) rabbit monoclonal antibody (#3597, Cell Signaling Technology, Beverly, MA) and anti-C/EBP homologous protein (CHOP) rabbit polyclonal antibody (sc-575, Santa Cruz Biotechnology, Santa Cruz, CA) and β-actin (Cell Signaling Technology, Beverly, MA) as a loading control. After washing, immunoreactivity was detected using ECL chemiluminescence reagents (ECL plus, GE Healthcare Japan, Tokyo, Japan). Chemiluminescence signals were quantified with a LAS-3000 imaging system (Fujifilm, Tokyo, Japan). The mean signals from five Leprdb/+Smp30Y/+ mice were assigned a relative value of 1.0. The total protein concentration was determined with a Pierce® BCA Protein Assay Kit (Thermo Fisher Scientific Inc., Waltham, MA) using bovine serum albumin as a standard.
Measurement of vitamin C levels in the liver
Livers were homogenized in 14 vol 5.4% metaphosphate and 1 mM EDTA, and the homogenate was then centrifuged at 21,000×g for 15 min at 4°C. Vitamin C was analyzed by high-performance liquid chromatography (HPLC) using an Atlantis dC18 5 µm column (4.6×150 mm; Nihon Waters, Tokyo, Japan) [17]. The mobile phase was 50 mM phosphate buffer (pH 2.8), 0.2 g/L EDTA, and 2% methanol at a flow rate of 1.3 ml/min, and electrical signals were recorded using an electrochemical detector equipped with a glassy carbon electrode at +0.6 V.
Measurement of T-cho, TG and PL in the liver
Liver tissues were homogenized with 2 vol. of water using a handy homogenizer (Moji-mojikun; Nippon Genetics, Tokyo, Japan). Homogenates were added to a chloroform-methanol (2∶1; v/v) mixture, and centrifuged at 21,000×g for 10 min at 4°C. The supernatant organic phase was then collected, dried under nitrogen gas and resolubilized in 2-propanol. T-cho, TG and PL concentrations in total lipid extracts were determined using commercial enzymatic kits (Wako Pure Chemical Industries, Osaka, Japan).
Thiobarbituric acid reactive substances (TBARS) assay
Lipid peroxidation was estimated by the amounts of TBARS in the liver that were determined according to the method of Ohkawa et al. [18]. The livers were first homogenized in ice-cold 0.1 M phosphate buffer (pH 7.4). The homogenates were then centrifuged at 15,000×g for 30 min at 4°C and the supernatant was used for further assays. One volume of sample was mixed thoroughly with two volumes of stock solution (15% (w/v) trichloroacetic acid, 0.375% (w/v) thiobarbituric acid, and 0.25 mM HCl). The mixture was then heated for 30 min in a boiling water bath. After cooling, the flocculate precipitate was removed by centrifugation at 1,000×g for 10 min and the absorbance (OD 532 nm) of the sample was measured. The TBARS levels are expressed as the equivalent amounts of malondialdehyde produced from 1,1,3,3-tetramethoxypropane.
HPLC analysis of plasma lipoproteins
Plasma lipoproteins were analyzed using a HPLC service (LipoSEARCH) at Skylight Biotech Inc. (Akita, Japan), as previously described [19]. In brief, 10 µL of whole plasma was injected into two connected columns (300×7.8 mm) packed with TSKgel LipopropakXL (Tosoh, Tokyo, Japan) and lipoproteins were separated with 0.05 mol/L Tris-buffered acetate (pH 8.0) containing 0.3 mol/L sodium acetate, 0.05% sodium azide, and 0.005% Brij-35 at a flow rate of 0.7 mL/min. The column effluent was split equally into two lines by a P-460 MicroSplitter (Upchurch Scientific Inc., Oak Harbor, WA, USA); one effluent portion was mixed with cholesterol reagent (Determiner L TC; Kyowa Medex Co., Ltd., Tokyo, Japan) and the other with TG reagent (Determiner L TG; Kyowa Medex). We defined 5 VLDL subclasses (fraction no. 3–7, 30–80 nm), 6 LDL subclasses (fraction no. 8–13, 16–30 nm) and 7 HDL subclasses (fraction no. 14–20, 8–16 nm) using 20 component peaks categorized on the basis of lipoprotein particle size (diameter).
Histological and immunohistochemical examination of liver
To evaluate histological changes, fixed liver sections were subjected to hematoxylin-eosin (H&E) staining or immunohistochemical staining with an anti-4-hydroxy-2-nonenal (4-HNE) protein adducts monoclonal antibody (1∶40 dilution; Nikken SEIL, Shizuoka, Japan).
The H&E-stained specimens were anonymized, and five different areas per mouse were randomly selected by a researcher. These specimens were scored by two independent investigators blinded to sample identity according to the NASH activity score (NAS) [20] for the degree of steatosis (0–3), lobular inflammation (0–3) and hepatocellular ballooning (0–2). The average of the two investigators' scores was regarded as the score for each mouse.
4-HNE was detected by indirect immunoperoxidase staining using corresponding Histofine Simple Stain MAX-PO kits (Nichirei Biosciences, Tokyo, Japan) and 3, 3-diaminobenzidine (DAB) as a chromogenic substrate. After DAB staining, nuclei were counterstained with Mayer's hematoxylin. Two independent observers evaluated the intensity of 4-HNE immunostaining and assigned scores of 0, 1, 2, or 3 (negative (including faint staining), weak, moderate, or strong, respectively).
RNA isolation, first-strand cDNA synthesis, and gene expression analysis
Liver tissue was finely ground with a liquid nitrogen-cooled mortar and pestle and homogenized in ice-cold TRIzol reagent (Life Technologies, Carlsbad, CA, USA) before isolation of total RNA according to the manufacturer's instructions. Total RNA (0.5 µg) was reverse-transcribed using PrimeScript RT Master Mix (TaKaRa Bio Inc., Shiga, Japan) for first-strand cDNA synthesis with an oligonucleotide dT primer and random hexamer priming according to the manufacturer's recommendations.
The mRNA expression levels of the following proteins: acetyl-CoA carboxylase (ACC), fatty acid synthase (FAS), sterol regulatory element-binding protein 1c (SREBP1c), SREBP2, 3-hydroxy-3-methylglutaryl-CoA reductase (HMGCoAR), peroxisome proliferator-activated receptor-α (PPARα), medium-chain acyl-CoA dehydrogenase (MCAD), microsomal triglyceride transfer protein (MTP), apolipoprotein-B100 (ApoB100), LDL receptor (LDLR) and VLDL receptor (VLDLR), CD36 and spliced X-box binding protein 1 (sXBP1), all of which are involved in lipid and lipoprotein metabolism, were quantitated using real-time reverse transcription polymerase chain reaction (RT-PCR). RT-PCR was performed using a Thermal Cycler Dice Real Time System II (TaKaRa Bio Inc.) and real-time SYBR® Premix Ex Taq™ (TaKaRa Bio Inc.) according to the manufacturer's instructions. The specific primers for the target genes and β-actin are described in Table 1. The following PCR conditions were used: 1 cycle for 30 s at 95°C, followed by 40 cycles for 5 s at 95°C, and 30 s at 60°C. The product specificity generated for each primer set was examined for each fragment using a melting curve and gel electrophoresis. The relative expression levels of each targeted gene were normalized to β-actin threshold cycle (CT) values and quantified using the comparative threshold cycle 2−ΔΔCT method as previously described [21]. Signals from Leprdb/+Smp30Y/+ mice were assigned a relative value of 1.0. Five mice from each group were examined, and real time RT-PCR was run in duplicate for each sample.
Table 1. Primer sequences for use in real-time quantitative RT-PCR.
| Gene name | Sequence | Size (bp) | GenBank accession no. | |
| Acetyl-CoA carboxylase | Forward | 5′-AGCGACATGAACACCGTACTGAA-3′ | 106 | NM133360 |
| Reverse | 5′-TAGGGTCCCGGCCACATAAC-3′ | |||
| Fatty acid synthase | Forward | 5′-ATTGGCTCCACCAAATCCAAC-3′ | 90 | NM007988 |
| Reverse | 5′-CCCATGCTCCAGGGATAACAG-3′ | |||
| Sterol regulatory element-binding protein-1c | Forward | 5′-AGCCTGGCCATCTGTGAGAA-3′ | 132 | NM011480 |
| Reverse | 5′-CAGACTGGTACGGGCCACAA-3′ | |||
| Sterol regulatory element-binding protein-2 | Forward | 5′-CCCACTCAGAACACCAAGCAT-3 | 67 | NM033218 |
| Reverse | 5′-TGGCAGTAGCTCGCTCTCGT-3′ | |||
| HMGCoA reductase | Forward | 5′- TGTCCTTGATGGCAGCCTTG-3′ | 183 | NM 008255 |
| Reverse | 5′- CCGCGCTTCAGTTCAGTGTC-3′ | |||
| Peroxisome proliferator-activated receptor-α | Forward | 5′-CTCAGGGTACCACTACGGAGTTCAC-3′ | 111 | NM001113418 |
| Reverse | 5′-TGAATCTTGCAGCTCCGATCAC-3′ | |||
| Medium-chain acyl-CoA dehydrogenase | Forward | 5′- TGAGTGGCGGCCATTAAGA-3′ | 179 | NM007382 |
| Reverse | 5′- CGGCTTCCACAATGAATCCAG-3′ | |||
| Microsomal triglyceride transfer protein | Forward | 5′-CATCTCCACAGTGCAGTTCTCACA-3′ | 167 | NM001163457 |
| Reverse | 5′-GGAGTTCACATCCGGCCACTA-3′ | |||
| Apolipoprotein-B100 | Forward | 5′-CAGGTGGCCACAGCCAATAA-3′ | 161 | NM009693 |
| Reverse | 5′-ACTGCAGGTCTGGCTCAGGA-3′ | |||
| LDL receptor | Forward | 5′- CTAGCAAGTGGGTGTGCGATG-3′ | 100 | NM010700 |
| Reverse | 5′- CTGAATTGATTGGACTGACAGGTGA -3′ | |||
| VLDL receptor | Forward | 5′- TGGCGTGTGCAAGGCAGTA-3′ | 145 | NM013703 |
| Reverse | 5′- TGCAATGTCCGCATCGAGA-3′ | |||
| CD36 | Forward | 5′-CCCAGATGACGTGGCAAAGA-3′ | 172 | NM001159555 |
| Reverse | 5′-TCCAACAGACAGTGAAGGCTCAA-3′ | |||
| Spliced XBP1 | Forward | 5′-GCTGAGTCCGCAGCAGGT-3′ | 86 | NM013842 |
| Reverse | 5′-GAATCTGAAGAGGCAACAGTGTCA-3′ | |||
| Senescence marker protein-30 | Forward | 5′-GAGGCAGCCTGATGCTGGTAA-3′ | 92 | NM009060 |
| Reverse | 5′-GAGCTGCAGTTCACCCTGCATA-3′ | |||
| β-actin | Forward | 5′-CATCCGTAAAGACCTCTATGCCAAC-3′ | 171 | NM007393 |
| Reverse | 5′-ATGGAGCCACCGATCCACA-3′ |
Statistical analysis
Data are expressed as means ± SEM. Statistical differences between groups were determined by one-way analysis of variance (ANOVA) with Scheffe's post hoc test. A P value<0.05 was considered to be statistically significant.
Results
Generation of Leprdb/dbSmp30Y/− mice
As shown in Fig. 1A and B, we established Leprdb/dbSmp30Y/− mice, which were born at the expected Mendelian ratios and by 24 weeks of age had appearances that were indistinguishable from obese Leprdb/dbSmp30Y/+ mice. Western blot analysis of liver tissue from Leprdb/+Smp30Y/− and Leprdb/dbSmp30Y/− mice demonstrated that these animals lacked SMP30 protein (Fig. 1C). There were no significant differences found in the liver vitamin C concentration among any of the experimental groups (Fig. 1D).
Given the difference in SMP30 protein levels observed for Leprdb/+Smp30Y/+ and Leprdb/dbSmp30Y/+ mice (Fig. 1C), we next quantified the amounts of SMP30 mRNA and protein and found that they were decreased by 25% and 47%, respectively, in Leprdb/dbSmp30Y/+ mice as compared to Leprdb/+Smp30Y/+ mice (both P<0.01) (Fig. 2).
Figure 2. Decrease in hepatic SMP30 levels in Leprdb/db mice.

(A) SMP30 mRNA and (B) protein levels in livers from Leprdb/+Smp30Y/+ and Leprdb/dbSmp30Y/+ mice. Values are given as means ± SEM of five animals. † P<0.01.
Leprdb/dbSmp30Y/− mice had manifestations of metabolic syndrome
The physiological and blood biochemical parameters of 24 week old animals of the four groups are presented in Table 2. Compared with Leprdb/+Smp30Y/+ and Leprdb/+Smp30Y/− mice, the food intake of Leprdb/dbSmp30Y/− mice during the experimental period was significantly increased by 33% and 40%, respectively (P<0.001). Meanwhile, there were no significant differences between Leprdb/dbSmp30Y/− and Leprdb/dbSmp30Y/+ mice (P = 0.07). The body weight of Leprdb/dbSmp30Y/− mice was 79% and 87% higher than Leprdb/+Smp30Y/+ and Leprdb/+Smp30Y/− mice, respectively (P<0.001). There were no significant differences in body weight between Leprdb/dbSmp30Y/− and Leprdb/dbSmp30Y/+ mice. Likewise, the epididymal and subcutaneous fat weight in Leprdb/dbSmp30Y/− mice were significantly higher than those of Leprdb/+Smp30Y/+ and Leprdb/+Smp30Y/− mice (P<0.001), although no significant differences in epididymal or subcutaneous fat weight were observed between Leprdb/dbSmp30Y/− and Leprdb/dbSmp30Y/+ mice.
Table 2. Physiological, blood and biochemical parameters in four experimetal groups of mice.
| Leprdb/+Smp30Y/+ | Leprdb/+Smp30Y/− | Leprdb/dbSmp30Y/+ | Leprdb/dbSmp30Y/− | |
| Food intake (g/day/mouse) | 4.2±0.1 | 3.9±0.1 | 5.0±0.2§ , ¶ | 5.5±0.1§ , ¶ |
| Body weight (g) | 34.4±1.7 | 32.9±0.9 | 69.4±2.2§ , ¶ | 61.5±3.0§ , ¶ |
| Epididymal fat weight (g) | 1.12±0.18 | 1.10±0.06 | 2.60±0.13§ , ¶ | 2.42±0.18§ , ¶ |
| Subcutaneous fat weight (g) | 1.20±0.36 | 1.40±0.09 | 12.40±0.80§ , ¶ | 10.82±0.66§ , ¶ |
| Fasting blood glucose (mg/dL) | 71.4±5.8 | 82.2±4.3 | 162.0±15.2* , # | 239.8±26.3§ , ¶ , ** |
| Fasting plasma insulin (ng/mL) | 1.0±0.2 | 1.3±0.5 | 11.6±1.8§ , ¶ | 9.2±0.8§ , ¶ |
| Plasma triglyceride (mg/dL) | 33.4±3.7 | 34.2±3.5 | 66.6±16.9 | 78.8±15.6 |
| Plasma cholesterol (mg/dL) | 59.6±4.0 | 58.4±1.7 | 358.8±58.8§ , ¶ | 333.2±68.1§ , ¶ |
| Plasma phospholipid (mg/dL) | 110.8±11.0 | 125.6±5.2 | 537.2±44.4§ , ¶ | 522.2±53.8§ , ¶ |
| Plasma aspartate aminotransferase (IU/L) | 32.8±4.6 | 37.6±7.1 | 98.3±3.7 | 136.8±27.3† , ‡ |
| Plasma alanine aminotransferase (IU/L) | 11.2±1.5 | 28.8±14.0 | 86.3±7.6* | 111.0±26.1† , # |
P<0.05,
P<0.01 and
P<0.001 versus Leprdb/+Smp30Y/+.
P<0.05,
P<0.01 and
P<0.001 versus Leprdb/+Smp30Y/−.
P<0.05 versus Leprdb/dbSmp30Y/+.
Values are given as means ± SEM of five animals.
Fasting blood glucose concentrations were significantly higher in Leprdb/dbSmp30Y/− mice (239.8±26.3 mg/dL) than in Leprdb/+Smp30Y/+ (71.4±5.8 mg/dL, P<0.001), Leprdb/+Smp30Y/− (82.2±4.3 mg/dL, P<0.001) and Leprdb/dbSmp30Y/+ mice (162.0±15.2 mg/dL, P<0.05). Fasting plasma insulin concentrations in Leprdb/dbSmp30Y/− mice were also higher than in Leprdb/+Smp30Y/+ and Leprdb/+Smp30Y/− mice (P<0.001). There were no significant differences in fasting plasma levels between Leprdb/dbSmp30Y/− and Leprdb/dbSmp30Y/+ mice.
Although there was a non-significant increase in plasma TG levels in the two groups of Leprdb/db mice (Leprdb/dbSmp30Y/− and Leprdb/dbSmp30Y/+) compared with the two groups of Leprdb/+ mice (Leprdb/+Smp30Y/− and Leprdb/+Smp30Y/+), there were no significant differences in plasma TG levels among any of the experimental groups. Both plasma T-cho and PL concentrations in Leprdb/dbSmp30Y/− mice were significantly higher than those in Leprdb/+Smp30Y/+ and Leprdb/+Smp30Y/− mice (both P<0.001). There were no significant differences in plasma T-cho and PL concentrations between Leprdb/dbSmp30Y/− and Leprdb/dbSmp30Y/+ mice.
Plasma AST and ALT concentrations in Leprdb/dbSmp30Y/− mice (136.8±27.3 and 111.0±26.1 IU/L, respectively) were significantly higher than those of Leprdb/+Smp30Y/+ (32.8±4.6 IU/L, P<0.01 and 11.2±15 IU/L, P<0.01, respectively) and Leprdb/+Smp30Y/− (37.6±7.1 IU/L, P<0.01 and 28.8±14.0 IU/L, P<0.01, respectively) mice. There were no significant differences in plasma AST and ALT concentrations between Leprdb/dbSmp30Y/− and Leprdb/dbSmp30Y/+ mice.
Leprdb/dbSmp30Y/− mice show decreased HDL-cho and increased small dense LDL-cho levels and increased LDL-cho/HDL-cho ratios
HPLC analysis of the plasma lipoprotein profile with cholesterol reagents is shown in Fig. 3A. Either with or without Smp30, the levels of VLDL- (Fr. 3–7), LDL- (Fr. 8–11), very-small particle size LDL- (Fr. 12–13) and HDL-cho (Fr.14–20) as well as the LDL-cho/HLD-cho ratio were significantly higher in Leprdb/db mice (Leprdb/dbSmp30Y/+ and Leprdb/dbSmp30Y/− mice) compared to Leprdb/+ mice (Leprdb/+Smp30Y/+ and Leprdb/+Smp30Y/− mice) (P<0.001) (Fig. 3B–F). There were no significant differences in VLDL-cho levels in Leprdb/dbSmp30Y/− and Leprdb/dbSmp30Y/+ mice (Fig. 3B). Although there were no significant differences in LDL-cho (Fr. 8–11) levels in Leprdb/dbSmp30Y/− and Leprdb/dbSmp30Y/+ mice (Fig. 3C), the very-small sized LDL-cho particles of Fr.12 and Fr.13 that correspond to small dense LDL-cho (sdLDL-cho) in Leprdb/dbSmp30Y/− mice (78.6±3.2 mg/dL) were significantly higher than in Leprdb/dbSmp30Y/+ mice (66.3±3.5 mg/dL, P<0.05) (Fig. 3D), while HDL-cho levels in Leprdb/dbSmp30Y/− mice (173.0±6.1 mg/dL) were significantly lower than that of Leprdb/dbSmp30Y/+ mice (221.0±4.8 mg/dL, P<0.001) (Fig. 3E). The LDL-cho/HDL-cho ratio in Leprdb/dbSmp30Y/− mice was significantly higher than that in Leprdb/dbSmp30Y/+ mice (P<0.001) (Fig. 3F). Likewise, the LDL-cho/HDL-cho ratio in Leprdb/+Smp30Y/− mice was significantly higher than that of Leprdb/+Smp30Y/+ mice (P<0.01) (Fig. 3F).
Figure 3. Increase in small dense LDL-cho and decrease in HDL-cho in plasma from Leprdb/dbSmp30Y/− mice.

(A) HPLC lipoprotein profile with cholesterol reagent and (B–F) cholesterol content in four subfractions according particle size, VLDL; Fr. 3–7, LDL; Fr. 8–11, small dense LDL; Fr. 12–13, HDL; Fr. 14–20 and LDL/HDL ratio in the four experimental groups. Blue dashed line and lane 1, Leprdb/+Smp30Y/+ mice; Red dashed line and lane 2, Leprdb/+Smp30Y/− mice; Blue line and lane 3, Leprdb/dbSmp30Y/+ mice; Red line and lane 4; Leprdb/dbSmp30Y/−mice. Values are given as means ± SEM of five animals. † P<0.01 and § P<0.001 versus Leprdb/+Smp30Y/+, ¶ P<0.001 versus Leprdb/+Smp30Y/−, ** P<0.05 and †† P<0.001 versus Leprdb/dbSmp30Y/+.
Increased hepatic lipid (TG, T-cho and PL) and lipid peroxidation (TBARS) levels in Leprdb/dbSmp30Y/− mice
As shown in Fig. 4A–D, hepatic TG, T-cho, PL and TBARS concentrations were significantly higher in Leprdb/dbSmp30Y/− mice than Leprdb/+Smp30Y/+ or Leprdb/+Smp30Y/− mice (P<0.001). Compared with Leprdb/dbSmp30Y/+ mice, hepatic TG, T-cho and PL concentrations were lower and TBARS concentrations were higher in Leprdb/dbSmp30Y/− mice.
Figure 4. Increase in TBARS in livers from Leprdb/dbSmp30Y/− mice.
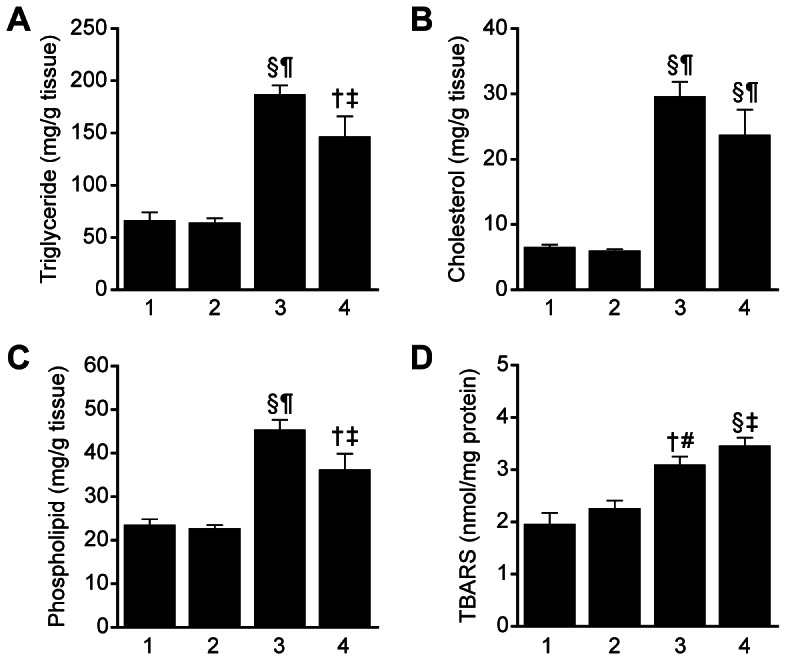
(A) Triglyceride, (B) Cholesterol, (C) Phospholipid and (D) TBARS content in livers from the four experimental groups. Lane 1, Leprdb/+Smp30Y/+ mice. Lane 2, Leprdb/+Smp30Y/− mice. Lane 3, Leprdb/dbSmp30Y/+ mice. Lane 4, Leprdb/dbSmp30Y/− mice. † P<0.01 and § P<0.001 versus Leprdb/+Smp30Y/+, ¶ P<0.001 versus Leprdb/+Smp30Y/−, ** P<0.05 and †† P<0.001 versus Leprdb/dbSmp30Y/+. Values are given as means ± SEM of five animals.
Leprdb/dbSmp30Y/− mice show fatty liver accompanied by inflammatory cells and oxidative stress despite being fed a standard diet
As shown in Fig. 5A, hepatic histological examination revealed increased steatosis in Leprdb/dbSmp30Y/+ mice and increased steatosis accompanied by inflammatory cells in Leprdb/dbSmp30Y/− mice. In contrast, no histological abnormalities were observed for both Leprdb/+Smp30Y/+ and Leprdb/+Smp30Y/− animals. Fibrosis was not observed in any of the groups.
Figure 5. Increased steatosis, inflammation and oxidative stress in liver sections from Leprdb/dbSmp30Y/− mice.

Representative images of (A) hematoxylin/eosin staining and (B) 4-HNE immunostaining in liver sections from Leprdb/+Smp30Y/+, Leprdb/+Smp30Y/−, Leprdb/dbSmp30Y/+ and Leprdb/dbSmp30Y/− mice. Scale bar is 100 µm. * P<0.05, † P<0.01 and § P<0.001 versus Leprdb/+Smp30Y/+, # P<0.05, ‡ P<0.01 and ¶ P<0.001 versus Leprdb/+Smp30Y/−, ** P<0.05 versus Leprdb/dbSmp30Y/+. Values are given as mean ± SEM of five animals.
Regarding the NASH activity score, 24 week old Leprdb/dbSmp30Y/− mice had a total score of 4.4±0.2, which was significantly higher than that of Leprdb/dbSmp30Y/+ mice (3.2±0.4, P<0.05). In immunohistochemical examinations using an anti-4HNE antibody, the 4-HNE intensity scores were significantly higher in Leprdb/dbSmp30Y/− mice (1.8±0.2) than in Leprdb/dbSmp30Y/+ (1.2±0.2, P<0.05), Leprdb/+Smp30Y/− (0.8±0.2, P<0.01) and Leprdb/+Smp30Y/+ (0.2±0.2, P<0.001) (Fig. 5B) mice.
Altered hepatic expression of lipid/lipoprotein metabolism-related genes in Leprdb/dbSmp30Y/− mice
Next, we investigated the hepatic expression of genes related to lipid/lipoprotein metabolism in Leprdb/dbSmp30Y/− mice (Fig. 6). Regarding lipogenesis genes, hepatic expression of ACC, FAS and SREBP1c were markedly higher in Leprdb/db mice (Leprdb/dbSmp30Y/+ and Leprdb/dbSmp30Y/−) than in Leprdb/+ mice (Leprdb/+Smp30Y/+ and Leprdb/+Smp30Y/−) with or without the SMP30 gene. SREBP1c mRNA levels in Leprdb/dbSmp30Y/− mice were significantly lower than that in Leprdb/dbSmp30Y/+ mice (P<0.05), although there was no significant difference in ACC and FAS mRNA expression between Leprdb/+Smp30Y/+ mice and Leprdb/+Smp30Y/− mice or between Leprdb/dbSmp30Y/+ mice and Leprdb/dbSmp30Y/− mice. On the other hand, independent of the SMP30 gene, SREBP2 and HMGCoAR mRNA levels were significantly lower in Leprdb/db mice than those in Leprdb/+. There were no significant differences in SREBP2 and HMGCoAR expression levels between Leprdb/dbSmp30Y/+ and Leprdb/dbSmp30Y/− mice or between Leprdb/+Smp30Y/+ and Leprdb/+Smp30Y/− mice.
Figure 6. Altered hepatic expression of lipid/lipoprotein metabolism-related genes in Leprdb/dbSmp30Y/− mice.

Gene expression levels of (A) ACC, (B) FAS, (C) SREBP1c, (D) SREBP2, (E) HMGCoAR, (F) PPARα, (G) MCAD, (H) MTP, (I) ApoB100, (J) LDLR (K) VLDLR, and (L) CD36 in the livers. Lane 1: Leprdb/+Smp30Y/+, lane 2: Leprdb/+Smp30Y/−, lane 3: Leprdb/dbSmp30Y/+ and lane 4: Leprdb/dbSmp30 Y/− mice. mRNA for each gene was measured using real time RT-PCR and normalized to β-actin. The values from Leprdb/+ Smp30Y/+ mice were assigned a relative value of 1.0. Values are given as means ± SEM of five animals. * P<0.05, † P<0.01 and § P<0.001 versus Leprdb/+Smp30Y/+, # P<0.05, ‡ P<0.01 and ¶ P<0.001 versus Leprdb/+Smp30Y/−, ** P<0.05 versus Leprdb/dbSmp30Y/+.
mRNA levels of PPARα, which is involved in fatty acid catabolism, were significantly lower in Leprdb/dbSmp30Y/− mice than in Leprdb/+Smp30Y/+ (P<0.001), Leprdb/+Smp30Y/− (P<0.001) or Leprdb/dbSmp30Y/+ (P<0.05) mice. Importantly, PPARα mRNA levels were significantly lower in Leprdb/+Smp30Y/− mice compared to Leprdb/+Smp30Y/+ mice (P<0.05). No significant difference was observed in MCDA mRNA levels between Leprdb/dbSmp30Y/+ and Leprdb/dbSmp30Y/− mice or between Leprdb/+Smp30Y/+ and Leprdb/+Smp30Y/− mice.
MTP and ApoB100 are key genes for VLDL secretion. Both MTP and ApoB100 mRNA levels were significantly lower in Leprdb/dbSmp30Y/− mice than in Leprdb/+Smp30Y/+ and Leprdb/+Smp30Y/− mice (P<0.001). Compared with Leprdb/dbSmp30Y/+ mice, MTP mRNA expression levels in Leprdb/dbSmp30Y/− mice were decreased, although the change did not reach statistical significance. There was no significant difference in ApoB100 mRNA levels between Leprdb/dbSmp30Y/+ and Leprdb/dbSmp30Y/− mice.
Interestingly, hepatic LDLR mRNA levels were significantly lower in Leprdb/dbSmp30Y/− mice than in Leprdb/+Smp30Y/+ (P<0.05) and Leprdb/dbSmp30Y/+ (P<0.05) mice, but not Leprdb/+Smp30Y/− mice. Hepatic VLDLR mRNA levels were significantly higher in Leprdb/dbSmp30Y/− mice than in Leprdb/+Smp30Y/+ (P<0.001) and Leprdb/+Smp30Y/− mice (P<0.001), and were significantly lower than in Leprdb/dbSmp30Y/+ mice (P<0.05). CD36 mRNA levels were significantly higher in Leprdb/dbSmp30Y/− mice than in Leprdb/+Smp30Y/+ (P<0.001), Leprdb/+Smp30Y/− (P<0.001) or Leprdb/dbSmp30Y/+ (P<0.05) mice.
Leprdb/dbSmp30Y/− mice show increased endoplasmic reticulum (ER) stress
The high plasma sdLDL-cho, high liver TBARS content and low mRNA expression levels of fatty acid β oxidation and lipid secretion genes in Leprdb/dbSmp30Y/− mice prompted us to analyze hepatic levels of ER stress markers. No significant differences in spliced XBP1 mRNA expression were observed among the four experimental mice groups (Fig. 7A). Phosphorylated eIF2α (phos-eIF2α) levels in Leprdb/dbSmp30Y/− mice were significantly (two-fold) higher (P<0.01) compared with Leprdb/+Smp30Y/+ mice (Fig. 7B). Also, there was a non-significant increase in phos-eIF2α levels in Leprdb/dbSmp30Y/− mice compared with Leprdb/+Smp30Y/− or Leprdb/dbSmp30Y/+ mice (Fig. 7B). On the other hand, CHOP levels were significantly higher in Leprdb/dbSmp30Y/− than in Leprdb/+Smp30Y/+ (P<0.01), Leprdb/+Smp30Y/− (P<0.01) or Leprdb/dbSmp30Y/+ (P<0.05) mice.
Figure 7. Increase in endoplasmic reticulum (ER) stress in livers from Leprdb/dbSmp30Y/− mice.

(A) Spliced XBP1 mRNA expression, (B) Phosphorylated eIF2α protein and (C) CHOP protein levels in livers from the four experimental groups. Values are given as means ± SEM of five animals. * P<0.05, † P<0.01 and § P<0.001 versus Leprdb/+Smp30Y/+, # P<0.05 versus Leprdb/+Smp30Y/−, ** P<0.05 versus Leprdb/dbSmp30Y/+.
Discussion
The two-hit theory proposed by Day and James [22], in which the initial trigger is the hepatic accumulation of excessive fat, followed by the second hit of oxidative stress, is widely advocated as a pathogenic mechanism for NASH. Therefore, it is of great interest to study in greater detail the role of SMP30 in relation to the pathogenic mechanism for NAFLD/NASH in SMP30-KO mice on a Leprdb/db background. Interestingly, we observed first that SMP30 levels in Leprdb/dbSmp30Y/+ mice were significantly lower than in Leprdb/+Smp30Y/+ mice, which suggests that this decrease is related to the development of obesity and obesity-related disorders in Leprdb/dbSmp30Y/+ mice. The mechanism(s) of SMP30 reduction in Leprdb/dbSmp30Y/+ mouse livers is unknown. Liver SMP30 protein levels were reported to decrease following liver injury in animals treated with carbon tetrachloride [23], lipopolysaccharide (LPS) [24] or D-galactosamine/LPS [25]. Furthermore, we recently reported that 17β-estradiol attenuates saturated fatty acid diet-induced apoptotic liver injury in ovariectomized mice by up-regulating hepatic SMP30 [14]. Thus, the lipid deposition accompanied by increased oxidative and ER stress in Leprdb/dbSmp30Y/− mice might result from decreased liver SMP30 levels and in turn exacerbate liver damage via decreased Ca2+-pumping activity and anti-oxidative effects of SMP30. Further study will be required to reveal the mechanisms of SMP30 suppression in obesity and obesity-related diseases.
A noteworthy finding of this study is that plasma levels of LDL-cho, in particular the smaller sized particles of Fr.no. 12 and Fr.no. 13 that correspond to small dense LDL-cho (sdLDL-cho) and the LDL-cho/HDL-cho ratio in Leprdb/dbSmp30Y/− mice, were significantly higher than those in Leprdb/dbSmp30Y/+ mice, although plasma levels of T-cho in Leprdb/dbSmp30Y/− and Leprdb/dbSmp30Y/+ mice were similar. A significantly increased LDL-cho/HDL-cho ratio was also found in Leprdb/+Smp30Y/− mice compared with Leprdb/+Smp30Y/+ mice. These results indicate that the SMP30 deficiency contributes to increases in plasma sdLDL-cho and decreases in HDL-cho regardless of leptin receptor mutation followed by hyperphagia and obesity. In a human study an association between fatty liver and increased sdLDL-cho was reported [26]–[28]. Furthermore, we recently reported that in patients with histologically diagnosed NAFLD/NASH, serum sdLDL-cho levels in patients with NAS ≥5 were significantly higher than those in patients with NAS≤2, and sdLDL-cho was significantly and inversely correlated with hepatic SMP30 levels [29]. However, we do not presently have an explanation for the observed increase in sdLDL-cho in Leprdb/dbSmp30Y/− mice. In humans, TG-rich VLDL (large VLDL1) can be a precursor of sdLDL-cho, i.e., large VLDL1 particles are converted to sdLDL particles by cholesteryl ester transport protein (CETP) and hepatic lipase (HL), the levels of which are commonly increased in type 2 diabetes [30]. Unlike humans, however, mice do not express CETP, and as such, cholesterol is mainly present in HDL. Qiu et al. reported that HL-deficient mice have sdLDLs, but TG enrichment was not observed in these mice [31]. In this study, no difference was observed in the mRNA expression of HL between Leprdb/dbSmp30Y/− and Leprdb/dbSmp30Y/+ mice or between Leprdb/+Smp30Y/− and Leprdb/+Smp30Y/+ mice (data not shown). Recently, we demonstrated that testosterone-deficient mice fed a high-fat diet showed markedly decreased serum TG and TG-VLDL levels and markedly increased serum sdLDL-cho levels, likely due to altered expression of genes involved in hepatic assembly and lipid secretion [32]. Further work will be required to elucidate the molecular mechanism for this increase in sdLDL-cho in SMP30-KO mice.
An additional notable finding of this study was that although Leprdb/dbSmp30Y/− mice showed no advanced stage NASH including fibrosis, compared with Leprdb/dbSmp30Y/+ mice Leprdb/dbSmp30Y/− mice had increased NAS activity and inflammation scores as well as enhanced oxidative stress. Hepatic steatosis results from an imbalance in lipid homeostasis in the liver, where fat uptake, de novo lipogenesis, fatty acid oxidation and fat export occur. Compared with Leprdb/dbSmp30Y/+ mice, RT-PCR of lipid homeostasis-related genes in the liver revealed that the expression levels of these genes, in particular PPARα and SREBP-1c (but not CD36), are reduced in Leprdb/dbSmp30Y/− mice. CD36, a member of the class B scavenger receptor family of cell surface proteins, is abundantly expressed in monocytes/macrophages. Therefore, compared with Leprdb/dbSmp30Y/+ mice, the increase in CD36 mRNA seen in Leprdb/dbSmp30Y/− mice is likely a partial reflection of the increase in the number of inflammatory cells. PPARα mRNA levels were significantly lower not only in Leprdb/+Smp30Y/− mice compared to Leprdb/+Smp30Y/+ mice, but also in Leprdb/dbSmp30Y/− mice compared to Leprdb/dbSmp30Y/+ mice, suggesting that decreased hepatic SMP30 mRNA expression is associated with mitochondrial and peroxisomal fatty acid β-oxidation.
Mitochondrial dysfunction is known to cause increases in oxidative stress, and indeed we showed that the levels of TBARS and 4-HNE rose in the livers of Leprdb/dbSmp30Y/− mice. When SMP30-KO mice are fed a vitamin C-deficient diet they do not thrive and display symptoms of scurvy such as bone fractures and rachitic rosary before dying around three months after beginning the deficient diet. As such, Leprdb/+Smp30Y/− and Leprdb/dbSmp30Y/− mice were given vitamin C-supplemented water in this study. Vitamin C is known to play an important role in the structure and function of mitochondria and endoplasmic reticulum [33]. Harrison et al. reported that vitamin E and vitamin C treatment improves fibrosis but not necroinflammation in NASH patients [34]. Therefore, vitamin C supplementation would be one reason that fibrosis was not observed in Leprdb/dbSmp30Y/− mice in this study.
In this study, Leprdb/+Smp30Y/− mice exhibited no phenotypes for lipid accumulation and mitochondrial damage in the liver as was previously reported [10]. However, in the previous study, mice were fed an autoclaved CRF-1 diet containing <55 mg of vitamin C per kg and tap water, and thus the liver vitamin C level in Smp30Y/− mice was about 6% that of Smp30Y/+ mice [8]. Although lipid accumulation and mitochondrial damage were observed in livers of Smp30Y/− mice at 12 months [10], it is unclear whether the phenotypes were caused by SMP30 deficiency (loss of unknown function except for vitamin C biosynthesis) and/or by vitamin C deficiency. Thus, the lack of steatosis in Leprdb/+Smp30Y/− mice might result from their younger age (24 weeks) or the vitamin C supplementation in the current study.
Although further studies will be required to define the exact molecular mechanism of the altered lipid homeostasis and liver damage caused by decreases in SMP30 levels, our data strongly suggest that SMP30 is closely associated with NAFLD pathogenesis and might be a possible therapeutic target for NAFLD.
Funding Statement
This study is supported by the Japan Society for the Promotion of Science (http://www.jsps.go.jp/english/e-grants/index.html) KAKENHI Grant Number 24380073 (A. Ishigami), 23590441 (N. Maruyama), 23790122 (Y. Kondo) and 23591317 (G. Hasegawa). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Ong JP, Younossi ZM (2007) Epidemiology and natural history of NAFLD and NASH. Clin Liver Dis 11: 1–16, vii. [DOI] [PubMed] [Google Scholar]
- 2. Charlton M (2004) Nonalcoholic fatty liver disease: a review of current understanding and future impact. Clin Gastroenterol Hepatol 2: 1048–58. [DOI] [PubMed] [Google Scholar]
- 3. Takahashi Y, Soejima Y, Fukusato T (2012) Animal models of nonalcoholic fatty liver disease/nonalcoholic steatohepatitis. World J Gastroenterol 18: 2300–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Anstee QM, Goldin RD (2006) Mouse models in non-alcoholic fatty liver disease and steatohepatitis research. Int J Exp Pathol 87: 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Rinella ME, Elias MS, Smolak RR, Fu T, Borensztajn J, et al. (2008) Mechanisms of hepatic steatosis in mice fed a lipogenic methionine choline-deficient diet. Lipid Res 49: 1068–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Fujita T, Uchida K, Maruyama N (1992) Purification of senescence marker protein-30 (SMP30) and its androgen-independent decrease with age in the rat liver. Biochim Biophys Acta 1116: 122–128. [DOI] [PubMed] [Google Scholar]
- 7. Fujita T, Inoue H, Kitamura T, Sato N, Shimosawa T, et al. (1998) Senescence marker protein-30 (SMP30) rescues cell death by enhancing plasma membrane Ca2+-pumping activity in HepG2 cells. Biochem Biophys Res Commun 250: 374–380. [DOI] [PubMed] [Google Scholar]
- 8. Kondo Y, Inai Y, Sato Y, Handa S, Kubo S, et al. (2006) Senescence marker protein 30 functions as gluconolactonase in L-ascorbic acid biosynthesis, and its knockout mice are prone to scurvy. Proc Natl Acad Sci USA 103: 5723–5728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ishigami A, Fujita T, Handa S, Shirasawa T, Koseki H, et al. (2002) Senescence marker protein-30 knockout mouse liver is highly susceptible to tumor necrosis factor-α- and Fas-mediated apoptosis. Am J Pathol 161: 1273–1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ishigami A, Kondo Y, Nanba R, Ohsawa T, Handa S, et al. (2004) SMP30 deficiency in mice causes an accumulation of neutral lipids and phospholipids in the liver and shortens the life span. Biochem Biophys Res Commun 315: 575–580. [DOI] [PubMed] [Google Scholar]
- 11. Hasegawa G, Yamasaki M, Kadono M, Tanaka M, Asano M, et al. (2010) Senescence marker protein-30/gluconolactonase deletion worsens glucose tolerance through impairment of acute insulin secretion. Endocrinology 151: 529–536. [DOI] [PubMed] [Google Scholar]
- 12. Kondo Y, Sasaki T, Sato Y, Amano A, Aizawa S, et al. (2008) Vitamin C depletion increases superoxide generation in brains of SMP30/GNL knockout mice. Biochem Biophys Res Commun 377: 291–296. [DOI] [PubMed] [Google Scholar]
- 13. Handa S, Maruyama N, Ishigami A (2009) Over-expression of Senescence Marker Protein-30 decreases reactive oxygen species in human hepatic carcinoma Hep G2 cells. Biol Pharm Bull 32: 1645–1648. [DOI] [PubMed] [Google Scholar]
- 14. Fukui M, Senmaru T, Hasegawa G, Yamazaki M, Asano M, et al. (2011) 17β-Estradiol attenuates saturated fatty acid diet-induced liver injury in ovariectomized mice by up-regulating hepatic senescence marker protein-30. Biochem Biophys Res Commun 415: 252–257. [DOI] [PubMed] [Google Scholar]
- 15. Sato T, Seyama K, Sato Y, Mori H, Souma S, et al. (2006) Senescence marker protein-30 protects mice lungs from oxidative stress, aging, and smoking. Am J Respir Crit Care Med 174: 530–537. [DOI] [PubMed] [Google Scholar]
- 16. Iwama M, Shimokado K, Maruyama N, Ishigami A (2011) Time course of vitamin C distribution and absorption after oral administration in SMP30/GNL knockout mice. Nutrition 27: 471–478. [DOI] [PubMed] [Google Scholar]
- 17. Sato Y, Uchiki T, Iwama M, Kishimoto Y, Takahashi R, et al. (2010) Determination of dehydroascorbic acid in mouse tissues and plasma by using tris(2-carboxyethyl)phosphine hydrochloride as reductant in metaphosphoric acid/ethylenediaminetetraacetic acid solution. Biol Pharm Bull 33: 364–369. [DOI] [PubMed] [Google Scholar]
- 18. Ohkawa H, Ohishi N, Yagi K (1979) Assay for lipid peroxides in animal tissues by thiobarbituric acid reaction. Anal Biochem 95: 351–358. [DOI] [PubMed] [Google Scholar]
- 19. Usui S, Hara Y, Hosaki S, Okazaki M (2002) A new on-line dual enzymatic method for simultaneous quantification of cholesterol and triglycerides in lipoproteins by HPLC. J Lipid Res 43: 805–814. [PubMed] [Google Scholar]
- 20. Kleiner DE, Brunt EM, Van Natta M, Behling C, Contos MJ, et al. (2005) Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology 41: 1313–1321. [DOI] [PubMed] [Google Scholar]
- 21. Livak KJ, Schmittgen TD (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2(−Delta Delta C(T)) Method. Methods 25: 402–40. [DOI] [PubMed] [Google Scholar]
- 22. Day CP, James OF (1998) Steatohepatitis: a tale of two “hits”? Gastroenterology 14: 842–845. [DOI] [PubMed] [Google Scholar]
- 23. Ishigami T, Fujita T, Simbula G, Columbano A, Kikuchi K, et al. (2001) Regulatory effects of senescence marker protein 30 on the proliferation of hepatocytes. Pathol Int 51: 491–497. [DOI] [PubMed] [Google Scholar]
- 24. Jung KJ, Ishigami A, Maruyama N, Takahashi R, Goto S, et al. (2004) Modulation of gene expression of SMP-30 by LPS and calorie restriction during aging process. Exp Gerontol 39: 1169–1177. [DOI] [PubMed] [Google Scholar]
- 25. Lv S, Wang JH, Liu F, Gao Y, Fei R, et al. (2008) Senescence marker protein 30 in acute liver failure: validation of a mass spectrometry proteomics assay. BMC Gastroenterol 8: 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Toledo FG, Sniderman AD, Kelley DE (2006) Influence of hepatic steatosis (fatty liver) on severity and composition of dyslipidemia in type 2 diabetes. Diabetes Care 29: 1845–1850. [DOI] [PubMed] [Google Scholar]
- 27. Cali AM, Zern TL, Taksali SE, de Oliveira AM, Dufour S, et al. (2007) Intrahepatic fat accumulation and alterations in lipoprotein composition in obese adolescents: a perfect proatherogenic state. Diabetes Care 30: 3093–3098. [DOI] [PubMed] [Google Scholar]
- 28. Sugino I, Kuboki K, Matsumoto T, Murakami E, Nishimura C, et al. (2011) Influence of fatty liver on plasma small, dense LDL-cholesterol in subjects with and without metabolic syndrome. J Atheroscler Thromb 18: 1–7. [DOI] [PubMed] [Google Scholar]
- 29. Park H, Ishigami A, Shima T, Mizuno M, Maruyama N, et al. (2010) Hepatic senescence marker protein-30 is involved in the progression of nonalcoholic fatty liver disease. J Gastroenterol 45: 426–434. [DOI] [PubMed] [Google Scholar]
- 30. Adiels M, Borén J, Caslake MJ, Stewart P, Soro A, et al. (2005) Overproduction of VLDL1 driven by hyperglycemia is a dominant feature of diabetic dyslipidemia. Arterioscler Thromb Vasc Biol 25: 1697–1703. [DOI] [PubMed] [Google Scholar]
- 31. Qiu S, Bergeron N, Kotite L, Krauss RM, Bensadoun A, et al. (1998) Metabolism of lipoproteins containing apolipoprotein B in hepatic lipase-deficient mice. J Lipid Res 39: 1661–8. [PubMed] [Google Scholar]
- 32. Senmaru T, Fukui M, Okada H, Mineoka Y, Yamazaki M, et al. (2013) Testosterone deficiency induces markedly decreased serum triglycerides, increased small dense LDL, and hepatic steatosis mediated by dysregulation of lipid assembly and secretion in mice fed a high-fat diet. Metabolism 2013 Jan 16.[Epub ahead of print]. [DOI] [PubMed] [Google Scholar]
- 33. Kim JC (1997) Ultrastrutural studies of vascular and muscular changes in ascorbic acid deficient guinea-pigs. Lab Anim 11: 113–117. [DOI] [PubMed] [Google Scholar]
- 34. Harrison SA, Torgerson S, Hayashi P, Ward J, Schenker S (2003) Vitamin E and vitamin C treatment improves fibrosis in patients with nonalcoholic steatohepatitis. Am J Gastroenterol 98: 2485–2490. [DOI] [PubMed] [Google Scholar]