

Abstract
Elevated levels of serum amyloid A (SAA) is a risk factor for cardiovascular diseases, however, the role of SAA in the pathophysiology of atherosclerosis remains unclear. Here we show that SAA induced macrophage foam cell formation. SAA-stimulated foam cell formation was mediated by c-jun N-terminal kinase (JNK) signaling. Moreover, both SAA and SAA-conjugated high density lipoprotein stimulated the expression of the important scavenger receptor lectin-like oxidized low-density lipoprotein receptor 1 (LOX1) via nuclear factor-κB (NF-κB). A LOX1 antagonist carrageenan significantly blocked SAA-induced foam cell formation, indicating that SAA promotes foam cell formation via LOX1 expression. Our findings therefore suggest that SAA stimulates foam cell formation via LOX1 induction, and thus likely contributes to atherogenesis.
Keywords: Serum amyloid A, Foam cell, Atherosclerosis, Lectin-like oxidized low-density lipoprotein, receptor 1
1. Introduction
Atherosclerosis is a chronic inflammatory disease affecting arterial blood vessels. Several types of inflammatory cells are involved in the atherogenic process, including monocyte/macrophages and smooth muscle cells [1]. Circulating monocytes migrate into the intima region in response to chemokine (C–C motif) ligand 2 (CCL2) [2] where macrophage colony-stimulating factor stimulates their differentiation into macrophages [3]. Macrophages engulf modified low-density lipoprotein (LDL), resulting in foam cell formation [4]. Foam cells produce diverse growth factors and proinflammatory mediators including tumor necrosis factor (TNF)-α [4,5]. These inflammatory mediators induce proliferation of smooth muscle cells, which induce plaque formation [6]. Given the importance of foam cell formation in the pathogenesis of atherosclerosis [5,7], understanding the underlying molecular mechanisms of foamy macrophage generation may lead to novel targets for therapeutic intervention. Previous studies have reported that several Toll-like receptors (TLRs) including TLR2, TLR4, and TLR9 are involved in foam cell formation: TLR ligands pam3csk, lipopolysaccharide (LPS), and CpG ligands can independently stimulate foam cell formation from macrophages [8-10]. Targeting the TLRs may, however, have undesirable effects on host immune defense. Thus the identification of additional stimuli and their receptors involved in foam cell formation may provide superior targets for therapeutic intervention.
Serum amyloid A (SAA), a major acute-phase protein, is released into blood circulation in response to infection or injury [11]. SAA is released after liver cells are stimulated by several pro-inflammatory cytokines, such as interleukin (IL)-1β or tumor necrosis factor (TNF)-α [12,13]. Circulating SAA levels are elevated approximately 1000-fold during acute-phase reactions compared with baseline conditions [12]. SAA has cytokine-like properties and plays a number of immunomodulatory roles. SAA induces proinflammatory cytokine and chemokine production in several cell types such as rheumatoid synoviocytes, intestinal epithelial cells, monocytes, and neutrophils [13-16]. SAA has also been suggested to play a role in the metabolism of high density lipoproteins (HDL), and as an impeder of the protective function of HDL on the development of atherosclerosis [17]. We previously demonstrated that SAA stimulates many atherogenic inflammatory markers, including CCL2, matrix metalloproteinase (MMP)-9 in monocytes and endothelial cells [18-21]. Importantly, circulating SAA levels are significantly elevated in patients with chronic vascular disease reaching around 11–15 μg/ml or in animals fed with high fat diet reaching around 28.5–34.8 μg/ml [22,23]. However it is unclear whether elevated SAA levels reflect underlying cardiovascular disease or can contribute directly to the pathophysiology of atherogenesis. Here we show that SAA directly stimulates foamy macrophage formation and thus implicate SAA as causal agents in atherogenesis.
2. Materials and methods
2.1. Materials
Recombinant human SAA (catalog number 300-13, produced in Escherichia coli, endotoxin level <0.1 ng/μg) was purchased from Peprotech (Rocky Hill, NJ, USA). Naïve LDL, LPS (from E. coli 055:B5, catalog number L2880) and carrageenan (type III kappa) were purchased from Sigma–Aldrich (St. Louis, MO, USA). All antibodies against the phospho-MAPKs were purchased from Cell Signaling Technology (Beverly, MA, USA), and the anti-lectin-like oxLDL receptor 1 (LOX1) antibody (catalog number AF1564) was purchased from R&D Systems (Minneapolis, MN, USA).
2.2. Cell culture and generation of bone marrow-derived macrophages
Raw264.7 cells were maintained in DMEM with 10% heat-inactivated fetal calf serum under standard incubator conditions (humidified atmosphere, 95% air, 5% CO2, and 37 °C). Bone marrow cells were isolated by flushing the femurs and tibias of wild-type ICR mice 5–8 weeks of age with ice-cold PBS. Bone marrow progenitor cells were cultured in 10% FBS containing α-MEM with 30 ng/ml M-CSF under standard incubator conditions for 3 days. The non-adherent cells were removed, and 10% FBS containing α-MEM with 30 ng/ml M-CSF was added, and the cells maintained for 2–3 days.
2.3. Foam cell formation and oil red O staining
Raw264.7 cells and mouse bone marrow-derived macrophages (1 × 104) were seeded on 96-well plates and cultured overnight. Cells were stimulated with LDL (50 μg/ml) plus vehicle, SAA, or LPS for 24 h. After washing with PBS, the cells were fixed with 4% formaldehyde for 10 min at room temperature. After washing with distilled water 3 times, the fixed cells were stained with oil Red-O solution for 20 min. The stained cells were detected by light microscopy and total cells and foam cells were counted.
2.4. Western blot analysis
Raw264.7 cells were stimulated with SAA for various times. After stimulation, the cells were lysed in lysis buffer (20 mM HEPES [pH7.2], 10% glycerol, 150 mM NaCl, 1% Triton X-100, 50 mM NaF, 1 mM Na3VO4, 10 μg/ml leupeptin, 10 μg/ml aprotinin, and 1 mM PMSF). Soluble proteins were separated on 10% SDS–polyacrylamide gels and blotted onto a nitrocellulose membrane. Subsequently, the membranes were incubated with specific antibodies against target protein, and antigen–antibody complexes were visualized by enhanced chemiluminescence.
2.5. Reverse transcription polymerase chain reaction (RT-PCR) analysis
Raw264.7 cells or mouse bone marrow-derived macrophages (1 × 106) were stimulated with SAA for the indicated times. Total RNA was isolated by using Trizol reagent (Invitrogen, Carlsbad, CA, USA) and 1 μg of total RNA was used as a template for cDNA using the Bioneer Reverse Transcriptase System. The primers used for the RT-PCR analyses have been reported previously. The sequences of the primers were as follows: LOX1: sense, 5′-AGGTCCTTGTCCACAAGACTGG-3′; antisense, 5′-ACGCCCCTGGTCTTAAAGAATTG-3′. actin: sense, 5′-TTCTTTGCAGCTCCTTCGTTGCCG-3′; antisense, 5′-TGGATGGCTACGTACATGGCTGGG-3′. GAPDH: sense, 5′-GACATCAAGAAGGTGGTGAA-3′; antisense, 5′-TGTCATACCAGGAAATGAGC-3′. cDNA was subjected to 35 PCR cycles at 94 °C (denaturation, 30 s), 55–65 °C (annealing, 30 s), and 72 °C (extension, 30 s). PCR products were electrophoresed on a 1.5% agarose gel and visualized by ethidium bromide staining.
2.6. Transfection of oilgodeoxynucleotides (ODN)
The sequence of phosphorothionate double-stranded decoy ODN against the NF-κB binding site was as follow: NF-κB decoy ODN (5′-CCTTGAAGGGATTTCCCTCC-3′/3′-GGAACTTCCCTAAAGGGAGG-5′), scrambled NF-κB ODN (5′-TTGCCGTACCTGACTTAGCC-3′/3′-AACGGCATGGACTGAATCGG-5′). Single-stranded ODN was annealed for 2 h while the temperature was decreased from 80 to 25 °C. Cells (3 × 105) were seeded on a 24-well plate and cultured overnight. The cells were transfected with 0.2 μg of NF-κB decoy ODN and scrambled NF-κB ODN using Lipofectamine 2000 reagent.
2.7. Luciferase assay
NF-κB reporter constructs were purchased from Clontech (Palo Alto, CA, USA). Raw264.7 cells were transfected with 2 μg of plasmid construct by the Lipofectamine method (Invitrogen). After transfection, cells were stimulated with 1 μM SAA for 24 h and lysed with lysis buffer; 5 μl of cell lysate was mixed with 25 μl of luciferase activity assay reagent, and the luminescence produced for 5 s was measured using Luminoskan (Labsystems).
2.8. Synthesis of reconstituted HDL and HDL-conjugated SAA
Human apolipoprotein A-I (apoA-I) was expressed and purified according to a previous report [24]. Discoidal reconstituted HDL was prepared with the purified apoA-I (at least 95% purity) as described previously [25]. HDL-SAA was prepared with apoA-I and SAA via sodium cholate dialysis with a POPC: cholesterol: apoA-I: SAA: sodium cholate molar ratio of 95:5:1:1:150. All HDL showed a similar low range of residual endotoxin level between 3.1–3.3 EU/mL based on endotoxin quantification using a commercially available test kit (BioWhittaker, Walkersville, MD, USA).
2.9. Data analysis
Results are expressed as mean ± SE. The Student’s t-test was used to compare individual treatments with their respective control values. Statistical significance was set at p < 0.05.
3. Results
3.1. SAA promotes foam cell formation
To investigate the effect of SAA on foam cell formation, we stimulated the mouse macrophage model cell line Raw264.7 with SAA and LDL for 24 h. SAA treatment significantly induced foam cell formation in a concentration-dependent manner (Fig. 1A and B). The increase in foam cell formation was apparent with exposure to 0.1–2 μM SAA (Fig. 1B). As a positive control, treatment with LPS and LDL also induced foam cell formation (Fig. 1A and C), which is consistent with a previous report [9]. Because the recombinant SAA was produced in E. coli, we examined the possible contribution of contaminating LPS to SAA-induced foam cell formation using polymyxin B, a potent LPS inhibitor. While polymyxin B inhibited exogenously added LPS-induced foam cell formation, it had no effect on SAA-induced foam cell formation (Fig. 1C), indicating that the effect of SAA was not mediated by contaminating LPS.
Fig. 1.

SAA stimulates foam cell formation. Raw264.7 cells were stimulated with, 1 μM SAA (A) or various concentrations of SAA (0, 0.01, 0.1, 0.5, 1, and 2 μM) (B) in the presence of 50 μg/ml LDL for 24 h. The cells were stained with Oil-Red O, and stained cells were detected by microscopy. Total cells and foam cells were counted. 1 μg/ml LPS was used as a positive control. NT: no treatment. (C) Raw264.7 cells were pre-incubated with LPS-inhibitor polymyxin B (PolB, 1 μg/ml) prior to addition of LDL (50 μg/ml) and SAA (1 μM) or LPS (1 μg/ml). Cells were stained with Oil-Red O, and total cells and foam cells were counted. All experiments were repeated three (B) or eight (C) times independently, and the results are presented as mean ± SE. *p < 0.05 and **p < 0.01.
3.2. SAA increases LOX1 expression
LOX1 is one of the major modified LDL scavenging receptors and its overexpression is important in foam cell formation [26]. Therefore, we asked if SAA induced LOX1 expression in our macrophage model cell line. SAA significantly induced LOX1 mRNA expression after 1–3 h (Fig. 2A), which was followed by robust SAA-induced protein expression 3–6 h after stimulation (Fig. 2B). To investigate the role of LOX1 on SAA-induced foam cell formation, a LOX1-selective inhibitor was used. Preincubation of Raw264.7 cells with LOX1 antagonist, κ-carrageenan prior to the addition of SAA caused significant inhibition of SAA-induced foam cell formation, indicating that SAA-induced foam cell formation is mediated by LOX1 expression (Fig. 2C). The LOX1 antagonist also blocked foam cell formation induced by oxLDL (Fig. 2C). We also investigated the effect of SAA on the upregulation of LOX1 in primary macrophages. SAA also strongly increased LOX1 mRNA expression in mouse bone marrow-derived primary macrophages (Fig. 2D). SAA also stimulated mouse primary macrophages foam cell formation, which was also markedly inhibited by LOX1 antagonist, κ-carrageenan (Fig. 2E).
Fig. 2.
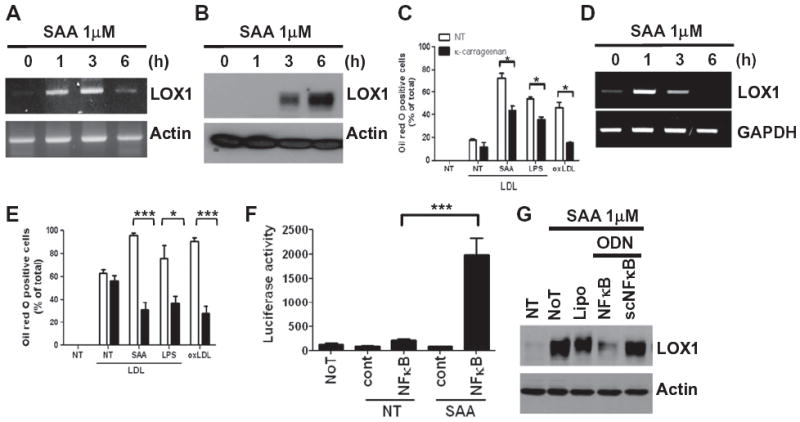
SAA stimulates LOX1 expression. Raw264.7 cells (A and B) or mouse bone marrow-derived macrophages (D) were stimulated with 1 μM SAA for the indicated time periods. (A and D) Cells were harvested for RNA preparation, and RT-PCR was performed using specific LOX1 and actin primers as a loading control. (B) The LOX1 expression level was determined by Western blot. Raw264.7 cells (C) or mouse bone marrow-derived macrophages (E) were pre-incubated with κ-carrageenan (250 μg/ml) for 15 min prior to LDL alone, SAA (1 μM) plus LDL (50 μg/ml), LPS (1 μg/ml) plus LDL (50 μg/ml) or oxLDL treatment (50 μg/ml). (F) Raw264.7 cells were transfected with a NF-κB-driven reporter construct using the Lipofectamine 2000 reagent. Transfected cells were stimulated with 1 μM SAA for 24 h, and luciferase activity was measured in cell lysates. Emitted luminescence is expressed in arbitrary luminescent units. (G) Raw264.7 cells were transfected with NF-κB decoy ODN and scrambled ODN and stimulated with 1 μM SAA for 6 h. LOX1 protein expression was determined by Western blot. NT: no treatment, NoT: no transfection, cont: luciferase vector control without NF-κB reporter, Lipo: lipofectamin only, ODN: oligodeoxynucleotide, NF-κB: decoy NF-κB binding site ODN, scNF-κB: scramble control for NF-κB ODN. Data are presented as representative of at least three independent experiments (A, B, D and G). For (C, E and F) the results are presented as mean ± SE of at least two independent experiments performed in duplicate. *p < 0.05 and ***p < 0.001.
Since others have reported that LOX1 induction (by LPS) is dependent on NF-κB activation [27], we next asked if the induction of LOX1 by SAA also acted through NF-κB. SAA dramatically increased NF-κB-dependent luciferase activity in Raw264.7 cells transfected with a NF-κB luciferase reporter construct (Fig. 2F). Furthermore, transfection of Raw264.7 cells with a double-stranded decoy oligodeoxynucleotide (ODN) against the NF-κB binding site dramatically inhibited SAA-induced LOX1 expression, compared with scrambled NF-κB decoy ODN (Fig. 2G). These results indicate that NF-κB activation is required for SAA-induced LOX1 expression.
3.3. JNK regulates SAA-induced foam cell formation
We next sought to define the intracellular signaling pathway triggered by SAA. MAPKs such as ERK, p38 MAPK, and JNK transduce extracellular signals to the nuclei in many cell types [28]. SAA induced robust ERK, p38 MAPK, and JNK phosphorylation, although ERK phosphorylation was induced early (2–5 min), while p38 MAPK and JNK later (10–30 min) (Fig. 3A). To determine the role of individual MAPKs on SAA-induced foam cell formation, Raw264.7 cells were pre-incubated with either PD98059 (a selective MEK inhibitor), SB203580 (a selective p38 MAPK inhibitor), or SP600125 (a selective JNK inhibitor) prior to SAA treatment. SAA-induced foam cell formation was dramatically inhibited by SP600125, but not by PD98059 or SB203580, indicating that JNK is essential for SAA-induced foam cell formation (Fig. 3B). Moreover, the JNK inhibitor SP600125 inhibited SAA-induced LOX1 expression in a concentration-dependent manner (Fig. 3C). Previous reports demonstrated that JNK induces NF-κB activation [29,30]. In this study, we also observed that preincubation of Raw264.7 cells with JNK inhibitor SP600125 prior to addition of SAA abolished SAA-induced NF-κB activity (Fig. 3D). The result suggests that JNK mediates NK-κB activity, resulting in foam cell formation downstream of SAA-induced signaling.
Fig. 3.
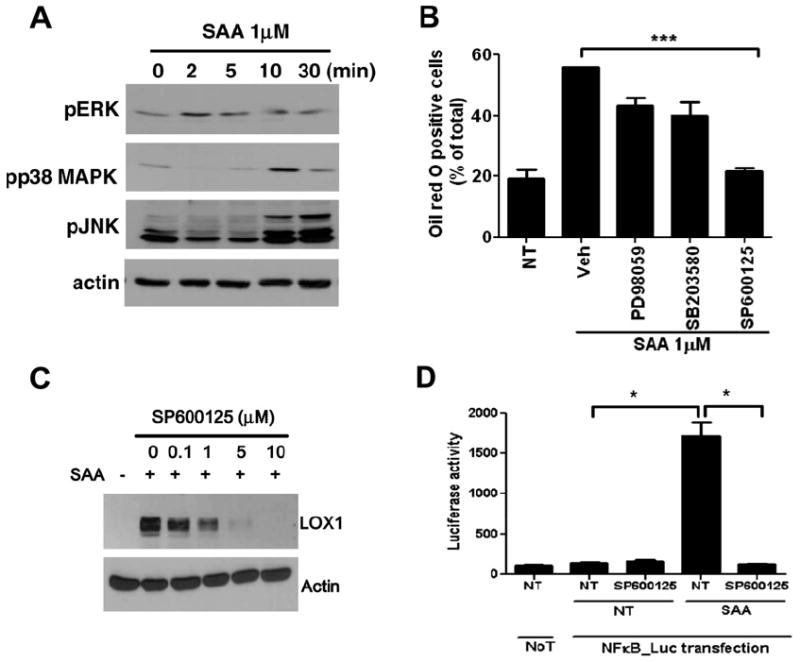
SAA-induced foam cell formation requires JNK activity. (A) Raw264.7 cells were stimulated with 1 μM SAA for the indicated time periods. (B) Raw264.7 cells were pre-incubated with vehicle, MEK inhibitor (PD98059, 50 μM) for 60 min, p38 MAPK inhibitor (SB203580, 20 μM), or JNK inhibitor (SP600125, 20 μM) for 15 min prior to SAA treatment. (A) Phospho-ERK, -p38 MAPK, and -JNK levels were determined by Western blot. (B) The cells were stained with Oil-Red O, and total cells and foam cells were counted. (C) Raw264.7 cells were pre-incubated with various concentrations of the JNK inhibitor SP600125 (0, 0.1, 1, 5, and 10 μM), prior to SAA treatment for 6 h. The LOX1 expression level was determined by Western blot. (D) Raw264.7 cells were transfected with a NF-κB-driven reporter construct. Transfected cells were stimulated with 1 μM SAA for 24 h in the absence or presence of SP600125 (10 μM), and luciferase activity was measured in cell lysates. Emitted luminescence is expressed in arbitrary luminescent units. A and C are representative blots. Results are presented as mean ± SE (B and D). *p < 0.05 and ***p < 0.001.
3.4. HDL-conjugated SAA also stimulates foam cell formation
Because SAA is conjugated to HDL under pathophysiological conditions [31], we tested whether SAA-HDL also stimulated foam cell formation. Both SAA and HDL-conjugated SAA induced foam cell formation, although SAA-HDL was 40% less efficient (Fig. 4A). apo-HDL alone did not stimulate foam cell formation (Fig. 4A). Similarly, SAA-HDL (but not HDL alone) stimulated LOX1 expression, although SAA-HDL was 50% less efficient (Fig. 4B).
Fig. 4.

SAA-conjugated HDL stimulates foam cell formation and LOX1 expression. (A) Raw264.7 cells were stimulated with SAA, HDL containing apoA-I (apo-HDL) or HDL containing SAA (SAA-HDL) for 24 h. Cells were stained with Oil Red O, and total cells and foam cells were counted. (B) Raw264.7 cells were stimulated with SAA (1 μM), HDL containing apoA-I (apo-HDL) (1 μM) or HDL containing SAA (SAA-HDL) (1 μM) for 6 h. LOX1 expression levels were determined by Western blot. All experiments were repeated independently three times, and the results are presented as the mean ± SE for (A); B is a representative blot. ***p < 0.001.
4. Discussion
Foam cell formation is a critical step in the pathogenesis of atherosclerosis. Here, we demonstrated that one of the acute phase reactants, SAA, strongly induces foam cell formation. Elevated levels of circulating SAA are present in atherosclerosis patients, as well as patients with other chronic diseases [32]. SAA may contribute at many levels to the pathogenesis of atherosclerosis. We reported previously that SAA stimulates CCL2 production and MMP9 expression. CCL2 (a potent monocyte chemoattractant) is a key molecule in recruiting monocytes into the intima region, and MMP9 triggers atherosclerotic plaque rupture and subsequent myocardial ischemia [18-21]. Taken together, we suggest that SAA plays an essential role in the pathogenesis of atherosclerosis by recruiting monocytes into the intima area, inducing foam cell formation by macrophages, and stimulating lethal plaque rupture.
Macrophages take up modified LDLs which include oxidized LDL and aggregated LDL [33]. For the uptake of modified LDL, several scavenger receptors including LOX1, CD36, and SR-A play key roles [26,34,35]. Among these scavenger receptors, LOX1 is one of the major scavenger receptors for oxLDL uptake, and LOX1 expression is upregulated by several inflammatory signals [26,36]. Stimulating macrophages with certain TLR ligands induces LOX1 expression [37,38]. LPS, for example, is a potent TLR4 ligand that stimulates LOX1 upregulation [39]. In this study we demonstrated that SAA stimulated LOX1 upregulation (Fig. 2A and B), and an antagonist for LOX1 (κ-carrageenan) significantly inhibited SAA-stimulated foam cell formation (Fig. 2C). The results indicates that SAA-induced foam cell formation is mediated by LOX1 expression.
HDL inhibits foam cell formation by facilitating cholesterol efflux from macrophages by up regulation of ATP-binding cassette transporter 1 [40]. Although less efficient that SAA alone, in this study we demonstrated that SAA-conjugated HDL also stimulates foam cell formation and LOX1 expression (Fig. 4). This result suggests that SAA impedes the protective function of HDL on the development of atherosclerosis. When SAA content increased in HDL during the acute inflammatory phase of HDL, it has been reported that HDL lost its anti-inflammatory and antioxidant activity [41]. Previously we also demonstrated that SAA-conjugated HDL also stimulates monocytes, resulting in CCL2 production and chemotactic migration of the cells [18]. These reports support our notion that SAA has inhibitory effect on HDL functioning against atherosclerosis.
In conclusion, SAA stimulates monocytes or macrophages resulting in vascular inflammation by enhancing foam cell formation via upregulation of LOX1 expression. Since LOX1 expression and foam cell formation is critical step on the pathogenesis of atherosclerosis, we suggest that SAA may be important new targets for the development of therapeutic agents against atherosclerosis.
Acknowledgments
This research was supported by National Research Foundation of Korea(NRF) Grant funded by the Korean government (MEST) (Nos. 2012R1A2A2A01007751, 2011-0014476) and a Grant (Code # 7-19-42) from Rural Development Administration, Republic of Korea. B. Zabel was supported by National Institutes of Health Grant AI-079320.
References
- 1.Li AC, Glass CK. The macrophage foam cell as a target for therapeutic intervention. Nat Med. 2002;8:1235–1242. doi: 10.1038/nm1102-1235. [DOI] [PubMed] [Google Scholar]
- 2.Valente AJ, Graves DT, Vialle-Valentin CE, Delgado R, Schwartz CJ. Purification of a monocyte chemotactic factor secreted by nonhuman primate vascular cells in culture. Biochemistry. 1988;27:4162–4168. doi: 10.1021/bi00411a039. [DOI] [PubMed] [Google Scholar]
- 3.Stanley ER, Berg KL, Einstein DB, Lee PS, Pixley FJ, Wang Y, Yeung YG. Biology and action of colony-stimulating factor-1. Mol Repord Dev. 1997;46:4–10. doi: 10.1002/(SICI)1098-2795(199701)46:1<4::AID-MRD2>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]
- 4.Ross R. The pathogenesis of atherosclerosis: a perspective for the 1990s. Nature. 1993;362:801–809. doi: 10.1038/362801a0. [DOI] [PubMed] [Google Scholar]
- 5.Cascieri MA. The potential for novel anti-inflammatory therapies for coronary artery disease. Nat Rev Drug Discov. 2002;1:122–130. doi: 10.1038/nrd723. [DOI] [PubMed] [Google Scholar]
- 6.Rudijanto A. The role of vascular smooth muscle cells on the pathogenesis of atherosclerosis. Acta Med Indones. 2007;39:86–93. [PubMed] [Google Scholar]
- 7.Lusis AJ. Atherosclerosis. Nature. 2000;407:233–241. doi: 10.1038/35025203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cao F, Castrillo A, Tontonoz P, Re F, Byme GI. Chlamydia pneumobniae-induced macrophage foam cell formation is mediated by toll-like receptor 2. Infect Immun. 2007;75:753–759. doi: 10.1128/IAI.01386-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Funk JL, Feingold KR, Moser AH, Grunfeld C. Lipopolysaccharide stimulation of RAW264.7 macrophages induces lipid accumulation and foam cell formation. Atherosclerosis. 1993;98:67–82. doi: 10.1016/0021-9150(93)90224-i. [DOI] [PubMed] [Google Scholar]
- 10.Lee JG, Lim EJ, Park DW, Lee SH, Kim JR, Baek SH. A combination of LOX-1 and Nox1 regulates TLR9-mediated foam cell formation. Cell Signal. 2008;20:2266–2275. doi: 10.1016/j.cellsig.2008.08.022. [DOI] [PubMed] [Google Scholar]
- 11.Uhlar CM, Whitehead AS. Serum amyloid A, the major vertebrate acute-phase reactat. Eur J Biochem. 1999;265:501–523. doi: 10.1046/j.1432-1327.1999.00657.x. [DOI] [PubMed] [Google Scholar]
- 12.Jensen LE, Whitehead AS. Regulation of serum amyloid A protein expression during the acute-phase response. Biochem J. 1988;334:489–503. doi: 10.1042/bj3340489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Urieli-Shoval S, Linke RP, Matzner Y. Expression and function of serum amyloid A, a major acute-phase protein, in normal and disease states. Curr Opin Hematol. 2000;7:64–69. doi: 10.1097/00062752-200001000-00012. [DOI] [PubMed] [Google Scholar]
- 14.Koga T, Torigoshi T, Motokawa S, Miyashita T, Maeda Y, Nakamura M, Komori A, Aiba Y, Uemura T, Yatsuhashi H, Ishibashi H, Eguchi K, Migita K. Serum amyloid A-induced IL-6 production by rheumatoid synoviocytes. FEBS Lett. 2008;582:579–585. doi: 10.1016/j.febslet.2008.01.022. [DOI] [PubMed] [Google Scholar]
- 15.Jijon HB, Madsen KL, Walker JW, Allard B, Jobin C. Serum amyloid A activates NF-kappaB and proinflammatory gene expression in human and murine intestinal epithelial cells. Eur J Immunol. 2005;35:718–726. doi: 10.1002/eji.200425688. [DOI] [PubMed] [Google Scholar]
- 16.Furlaneto CJ, Campa A. A novel function of serum amyloid A: a potent stimulus for the release of tumor necrosis factor-alpha, interleukin-1beta, and interleukin-8 by human blood neutrophil. Biochem Biophys Res Commun. 2000;268:405–408. doi: 10.1006/bbrc.2000.2143. [DOI] [PubMed] [Google Scholar]
- 17.Malle E, Steinmetz A, Raynes JG. Serum amyloid A (SAA): an acute phase protein and apolipoprotein. Atherosclerosis. 1993;102:131–146. doi: 10.1016/0021-9150(93)90155-n. [DOI] [PubMed] [Google Scholar]
- 18.Lee HY, Kim SD, Shim JW, Lee SY, Lee H, Cho KH, Yun J, Bae YS. Serum amyloid A induces CCL2 production via formyl peptide receptor-like 1-mediated signaling in human monocytes. J Immunol. 2008;81:4332–4339. doi: 10.4049/jimmunol.181.6.4332. [DOI] [PubMed] [Google Scholar]
- 19.Lee HY, Kim MK, Park KS, Bae YH, Yun J, Park JI, Kwak JY, Bae YS. Serum amyloid A stimulates matrix-metalloproteinase-9 upregulation via formyl peptide receptor like-1-mediated signaling in human monocytic cells. Biochem Biophys Res Commun. 2005;330:989–998. doi: 10.1016/j.bbrc.2005.03.069. [DOI] [PubMed] [Google Scholar]
- 20.Lee HY, Kim SD, Shim JW, Yun J, Kim K, Bae YS. Activation of formyl peptide receptor like-1 by serum amyloid A induces CCL2 production in human umbilical vein endothelial cells. Biochem Biophys Res Commun. 2009;380:313–317. doi: 10.1016/j.bbrc.2009.01.068. [DOI] [PubMed] [Google Scholar]
- 21.Lee HY, Kim SD, Shim JW, Kim HJ, Yun J, Baek SH, Kim K, Bae YS. A pertussis toxin sensitive G-protein-independent pathway is involved in serum amyloid A-induced formyl peptide receptor 2-mediated CCL2 production. Exp Mol Med. 2010;42:302–309. doi: 10.3858/emm.2010.42.4.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Glurich I, Grossi S, Albini B, Ho A, Shah R, Zeid M, Baumann H, Genco RJ, De Nardin E. Systemic inflammation in cardiovascular and periodontal disease: comparative study. Clin Diagn Lab Immunol. 2002;9:425–432. doi: 10.1128/CDLI.9.2.425-432.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lewis KE, Kirk EA, McDonald TO, Wang S, Wight TN, O’Brien KD, Chait A. Increase in serum amyloid A evoked by dietary cholesterol is associated with increased atherosclerosis in mice. Circulation. 2004;110:540–545. doi: 10.1161/01.CIR.0000136819.93989.E1. [DOI] [PubMed] [Google Scholar]
- 24.Cho KH, Jonas A. A key point mutation (V156E) affects the structure and functions of human apolipoprotein A-1. J Biol Chem. 2000;275:26821–26827. doi: 10.1074/jbc.M002841200. [DOI] [PubMed] [Google Scholar]
- 25.Han JM, Jeong TS, Lee WS, Choi I, Cho KH. Structural and functional properties of V156K and A158E mutants of apolipiprotein A-1 in the lipid-free and lipid-bound state. J Lipid Res. 2005;46:589–596. doi: 10.1194/jlr.M400468-JLR200. [DOI] [PubMed] [Google Scholar]
- 26.Kataoka H, Kume N, Miyamoto S, Minami M, Moriwaki H, Murase T, Sawamura T, Masaki T, Hashimoto N, Kita T. Expression of lectin-like oxidized low-density lipoprotein receptor-1 in human atherosclerotic lesions. Circulation. 1999;99:3110–3117. doi: 10.1161/01.cir.99.24.3110. [DOI] [PubMed] [Google Scholar]
- 27.Hermonat PL, Zhu H, Cao M, Mehta JL. LOX-1 transcription. Cardiovasc Drugs Ther. 2011;25:393–400. doi: 10.1007/s10557-011-6322-8. [DOI] [PubMed] [Google Scholar]
- 28.Johnson GL, Lapadat R. Mitogen-activated protein kinase pathways mediated by ERK, JNK, and p38 protein kinases. Science. 2002;298:1911–1912. doi: 10.1126/science.1072682. [DOI] [PubMed] [Google Scholar]
- 29.Wang J, An FS, Zhang W, Gong L, Wei SJ, Qin WD, Wang XP, Zhao YX, Zhang Y, Zhang C, Zhang MX. Inhibition of c-Jun N-terminal kinase attenuates low shear stress-induced atherogenesis in apolipoprotein E-deficient mice. Mol Med. 2011;17:990–999. doi: 10.2119/molmed.2011.00073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yang CM, Luo SF, Hsieh HL, Chi PL, Lin CC, Wu CC, Hsiao LD. Interleukin-1beta induces ICAM-1 expression enhancing leukocyte adhesion in human rheumatoid arthritis synovial fibroblasts: involvement of ERK, JNK, AP-1, and NF-kappaB. J Cell Physiol. 2010;224:516–526. doi: 10.1002/jcp.22153. [DOI] [PubMed] [Google Scholar]
- 31.Abbas A, Fadel PJ, Wang Z, Arbique D, Jialal I, Vongpatanasin W. Contrasting effects of oral versus transdermal estrogen on serum amyloid A (SAA) and high-density lipoprotein-SAA in postmenopausal women, Arterioscler. Thromb Vasc Biol. 2004;24:e164–e167. doi: 10.1161/01.ATV.0000140198.16664.8e. [DOI] [PubMed] [Google Scholar]
- 32.Fyfe AI, Rothenberg LS, DeBeer FC, Cantor RM, Rotter JI, Lusis AJ. Association between serum amyloid A proteins and coronary artery disease: evidence from two distinct arteriosclerotic processes. Circulation. 1997;96:2914–2919. doi: 10.1161/01.cir.96.9.2914. [DOI] [PubMed] [Google Scholar]
- 33.Miller YI, Choi SH, Fang L, Tsimikas S. Lipoprotein modification and macrophage uptake: role of pathologic cholesterol transport in atherogenesis. Subcell Biochem. 2010;51:229–251. doi: 10.1007/978-90-481-8622-8_8. [DOI] [PubMed] [Google Scholar]
- 34.Endemann G, Stanton LW, Maden KS, Bryant CM, White RT, Protter AA. CD36 is a receptor for oxidized low density lipoprotein. J Biol Chem. 1993;268:11811–11816. [PubMed] [Google Scholar]
- 35.Suzuki H, Kurihara Y, Takeya M, Kamada N, Kataoka M, Jishage K, Ueda O, Sakaguchi H, Higashi T, Suzuki T, Takashima Y, Kawabe Y, Cynshi O, Wada Y, Honda M, Kurihara H, Aburatani H, Doi T, Matsumoto A, Azuma S, Noda T, Toyoda Y, Itakura H, Yazaki Y, Kodama T, et al. A role for macrophage scavenger receptors in atherosclerosis and susceptibility to infection. Nature. 1997;386:292–296. doi: 10.1038/386292a0. [DOI] [PubMed] [Google Scholar]
- 36.Liang M, Zhang P, Fu J. Up-regulation of LOX-1 expression by TNF-alpha promotes trans-endothelial migration of MDA-MB-231 breast cancer cells. Cancer Lett. 2007;258:31–37. doi: 10.1016/j.canlet.2007.08.003. [DOI] [PubMed] [Google Scholar]
- 37.Taront S, Dieudonné A, Blanchard S, Jeannin P, Lassalle P, Delneste Y, Gosset P. Implication of scavenger receptors in the interactions between diesel exhaust particles and immature or mature dendritic cells. Part Fibre Toxicol. 2009;6:9–22. doi: 10.1186/1743-8977-6-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Doyle SE, O’Connell RM, Miranda GA, Vaidya SA, Chow EK, Liu PT, Suzuki S, Suzuki N, Modlin RL, Yeh WC, Lane TF, Cheng G. Toll-like receptors induce a phagocytic gene program through p38. J Exp Med. 2004;199:81–90. doi: 10.1084/jem.20031237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang HY, Qu P, Lu S, Liu M, Jiang H. The effect of TLR4/NF-kappaB activation and LOX-1 on monocyte adhesion to endothelium. Zhonghua Xin Xue Guan Bing Za Zhi. 2005;33:827–837. [PubMed] [Google Scholar]
- 40.Nandi S, Ma L, Denis M, Karwatsky J, Li Z, Jiang XC, Zha X. ABCA1-mediated cholesterol efflux generates microparticles in addition to HDL through processes governed by membrane rigidity. J Lipid Res. 2009;50:456–466. doi: 10.1194/jlr.M800345-JLR200. [DOI] [PubMed] [Google Scholar]
- 41.Dufton N, Hannon R, Brancaleone V, Dalli J, Patel HB, Gray M, D’Acquisto F, Buckingham JC, Perretti M, Flower RJ. Anti-inflammatory role of the murine formyl-peptide receptor 2: ligand-specific effects on leukocyte responses and experimental inflammation. J Immunol. 2010;184:2611–2619. doi: 10.4049/jimmunol.0903526. [DOI] [PMC free article] [PubMed] [Google Scholar]